

Abstract
Escherichia coli cold shock protein, CspA, folds very rapidly (time constant, τ = 4 msec) by an apparent two-state mechanism. However, recent time-resolved infrared (IR) temperature-jump experiments indicate that the folding trajectory of CspA may be more complicated. The sole tryptophan of wild-type CspA (Trp11), which is used to monitor the folding process by fluorescence spectroscopy, is located in an unusual aromatic cluster on the surface of CspA within the nucleic acid binding site. To gain a more global picture of the folding kinetics of CspA and to determine if there are any previously undetected intermediates, we have introduced a second tryptophan at three different surface locations in the protein. The three mutations did not significantly alter the tertiary structure of CspA, although two of the substitutions were found to be slightly stabilizing. Two-state folding, as detected by stopped-flow fluorescence spectroscopy, is preserved in all three mutants. These results indicate that the fast folding of CspA is driven by a concerted mechanism.
Keywords: Folding kinetics, two-state folding, sheet assembly, aromatic cluster, stopped-flow fluorescence, cold shock protein
Although considerable progress has been made in understanding the relationship between the primary sequence of a protein and its three-dimensional structure, the protein folding problem is far from understood. A good starting point is to study the folding of small, single-domain proteins to gain insight into the basic mechanisms of protein folding. It has been shown that a number of small, single-domain proteins fold on the millisecond time scale without detectable intermediates (Jackson and Ferst 1991;Viguera et al. 1994; Schindler et al. 1995; Jackson 1998). The study of these small proteins with simple folding trajectories both individually and as a set has led to considerable progress in understanding the early events that nucleate folding and the relationship between topology and folding kinetics (Nölting and Andert 2000; Plaxco et al. 2000). The model for our studies, Escherichia coli CspA, is one such protein (Fig. 1 ▶).
Fig. 1.

Two views of E. coli CspA (Schindelin et al. 1994) showing the relative location of the basic and aromatic nucleic acid binding site (upper left) and residues Trp11, Tyr42, Ser52, and Thr68. This figure was rendered using the MOLSCRIPT program (Kraulis 1991).
CspA is a small β-sheet protein that lacks complicating folding factors such as disulfide bonds, cofactors, or cis prolines (Fig. 1 ▶; Schindelin et al. 1994; Feng et al. 1998). It folds rapidly and, according to equilibrium and rapid mixing fluorescence experiments, via a two-state mechanism involving only native and denatured states (Reid et al. 1998). In addition, all cold shock proteins studied to date fold through an unusually compact, native-like, transition-state ensemble (Schindler et al. 1995; Schindler and Schmid 1996; Perl et al. 1998; Reid et al. 1998).
CspA possesses a cluster of six aromatic residues on the surface of β-strands 1, 2, and 3 that contribute to an unusually large nonpolar surface patch (790 Å2). These residues, together with several lysine side chains, comprise the single-stranded nucleic acid binding site of CspA (Fig. 1 ▶; Newkirk et al. 1994; Schindelin et al. 1994; Feng et al. 1998). The aromatic residues within this cluster are important for function and influence the stability and folding of CspA (Hillier et al. 1998; Rodriguez et al. 2000). The single tryptophan of wild-type CspA (Trp11), used to monitor folding by fluorescence, is located within this aromatic cluster. The thermodynamic parameters derived from equilibrium experiments are consistent with stopped-flow fluorescence experiments indicating that CspA folds by a two-state process with no intermediates (Reid et al. 1998).
Evidence that the folding of CspA may be more complicated than a simple two-state process comes from our recent time-resolved infrared (IR) temperature-jump (T-jump) studies (Thorn Leeson et al. 2000). The relaxation dynamics of CspA, which occur on the time scale of tens to hundreds of microseconds at temperatures near the thermal denaturation midpoint of CspA (∼59°C), are followed by probing the changes in amide I absorption (from the C=O stretch vibration) of the polypeptide backbone. Two kinetic phases are detected in the amide I band at 1632 cm−1 (ascribed to β-sheet structure) above a threshold temperature jump of at least ΔT = +12°C. The time constants of these fast and slow phases differ by factors of 3 to 5, depending on the exact starting and final temperatures, with a significant amplitude contribution from both phases. We have proposed that these two phases result from a shift in the location of the folded state on the energy landscape and not from the existence of a third energy minimum (Thorn Leeson et al. 2000). Such a mechanism would explain the order dependence of the kinetics on the size of the temperature jump.
It may be expected that similar biphasic kinetics would be observed in rapid mixing experiments when the change in denaturant concentration is large. However, we only observed a single phase in all unfolding and refolding experiments at all starting and final concentrations of denaturant (Reid et al. 1998; Rodriguez et al. 2000). One potential reason for the difference in results between our stopped-flow fluorescence studies and our T-jump IR studies is that Trp11, the fluorescence probe of CspA, only provides information about the dynamics of the aromatic cluster within which it resides. Therefore, the question we wish to resolve is: does CspA fold through an all-or-none transition to acquire its overall topology, or do the subdomains of CspA fold on separate time scales?
