

Abstract
Rat corticotropin-releasing factor receptor 1 (rCRFR1) was produced either in transfected HEK 293 cells as a complex glycosylated protein or in the presence of the mannosidase I inhibitor kifunensine as a high mannose glycosylated protein. The altered glycosylation did not influence the biological function of rCRFR1 as demonstrated by competitive binding of rat urocortin (rUcn) or human/rat corticotropin-releasing factor (h/rCRF) and agonist-induced cAMP accumulation. The low production rate of the N-terminal domain of rCRFR1 (rCRFR1-NT) by transfected HEK 293 cells, was increased by a factor of 100 in the presence of kifunensine. The product, rCRFR1-NT-Kif, bound rUcn specifically (KD = 27 nM) and astressin (KI = 60 nM). This affinity was 10-fold lower than the affinity of full length rCRFR1. However, it was sufficiently high for rCRFR1-NT-Kif to serve as a model for the N-terminal domain of rCRFR1. With protein fragmentation, Edman degradation, and mass spectrometric analysis, evidence was found for the signal peptide cleavage site C-terminally to Thr23 and three disulfide bridges between precursor residues 30 and 54, 44 and 87, and 68 and 102. Of all putative N-glycosylation sites in positions 32, 38, 45, 78, 90, and 98, all Asn residues except for Asn32 were glycosylated to a significant extent. No O-glycosylation was observed.
Keywords: Corticotropin-releasing factor, CRF receptor, human embryonic kidney cells, scintillation proximity assay, amino-terminal domain, binding domain, disulfide bond structure, glycosylation structure
Corticotropin-releasing factor (CRF), a peptide 41 amino acids long (Spiess et al. 1981), is released from the hypothalamus into the hypophyseal portal system and stimulates ACTH secretion from the pituitary (Vale et al. 1981) as an endocrine response to stress. In addition to CRF, the CRF-like 40-amino acid peptide urocortin (Ucn) has been characterized (Vaughan et al. 1995; Donaldson et al. 1996). CRF and Ucn are distributed widely throughout the CNS of rodents and humans (Eckart et al. 1999), where they modulate various central functions such as locomotor activity, food intake, anxiety, and learning (Eckart et al. 1999; Radulovic et al. 1999). Furthermore, pathophysiological changes in the CRF system have been associated with several neuropsychiatric disorders such as major depression, panic disorder, anorexia nervosa, and Alzheimer's disease (Behan et al. 1996).
CRF and Ucn exert their biological actions by binding to two CRF receptor (CRFR) subtypes, CRFR1 and CRFR2. CRF receptors belong to the class of G protein-coupled receptors (GPCR) which possess four extracellular, four intracellular, and seven transmembrane domains (Radulovic et al. 1999). They are coupled to G proteins mainly stimulating the production of cAMP as second messenger. In different species, the CRFR1 precursor consists of 415 to 420 amino acids (Chang et al. 1993; Chen et al. 1993; Perrin et al. 1993; Vita et al. 1993; Yu et al. 1996; Dautzenberg et al. 1997; Myers et al. 1998; Palchaudhuri et al. 1998) and is expressed mainly in the brain and pituitary (Potter et al. 1994). Several splice variants of CRFR2 have been ideied: CRFR2α, CRFR2β, and CRFR2γ. They consist, depending upon the species, of 410–413 (α), 431–438 (β), and 397 (γ) amino acids (Kishimoto et al. 1995; Lovenberg et al. 1995; Perrin et al. 1995; Stenzel et al. 1995; Liaw et al. 1996; Dautzenberg et al. 1997; Kostich et al. 1998; Palchaudhuri et al. 1999). CRFR2 is found in discrete regions of the brain and peripheral organs (Chalmers et al. 1995; Stenzel et al. 1995).