To investigate whether CspA in the vicinity of Trp11 folds on the same time scale as the rest of the protein, we have introduced additional tryptophan probes at three different surface locations that are distant from the aromatic cluster of the protein (Fig. 1 ▶). Ser52, Thr68, and Tyr42 were each substituted with tryptophan to create three mutants referred to as S52W, T68W, and Y42W, respectively. Trp11 was retained in these three mutants to preserve the stability of CspA, which is largely determined at the aromatic cluster region (Hillier et al. 1998), and to preserve a method for measuring nucleic acid binding affinity and hence structural integrity. Ser52 and Thr68 are on β-strands 4 and 5, which are situated on the back side of CspA relative to the aromatic cluster nucleic acid binding site. Tyr42, the only tyrosine of CspA, is on loop L3 separating β-strands 1–3 from β-strands 4 and 5. We selected this site because of our findings with the steady-state and time-resolved IR data recorded at 1623 cm−1 (reflecting contributions from both the loop and β-sheet structure), which show very different kinetics and equilibrium melting behavior from those measured at 1632 cm−1 (reflecting mainly β-sheet structure; Thorn Leeson et al. 2000). Tyrosine has a unique vibrational mode at 1603 cm−1. Because the temperature dependence of the 1623 cm−1 component is similar to that of the tyrosine mode, we assigned this amide I component to loop L3 and speculated that the metastable state of CspA detected at T-jumps greater than 12°C may be attributed to some behavior of loop L3. A more intensely fluorescent probe such as tryptophan placed at this location may reveal similar behavior using stopped-flow fluorescence spectroscopy.
Results
Structural and functional characterization of the tryptophan mutants
The tryptophan substitutions introduced at positions 42, 52, and 68 did not significantly alter the native structure of CspA. The far-UV CD spectra of Y42W, S52W, T68W, and wild-type CspA are shown in Figure 2 ▶. The characteristic negative ellipticity at ∼218 nm expected for a β-sheet structure is masked by the ordered aromatic residues in wild-type CspA (Chatterjee et al. 1993; Reid et al. 1998). The CD spectra of Y42W, S52W, and T68W all exhibited positive ellipticity in the far-UV region similar to wild-type CspA, indicating there is little disruption of structure in the tryptophan mutants.
Fig. 2.

Far-UV–CD spectra of native wild-type CspA (filled circles), Y42W (plus signs), S52W (open squares), and T68W (open triangles). The protein concentration was approximately 50 μM, and the buffer contained 50 mM potassium phosphate, 100 mM KCl at pH 7.0. The spectra were recorded at 25°C in a 1-mm cuvette.
The affinity of single-stranded DNA to the tryptophan substitution mutants was determined using fluorescence quenching studies. Wild-type CspA was previously shown to bind to the oligonucleotide 5`-CTTGAGGTTAATCCA-3`. Binding quenches the fluorescence of Trp11, thereby allowing the determination of binding affinity as described (Hillier et al. 1998). The binding constants for Y42W (KD = 3 ± 1 μM), S52W (KD = 4 ± 1 μM), and T68W (KD = 6 ± 1 μM) were within error of the binding constant determined for wild-type CspA (KD = 6 ± 1 μM) (Hillier et al. 1998). These results imply that the mutations do not cause structural changes that are propagated to the nucleic acid binding region of CspA. As a point of reference, the somewhat conservative mutation of any one of the three phenylalanines to leucines in the nucleic acid binding site causes significant changes to the binding affinity (KDs in the range of 19–55 μM) (Hillier et al. 1998). Taken together, the far-UV CD and fluorescence binding studies results strongly indicate that the native state structures of the tryptophan mutants are similar to wild-type CspA.
Temperature- and denaturant-induced equilibrium unfolding
Fluorescence spectroscopy was used to probe the tryptophan environments during the equilibrium unfolding process. Monitoring the intensity and emission maximum of tryptophan reveals the relative polarity change of the probe during the unfolding process. Following excitation at 280 or 295 nm, the emission spectrum of wild-type CspA exhibits maximum emission intensity at 350 nm (Chatterjee et al. 1993; Reid et al. 1998). Upon unfolding, the emission intensity of Trp11 decreases about 50% along with a slight (4-nm) red shift (Chatterjee et al. 1993; Reid et al. 1998). The fluorescence intensities of Y42W, S52W, and T68W are approximately twofold larger than wild-type CspA, as expected for proteins containing an additional tryptophan (Fig. 3 ▶). The wavelengths of maximum emission for the mutants are all at 350 nm, indicating that the additional fluorophores at positions 42, 52, and 68 are all in relatively polar environments like the Trp11 in wild-type CspA. The fluorescence intensities of the denatured Y42W, S52W, and T68W mutants in 8 M urea are also twofold higher than denatured wild-type CspA with a slight red shift of the emission maximum to 356 nm (Fig. 3 ▶).
Fig. 3.