Two independent studies indicate that the N-terminal domain of CRFR1 is essential for ligand recognition. Dautzenberg et al. (1998) made use of the unusual binding properties of Xenopus leavis CRFR1 (xCRFR1) which binds ovine CRF (oCRF) and the amphibian CRF analog sauvagine (Svg) (Montecucchi and Henschen, 1981) with significantly lower affinity than hCRFR1 (Dautzenberg et al. 1997). In experiments with chimeric receptors of xCRFR1 and hCRFR1, it was shown that the N-terminal domain (NT) of xCRFR1 is responsible for the ligand selectivity of xCRFR1 (Dautzenberg et al. 1998). Perrin et al. (1998) constructed a chimeric receptor composed of the N-terminal part of rCRFR1-NT connected to the transmembrane and intracellular domains of the activin II B receptor (Perrin et al. 1998). This chimeric receptor bound rat Ucn (rUcn) and astressin (Ast), a peptidic CRFR antagonist (Gulyas et al. 1995). In the same study, it was observed that chimeras composed of rCRFR1 and the GPCR rat growth hormone-releasing factor receptor, which contained the N-terminal domain of rCRFR1, bound Ucn and Ast with high affinity. Therefore, it was suggested that only the N-terminal domain of rCRFR1 was required for high affinity binding of Ucn and Ast.
It is known that the extracellular cysteines of CRFR1 are critical for binding of CRF (Qi et al. 1997). Chemical reduction of the disulfide bonds of mouse CRFR1 (mCRFR1) decreased the specific binding of h/rCRF significantly (Qi et al. 1997). Additionally, several single and paired mutations of cysteine residues to serine or alanine were introduced and the biological activity of the mutated receptors was analyzed. On the basis of these data, a pattern of disulfide linkages was proposed (Qi et al. 1997).
The objective of this study was to develop a model of rCRFR1. Therefore, the N-terminal domain of rCRFR1 (rCRFR1-NT) was produced as a soluble protein in human embryonic kidney (HEK) 293 cells transfected with cDNA coding for rCRFR1-NT. The production of biologically functional full length rCRFR1 in these cells has been demonstrated (Dautzenberg et al. 1998). The yield of rCRFR1-NT produced by the transfected HEK 293 cells was increased significantly by the mannosidase I inhibitor kifunensine, which prevented formation of complex carbohydrate moieties. The suitability of the resulting high mannose glycosylated rCRFR1-NT (rCRFR1-NT-Kif) as a model for rCRFR1 was demonstrated by specific binding of rUcn and Ast. Furthermore, the role of the glycosylation type for high affinity binding and receptor functionality was investigated by two differently glycosylated forms of rCRFR1 produced in the presence or absence of kifunensine. We have used mass spectrometry coupled on-line to RP-HPLC for the analysis of the N-terminal processing sites, the disulfide linkages, and the glycosylation pattern of the purified protein rCRFR1-NT-Kif. Furthermore, the secondary structure domains of rCRFR1-NT were proposed by a prediction method.
Results
Influence of the glycosylation type on the pharmacologic properties of rCRFR1
The glycosylation type of rCRFR1 was changed by the mannosidase I inhibitor kifunensine, which was used in a concentration of 0.5 μg/ml in the medium of HEK 293 cells producing rCRFR1 (rCRFR1-Kif). The cells did not show morphological changes upon kifunensine treatment. The size of the receptor shifted from 65 kD for rCRFR1 (Fig. 1A ▶) to 50 kD for rCRFR1-Kif, whereas no significant changes in the production rates were detected (Fig. 1B ▶). After deglycosylation with PNGaseF, rCRFR1 and rCRFR1-Kif were detected as 37 kD proteins (Fig. 1A–B ▶). Thus, the 15 kD mass difference between rCRFR1 and rCRFR1-Kif was due to different asparagine-linked carbohydrates dependent on kifunensine treatment. By using EndoHf for deglycosylation, rCRFR1 was not deglycosylated, whereas rCRFR1-Kif was deglycosylated to a 37 kD protein. The known specificity of EndoHf for high-mannose and hybrid oligosaccharide structures (Maley et al. 1989) indicated the presence of complex type N-linked oligosaccharides for rCRFR1 produced in HEK 293 cells. Since kifunensine is known to prevent the formation of hybrid and complex type structures (Elbein et al. 1990), it was assumed that rCRFR1-Kif was N-glycosylated by high-mannose carbohydrates.