Fluorescence intensity spectra of native (0 M urea) and denatured (8 M urea) wild-type CspA (native, filled circles; denatured, open circles), Y42W (native, plus signs; denatured, times signs), S52W (native, filled squares; denatured, open squares), and T68W (native, filled triangles; denatured, open triangles). The excitation wavelength was 280 nm. The protein concentration was 3.5 μM, and the buffer contained 50 mM potassium phosphate, 100 mM KCl at pH 7.0 for all samples.
Figure 4 ▶ shows the fraction unfolded (monitored by the change in fluorescence emission intensity) as a function of urea concentration at 25°C for Y42W, S52W, T68W, and wild-type CspA. A single transition between the native and the denatured states is observed for all the tryptophan mutants and wild-type CspA. The meq values for Y42W, S52W, and T68W (0.72–0.77 kcal/mol−1 M−1) show relatively similar dependence of unfolding free energy on urea concentration to wild-type CspA (meq = 0.71 kcal/mol−1 M−1; Table 1). Because the meq value reflects the difference in the amount of denaturant bound to the denatured and native states, this reflects the difference in solvent accessibility between these two states (Schellman 1978). Therefore, the overall changes in solvent-exposed hydrophobic surface area upon unfolding of the tryptophan mutants are similar to wild-type CspA. The meq values for guanidinium chloride-dependent equilibrium unfolding of the tryptophan mutants are also very close to one another (Table 1).
Fig. 4.

Urea-induced denaturation of wild-type CspA (filled circles), Y42W (plus signs), S52W (open squares), and T68W (open triangles) expressed as the fraction unfolded. Denaturation was followed by fluorescence emission intensity at 350 nm (excitation at 280 nm). The protein concentration was 3.5 μM in 50 mM potassium phosphate, 100 mM KCl at pH 7.0, at 25°C.
Table 1.
Equilibrium thermodynamic parameters (25°C)
| ΔGH2O (kcal mol−1) Urea | ΔGH2O (kcal mol−1) GdmCl | Cm (M) Urea | Cm (M) GdmCl | meq (kcal mol−1 M−1) Urea | meq (kcal mol−1 M−1) GdmCl | Tm (°C) CD | |
| Wild-type CspA | 3.0 ± 0.2 | 3.1 ± 0.1 | 4.2 ± 0.1 | 1.4 ± 0.2 | 0.71 ± 0.04 | 2.2 ± 0.1 | 59 ± 1 |
| Y42W | 3.2 ± 0.1 | 3.2 ± 0.1 | 4.3 ± 0.2 | 1.5 ± 0.1 | 0.72 ± 0.02 | 2.3 ± 0.1 | 63 ± 1 |
| S52W | 2.8 ± 0.1 | 2.9 ± 0.2 | 3.8 ± 0.2 | 1.3 ± 0.1 | 0.74 ± 0.01 | 2.3 ± 0.1 | 56 ± 1 |
| T68W | 3.7 ± 0.1 | 3.8 ± 0.2 | 4.8 ± 0.2 | 1.7 ± 0.1 | 0.77 ± 0.02 | 2.3 ± 0.1 | 63 ± 1 |
The three mutants show fairly similar effects on the free energy of unfolding (ΔGH2O) and midpoints of denaturation (Cm) compared to wild-type CspA. The stability of Y42W (ΔGH2O = 3.2 kcal/mol−1) and S52W (ΔGH2O 2.8 kcal/mol−1) are somewhat comparable to wild-type CspA, and their Cm values reflect their modest differences. T68W has a slightly higher free energy of unfolding (ΔGH2O = 3.7 kcal/mol−1, Cm = 4.8 M urea) than wild-type CspA (ΔGH2O =3.0 kcal/mol−1, Cm = 4.2 M urea). The amount of additional stability observed for T68W (0.7 kcal/mol−1 by urea) is noteworthy considering CspA is a small protein and the substitution is nonconservative. Similar values of the free energy of unfolding were also observed for the change in fluorescence emission intensity upon titration with guanidinium chloride (GdmCl; Table 1).
Using CD, the thermal denaturation profiles of Y42W, S52W, and T68W were determined by monitoring the change in ellipticity at 222 nm, near a peak in the spectrum of folded CspA (Fig. 2 ▶), as a function of temperature. At this wavelength, there is a local maximum in the CD spectrum of wild-type CspA. All three mutants exhibited a single transition and showed reversibility to thermal denaturation. The midpoints of thermal denaturation of Y42W (Tm = 63°C), S52W (Tm = 56°C), and T68W (Tm = 63°C) compared to wild-type CspA (Tm = 59°C) were consistent with the stability trends observed for denaturant titrations.
Unfolding and folding kinetics of mutants determined by stopped-flow fluorescence
Stopped-flow fluorescence experiments using urea as a denaturant were performed to probe the effects the tryptophan substitutions had on the kinetics of folding. Unfolding and refolding of CspA were initiated by a rapid dilution of the concentrated native and unfolded proteins, respectively, into the final buffered urea concentrations. We chose urea for these experiments instead of GdmCl because in wild-type CspA the denaturation midpoint occurs at a higher concentration for urea (4.2 M) than for GdmCl (1.5 M) (Reid et al. 1998). This extends the concentration range within which refolding-dominated reactions can be observed.
The refolding and unfolding traces of Y42W, S52W, and T68W are described well by monoexponential functions of time (Fig. 5A,B,C ▶). This is true for refolding jumps of all magnitude of urea concentrations. The dependences of the natural logarithm of the refolding and unfolding rate constants plotted as a function of urea (commonly referred to as chevron plots) are shown in Figure 6 ▶. The extrapolated rate constants for refolding and unfolding were obtained from equation 1 and are shown in Table 2 (Tanford 1970; Matthews and Hurle 1987; Chen et al. 1992). Y42W and T68W refold faster than wild-type CspA with extrapolated rate constants of 459 sec−1 and 355 sec−1, respectively (vs. 264 sec−1 for wild-type CspA). The extrapolated unfolding rate is 0.69 sec−1 for T68W, which is slower than wild-type CspA (ku = 1.9 sec−1), whereas the unfolding rate for Y42W (1.7 sec−1) is similar to that for wild-type CspA. The free energies of unfolding for Y42W and T68W calculated from kinetic data (ΔG = 3.3 kcal/mol−1 and 3.7 kcal/mol−1, respectively) agree well with the values obtained from equilibrium experiments (Table 1). The S52W mutant has an extrapolated refolding rate constant of 247 sec−1, which is similar to that for wild-type CspA. The unfolding rate constant for S52W is 2.4 sec−1. The free energy of unfolding of S52W determined from kinetic data (ΔG = 2.8 ± 0.2 kcal/mol−1) is also in good agreement with equilibrium experiments. Within the limits of our instrument, the total amplitudes of the fluorescence changes were recovered in the refolding traces of all the mutants at low urea concentrations.
Fig. 5.
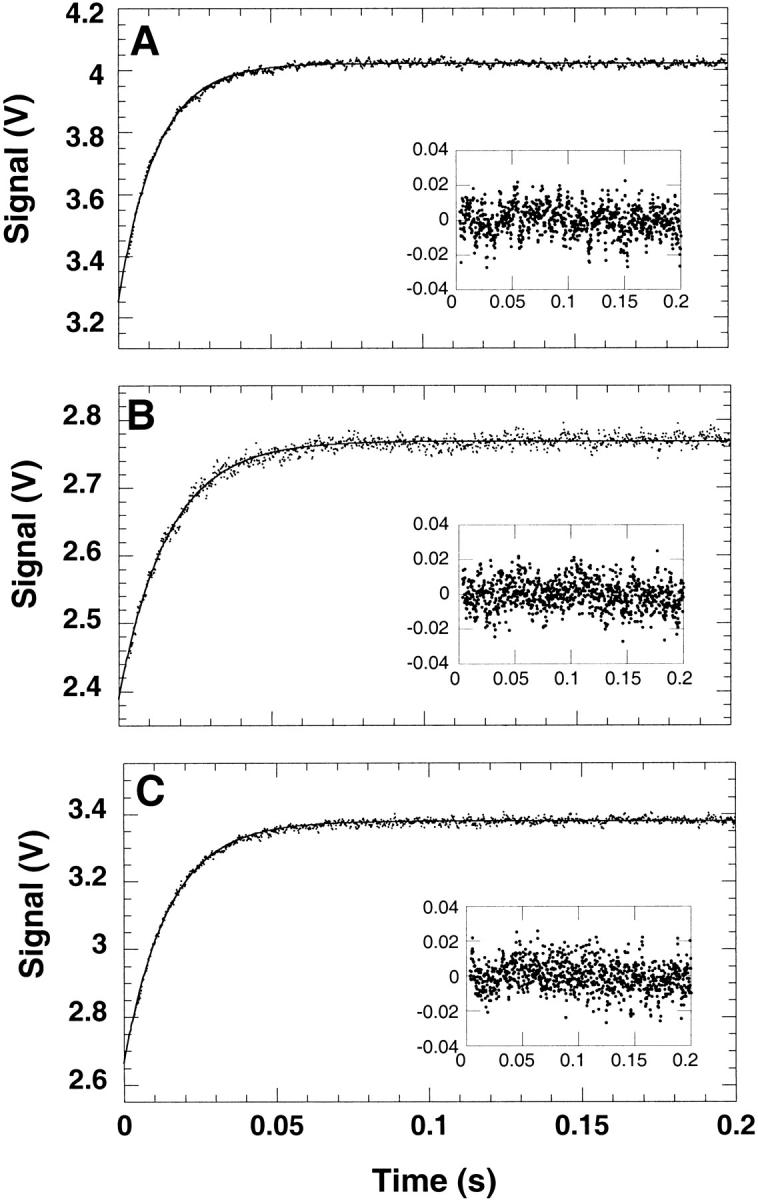
Representative refolding traces of (A) Y42W, (B) S52W, and (C) T68W. The insets show the residuals of the fit. Refolding was initiated by a 10-fold dilution of the mutants in Buffer A from 7 M urea to a final urea concentration of 2 M. The kinetics were measured by the change in fluorescence above 325 nm at a final protein concentration of 5 μM at 25°C. The observed apparent rate constants (λ) were 80 sec−1, 60 sec−1, and 66 sec−1 for Y42W, S52W, and T68W, respectively.