Fig. 1.
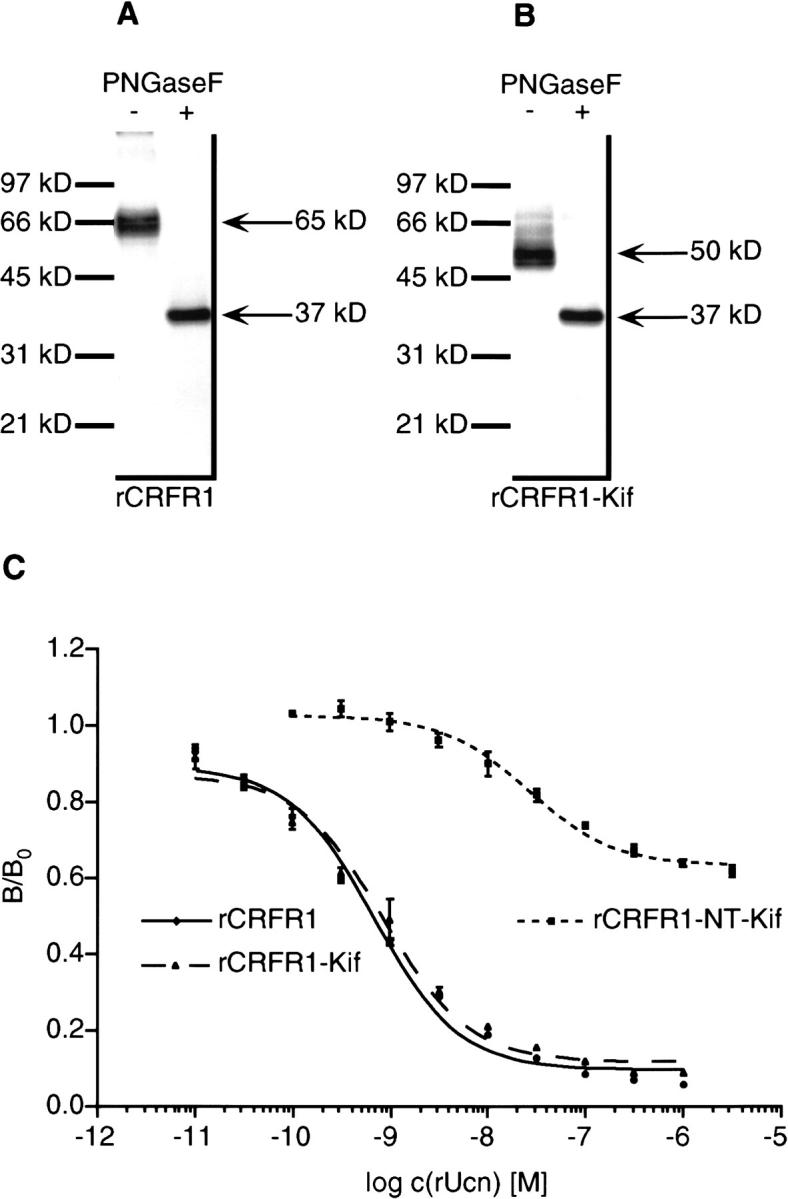
Western blot analysis of rCRFR1 and rCRFR1-Kif and binding of rUcn to rCRFR1, rCRFR1-Kif, and rCRFR1-NT-Kif. Membrane preparations with a total protein content of 11 μg, which were obtained from HEK 293 cells producing either rCRFR1 (A) or rCRFR1-Kif (B) were applied. The absence or presence of PNGaseF is indicated. (C) Competitive binding was performed using [125I-Tyr0]-rUcn as radioligand and increasing concentrations (10 pM–3.16 μM) of rUcn. Data represent duplicates from two independent experiments. Binding curves were normalized by total binding in absence of competitor [B0].