Fig. 6.

Urea dependence of the observed folding rates of wild-type CspA (filled circles), Y42W (plus signs), S52W (open squares), and T68W (open triangles). Curve fits were obtained as described in Materials and Methods.
Table 2.
Kinetic parameters obtained from fluorescence stopped-flow experiments (25°C)
| ΔGkin (kcal mol−1) | kf (s−1) | mf (M−1) | ku (s−1) | mu (M−1) | mkin (kcal mol−1 M−1) | α‡ | |
| Wild-type CspA | 2.9 ± 0.1 | 264 ± 16 | −0.87 ± 0.01 | 1.9 ± 0.2 | 0.14 ± 0.05 | 0.60 ± 0.07 | 0.86 ± 0.07 |
| Y42W | 3.3 ± 0.1 | 459 ± 05 | −0.92 ± 0.02 | 1.7 ± 0.1 | 0.20 ± 0.01 | 0.66 ± 0.02 | 0.82 ± 0.02 |
| S52W | 2.8 ± 0.1 | 247 ± 10 | −0.74 ± 0.01 | 2.4 ± 0.1 | 0.27 ± 0.02 | 0.60 ± 0.02 | 0.78 ± 0.02 |
| T68W | 3.7 ± 0.1 | 355 ± 04 | −0.91 ± 0.01 | 0.7 ± 0.1 | 0.27 ± 0.01 | 0.70 ± 0.02 | 0.73 ± 0.02 |
mkin = RT • (mu − mf).
The slopes of the dependence of the refolding and unfolding rates on denaturant concentration (i.e., the left and right branches of the chevron plot) are thought to correlate with the change of solvent exposure among the unfolded, transition, and folded states (Tanford 1968). A small slope in the unfolding reaction is observed for wild-type CspA (0.14 M−1). This small dependence of the unfolding rate on urea concentration indicates that there is little difference in solvent exposure between the transition and the native state for wild-type CspA. A quantitative estimate of the relative difference in solvent accessibilities of the transition state of a protein may be inferred from the value of α‡ (equation 2; Table 2). By this measure, the transition state ensemble of wild-type CspA is 86% native-like. Although the unfolding rates show more dependence on urea concentration for Y42W (mu = 0.20 M−1), T68W, and S52W (both mu = 0.27 M−1), these values are comparably small and similar to wild-type CspA. These small mu values translate into transition states of Y42W, S52W, and T68W that are more than 73% native-like (Table 2) and therefore have comparable compactness to wild-type CspA. The denaturant dependencies of the folding transitions as a whole (mkin), as calculated from the slopes of the unfolding and refolding branches of the chevron plot (Table 2), do not agree well with the equilibrium data (meq; Table 1). The poor agreement in equilibrium and kinetic m-values may stem from the error associated with fitting a curve to the shallow slopes of the unfolding branches of the chevron plots of CspA and its mutants.
Discussion
A growing number of small, single-domain proteins have been shown to fold rapidly and by an apparent two-state mechanism (Jackson 1998; Plaxco et al. 2000). In this respect, the observed two-state folding of CspA is not exceptional. However, because of the unusual location of the tryptophan fluorophore in the solvent-exposed aromatic cluster of CspA and the possibility that this cluster may be involved in the folding transition state of CspA (Rodriguez et al. 2000), the folding of CspA as a whole may not be reflected accurately by this intrinsic probe. In addition, our T-jump studies detect the presence of an intermediate that we speculate involves structural changes in loop L3 (Thorn Leeson et al. 2000). In this study, we introduced a second tryptophan in three separate surface locations that are remote from wild-type Trp11 (Fig. 1 ▶) to follow the folding of different regions of CspA.
The additional tryptophans at positions 42, 52, and 68 are good reporters of structure on loop L3 as well as the back side of CspA with respect to the aromatic cluster. In all three mutants, the substitution of the tryptophan residues resulted in minimal perturbations to the stability and the function of CspA. The additional tryptophan of all of the mutants and Trp11 contribute equally to the fluorescence emission intensity in both the native and denatured states. The emission maxima of Y42W, S52W, and T68W in the native states are similar to wild-type CspA and indicate that these introduced tryptophans are in partially solvent-exposed environments similar to Trp11.
We find no evidence of a three-state folding process for CspA, as all the tryptophan mutants are observed to fold by two-state mechanisms. The refolding and unfolding traces of Y42W, S52W, and T68W are described well by monoexponential processes (Fig. 5A,B,C ▶), and no intermediates were detected using stopped-flow fluorescence spectroscopy for all the mutants. If Trp42, Trp52, or Trp68 were reporting on different subdomains of CspA than Trp11, it would have been difficult to model the refolding traces using monoexponential functions (Fig. 5A,B,C ▶) because each tryptophan appears to contribute equally to the fluorescence emission intensity. In the T-jump experiments, the fast phase accounts for a significant fraction of the amplitude (20%–50%). If we were missing a faster phase within the mixing dead time of the stopped-flow experiments, we should detect a considerable loss of amplitude in the refolding traces. Within the limits of our experiment, the full amplitudes expected are accounted for in the refolding traces for all mutants when going from high to low urea concentration.
The rate constants for unfolding and refolding are also linearly dependent on the concentration of denaturant (Jackson and Fersht 1991) and show no "rollover" kinetics in the refolding arm of the chevron plots, indicative of a stable folding intermediate populated at low urea concentration. Although the inconsistency in the m-values from kinetic and equilibrium experiments could indicate the presence of intermediates, the poor agreement in m-values may stem from the difficulty of accurately estimating the uncharacteristically shallow slopes of the unfolding branches of the chevron plots of CspA and its mutants. In addition, the free energy of unfolding values obtained from equilibrium data are in excellent agreement with values obtained from kinetic experiments. Because the tryptophan substitutions report on different regions of CspA, and because we observe no major changes in the solvent accessibility of the transition-state ensembles of the tryptophan mutants compared to wild-type CspA, we can conclude that global folding of CspA follows a two state-folding mechanism with a compact native-like transition-state ensemble.
The two-state mechanism is in potential conflict with the findings of the T-jump studies on CspA, which detect two phases by IR spectroscopy for T-jumps larger than 12°C (Thorn Leeson et al. 2000). We can conceive of three hypotheses to explain the discrepancy: (1) The thermally denatured state is very different from the chemically denatured state, and refolding and unfolding between the native state and either of these denatured states follow completely different kinetics. There is some preliminary support for this hypothesis in that the thermally denatured state ensemble appears to be unusually compact (F. Gai, S. Gallagher, J. Trewhella, and R.B. Dyer, unpubl.). (2) The assembly of the tertiary structure of CspA (i.e., the side-chain contacts and overall topology) precedes the formation of a majority of the hydrogen bonds. The β-clam protein, CRABPI, sets a precedent for such a mechanism (Clark et al. 1997). (3) Because β-strands 2 and 3 contain the majority of the aromatic residues, the region between residues 11 and 42 could be a subdomain of CspA that folds independently from the rest of the protein. Our study did not extend the tryptophan probes to β-strands 2 and 3 because mutations in this particular region have been shown to decrease the function and stability of CspA (Hillier et al. 1998; Rodriguez et al. 2000). However, this possibility cannot be ruled out, especially considering the importance of this region to stability. Further investigation of the Y42W, S52W, and T68W mutants by T-jump fluorescence and IR spectroscopy may yield more details about the folding mechanisms of these mutants.
To summarize, we have shown that the folding of different regions of CspA can be followed by introducing tryptophan probes at solvent-exposed, surface positions in conjunction with fluorescence spectroscopy. Apparent two-state folding is observed for all the tryptophan mutants of CspA, suggesting that the tertiary structure of this small protein folds as a cooperative unit.
Materials and methods
GdmCl and urea analytical grade were purchased from ICN Biomedicals. All other buffer and purification reagents were of analytical grade and purchased from Fisher Scieic. Ni-NTA agarose used to purify the histidine-tagged CspA was purchased from QIAGEN. Tobacco Etch Virus (TEV) protease was cloned and produced in-house (B. Hillier and L. Gregoret, unpubl.). Protein concentration was determined using a UV-visible spectrophotometer. The extinction coefficients were calculated (Pace et al. 1995) to be 8437 M−1 cm−1 for wild-type CspA, 12,400 M−1 cm−1 for Y42W, and 14,000 M−1 cm−1 for T68W and S52W.
Cloning of Y42W, S52W, and T68W
Construction of the gene coding for CspA with six histidines at the amino terminus followed by a TEV protease cleavage site has been described previously (Hillier et al. 1998). Y42W, S52W, and T68W mutants were created using cassette mutagenesis. The mutagenic oligonucleotides were synthesized by standard phosphoramidite chemistry on a Millipore Expedite nucleic acid synthesizer. The gene sequences were verified using the dideoxy chain termination method (Sanger et al. 1977). The resulting plasmids were transformed into E. coli strain BL21 (DE3).
Protein purification
The proteins were purified under denaturing conditions using the QIAGEN Ni-NTA resin for purification of proteins containing histidine tags as described (Reid et al. 1998). The six-histidines tag was removed using the TEV protease as described (Hillier et al. 1998). The molecular masses of the proteins were confirmed using a MicroMass Quattro II electrospray mass spectrometer. The masses obtained for Y42W (7300 D), S52W (7370 D), and T68W (7360 D) were consistent with the expected masses.
DNA binding determined by fluorescence
The binding of the oligonucleotide (5`-CTTGAGGTTAATCCA-3`) to the tryptophan mutants was determined by measuring the quenching of the fluorescence of Trp11 using a LS50B Perkin Elmer fluorimeter. Excitation was at 295 nm, and the emission intensity was measured at 350 nm. The oligonucleotide was synthesized using a Millipore Expedite nucleic acid synthesizer. The extinction coefficient used to determine the oligonucleotide concentration was ɛ260 = 141.6 mM−1 cm−1. Concentrated DNA was titrated into a 1-mL cuvette in Buffer A (50 mM potassium phosphate, 100 mM KCl at pH 7.0) at 25°C, containing 3.5 μM protein. The observed intensity was corrected for dilution and for the inner filter effect using free tryptophan (Birdsall et al. 1983) as described previously (Hillier et al. 1998).
GdmCl and urea-induced denaturation followed by fluorescence
Fluorescence measurements were performed on a Perkin Elmer LS50B fluorometer by exciting the tryptophan at 280 nm and monitoring its emission at 350 nm. A 10-nm slit width was used for the excitation and emission beams employing a 1% transmission filter. Native samples containing 3.5 μM protein in Buffer A were placed in a 1-mL fluorescence quartz cuvette. Denatured samples were comprised of 3.5 μM protein in Buffer A with either 8 M urea or 6 M GdmCl. All experiments were performed at 25°C unless otherwise noted. GdmCl and urea titrations were performed as described (Reid et al. 1998). The relative change in fluorescence intensity with increasing urea concentration was determined using the Pace method (Pace 1986), which assumes a two-state transition. The values of ΔGH2O, the extrapolated free energy of unfolding in the absence of denaturant, and meq, the dependence of ΔG on urea concentration, were obtained by a six-parameter fit as described previously (Reid et al. 1998).
Circular dichroism studies
Far-UV–CD denaturation experiments were performed on an Aviv Associates model 60DS spectropolarimeter. Spectra were recorded from 195 to 260 nm using a 1-mm path length cuvette. Data were collected every 1 nm, with a 5-sec averaging time and 1.5-nm bandwidth. Three wavelength scans were averaged to obtain each spectrum. Protein concentrations were between 40 and 50 μM.
Thermal denaturation of wild-type CspA and the tryptophan mutants was monitored by measuring the change in ellipticity at 222 nm with increasing temperature and recorded as described (Reid et al. 1998). The temperature was increased at a constant rate of 0.33°C/min using a programmable water bath (Neslab). Thermal denaturation curves were fit using a nonlinear least-squares method assuming a two-state transition as described (Reid et al. 1998).
Stopped-flow fluorescence
All folding and unfolding reactions were followed at 25°C using a Biologic SFM-2 stopped-flow apparatus, and Biokine 3.0 software (Molecular Kinetics) was used for data acquisition. Refolding was initiated by rapid 10-fold dilution of concentrated protein in 7 M urea into Buffer A without urea and with varying concentrations of urea. Unfolding reactions were initiated by diluting the native protein in Buffer A into different buffered urea concentrations. The fluorescence change was monitored using a 325-nm cutoff filter following excitation at 280 nm using a 2-mm slit width. Final protein concentrations were between 3 and 6 μM. The apparent rate constants for the unfolding and refolding reactions were obtained from the average of at least 10 traces. These data were fit to a monoexponential time course using nonlinear least-squares regression as implemented by Kaleidagraph software (Synergy Software).
Analysis of folding kinetics
The extrapolated rate constants for unfolding, ku, and refolding, kf, were obtained assuming a two-state transition. The natural logarithms of ku and kf vary linearly with urea concentration according to equation 1 and reported in Table 2.
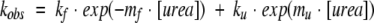 |
(1) |
The slopes of the unfolding and refolding rates as a function of urea concentration are mu and mf. These slopes are thought to be proportional to the change in solvent-accessible surface area between the native and transition states and between the denatured and transition states, respectively. If the folding and unfolding of a protein follow a two-state mechanism, the solvent accessibility of the transition state relative to the folded state is given by equation 2 and reported in Table 2 (Tanford 1970; Chen et al. 1989).
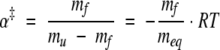 |
(2) |
Acknowledgments
We thank Brian Dyer for helpful discussions and Chuck Wilson for use of his laboratory's DNA synthesizer. This work was supported by National Institutes of Health grant GM52885-R29 and a Collaborative University/Los Alamos Research Grant from the Science and Technology Base at Los Alamos National Labs. D.V. is supported by a GAANN graduate fellowship from the U.S. Department of Education. The purchase of the mass spectrometer used in this study was made possible by a generous gift to UCSC from the W.M. Keck Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.16201.
References
- Birdsall, B., King, R.W., Wheeler, M.R., Lewis, C.A., Goode, S.R., Dunlap, R.B., and Roberts, G.C.K. 1983. Correction for light absorption in fluorescence studies of protein–ligand interactions. Analyt. Biochem. 132:353–361. [DOI] [PubMed] [Google Scholar]
- Chatterjee, S., Jiang, W., Emerson, S.D., and Inouye, M. 1993. The backbone structure of the major cold-shock protein CS7.4 of Escherichia coli in solution includes extensive β-sheet structure. J. Biochem. 114:663–669. [DOI] [PubMed] [Google Scholar]
- Chen, B., Baase, W.A., and Schellman, J.A. 1989. Low-temperature unfolding of a mutant of phage T4 lysozyme. 2. Kinetic Investigations. Biochemistry 28:691–699. [DOI] [PubMed] [Google Scholar]
- Chen, B.L., Baase, W.A., Nicholson, H., and Schellman, J.A. 1992. Folding kinetics of T4 lysozyme and nine mutants at 12 degrees C. Biochemistry 31:1464–1476. [DOI] [PubMed] [Google Scholar]
- Clark, P.L., Lie, Z.-P., Rizo, J., and Gierasch, L.M. 1997. Cavity formation before stable hydrogen bonding in the folding of a β-Clam protein. Nature Struct. Biol. 4:883–886. [DOI] [PubMed] [Google Scholar]
- Feng, W., Tejero, R., Zimmerman, D.E., Inouye, M., and Montelione, G.T. 1998. Solution NMR structure and backbone dynamics of the major cold-shock protein (CspA) from Escherichia coli: Evidence for conformational dynamics in the single-stranded RNA-binding site. Biochemistry 37:10881–10896. [DOI] [PubMed] [Google Scholar]
- Hillier, B.J., Rodriguez, H.M., and Gregoret, L.M. 1998. Coupling of protein stability and protein function in E. coli CspA. Folding & Design 3:87–93. [DOI] [PubMed] [Google Scholar]
- Jackson, S.E. 1998. How do small single-domain proteins fold? Folding & Design 3:R81–R91. [DOI] [PubMed] [Google Scholar]
- Jackson, S.E. and Fersht, A.R. 1991. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry 30: 10428–10435. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT—A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystal. 24:946–950. [Google Scholar]
- Matthews, C.R. and Hurle, M.R. 1987. Mutant sequences as probes of protein folding mechanisms. BioEssays 6: 254–257. [DOI] [PubMed] [Google Scholar]
- Newkirk, K., Feng, W., Jiang, W., Tejero, R., Emerson, S.D., Inouye, M., and Montelione, G.T. 1994. Solution NMR structure of the major cold shock protein (CspA) from Escherichia coli: Ideication of a binding epitope for DNA. Proc. Natl. Acad. Sci. USA 91:5114–5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nölting, B. and Andert, K. 2000. Mechanism of protein folding. PROTEINS: Structure, Function and Genetics 41: 288–298. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131:266–280. [DOI] [PubMed] [Google Scholar]
- Pace, C.N., Vajdos, F., Fee, L., Grimsley, G., and Gray, T. 1995. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4: 2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl, D., Welker, C., Schindler, T., Schroeder, K., Maraheil, M.A., Jaenicke, R., and Schmid, F.X. 1998. Conservation of rapid two-state folding in mesophilic, thermophilic, and hyperthermophilic cold shock proteins. Nature Struct. Biol. 5: 229–235. [DOI] [PubMed] [Google Scholar]
- Plaxco, K.W., Simons, K.T., Ruczinski, I., and Baker, D. 2000. Topology, stability, sequence, and length: Defining the determinants of two-state protein folding kinetics. Biochemistry 39: 11177–11183. [DOI] [PubMed] [Google Scholar]
- Reid, K.L., Rodriguez, H.M., Hillier, B.J., and Gregoret, L.M. 1998. Stability and folding properties of a model β-sheet protein, Escherichia coli CspA. Protein Sci. 7: 470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez, H.M., Vu, D.M., and Gregoret, L.M. 2000. Role of a solvent-exposed aromatic cluster in the folding of Escherichia coli CspA. Protein Sci. 9: 1993–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger, F., Nicklen, S., and Coulson, A.R. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74: 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellman, J.A. 1978. Solvent denaturation. Biopolymers 17: 1305–1322. [DOI] [PubMed] [Google Scholar]
- Schindelin, H., Jiang, W., Inouye, M., and Heinemann, U. 1994. Crystal structure of CspA, the major cold shock protein of Escherichia coli. Proc. Natl. Acad. Sci. USA 91:5119–5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler, T. and Schmid, F.X. 1996. Thermodynamic properties of an extremely rapid protein folding reaction. Biochemistry 35:16833–16842. [DOI] [PubMed] [Google Scholar]
- Schindler, T., Herrler, M., Maraheil, M.H., and Schmid, F.X. 1995. Extremely rapid protein folding in the absence of intermediates. Nature Struct. Biol. 2:663–673. [DOI] [PubMed] [Google Scholar]
- Tanford, C. 1968. Protein denaturation. Adv. Protein Chem. 23: 121–282. [DOI] [PubMed] [Google Scholar]
- Tanford C. 1970. Protein denaturation. C. Theoretical models for the mechanism of denaturation. Adv. Protein Chem. 24:1–95. [PubMed] [Google Scholar]
- Thorn Leeson, D., Gai, F., Rodriguez, H.M., Gregoret, L.M., and Dyer, R.B. 2000. Protein folding and unfolding on a complex energy landscape. Proc. Natl. Acad. Sci. USA 97:2527–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viguera, A.R., Martinez, J.C., Filimonov, V.V., Mateo, P.L., and Serrano, L. 1994. Thermodynamic and kinetic analysis of the SH3 domain of spectrin shows a two-state folding transition. Biochemistry 33: 2142–2150. [DOI] [PubMed] [Google Scholar]