The influence of the glycosylation type on binding affinities of rUcn, human/rat CRF (h/rCRF), and Ast was investigated. The scintillation proximity assay (SPA) utilizing commercially available scintillation beads coated with WGA was employed to determine the affinity of various CRF-like peptides to rCRFR1 and rCRFR1-NT. rCRFR1 which was produced in HEK 293 cells bound rUcn with a KD of 0.61 ± 0.05 nM and h/rCRF with a KD of 1.0 ± 0.2 nM (Table 1). No significant differences in affinity were found in comparison to rCRFR1-Kif (Fig. 1C ▶, Table 1). Additionally, the effect of the altered glycosylation type on intracellular cAMP accumulation was investigated by stimulation of the HEK 293 cells producing either rCRFR1 or rCRFR1-Kif when increasing concentrations of rUcn or h/rCRF were applied. No significant differences between both glycosylated receptor species with respect to efficacy and capacity of cAMP accumulation were observed (Table 2).
Table 1.
Binding properties of rCRFR1, rCRFR1-Kif, and rCRFR1-NT-Kif
| Peptide (radioligand used) | rCRFR1 KD or KI [nM] | rCRFR1-Kif KD or KI [nM] | rCRFR1-NT-Kif KD or KI [nM] |
| rUcn ([125I-Tyr0]-rUcn) | 0.61 ± 0.05 (n = 3) | 0.79 ± 0.17 (n = 3) | 27 ± 10 (n = 4) |
| h/rCRF ([125I-Tyr0]-h/rCRF) | 1.0 ± 0.2 (n = 3) | 1.2 ± 0.2 (n = 2) | no binding |
| Ast ([125I-Tyr0]-rUcn) | 4.9 ± 1.0 (n = 3) | 3.4 ± 0.4 (n = 3) | 60 ± 24 (n = 3) |
The affinity constants are mean ± sdv of n independent binding experiments performed in duplicate. KD values were calculated for rUcn and h/rCRF and KI values for Ast.
Table 2.
Stimulation of intracellular cAMP accumulation by h/rCRF and rUcn in HEK 293 cells producing rCRFR1 or rCRFR1-Kif
| Peptide | rCRFR1 | rCRFR1-Kif | ||
| EC50 [nM] | cAMP/cells | EC50 [nM] | cAMP/cells | |
| h/rCRF | 0.33 ± 0.06 | 90 ± 7 pmol/104 | 0.27 ± 0.04 | 83 ± 9 pmol/104 |
| rUcn | 0.89 ± 0.10 | 94 ± 3 pmol/104 | 0.50 ± 0.09 | 78 ± 14 pmol/104 |
The values are mean ± sdv of at least four independent determinations performed in duplicate.
Production of rCRFR1-NT
HEK 293 cells were transfected stably with cDNA coding for the first 121 amino acid residues of rCRFR1 fused with a His6 sequence. rCRFR1-NT was barely detectable in the medium by immunoblotting. After Ni-affinity purification, rCRFR1-NT was detected with a size of 40 kD by immunoblotting (Fig. 2A ▶, lane 1). However, no intracellular accumulation of rCRFR1-NT was found. By deglycosylation with PNGaseF, a 13 kD species was generated (Fig. 2A ▶, lane 2). The protein concentration was low when compared with the concentration of rCRFBP produced in identical cells using an identical promotor (Jahn et al. 2001).
Fig. 2.

Polyacrylamide gel analysis of rCRFR1-NT and rCRFR1-NT-Kif. For deglycosylation with PNGaseF, approximately 100 ng Ni-affinity-purified rCRFR1-NT (A) or 37.5 μL of medium containing rCRFR1-NT-Kif (B) were applied to SDS-PAGE followed by Western blot and immunodetection. The absence or presence of PNGaseF is indicated. (C) SDS-PAGE of affinity-purified rCRFR1-NT-Kif was performed by application of 37.5 μL medium of transfected HEK 293 cells (M), 37.5 μL supernatant after adsorption on Ni-affinity resin (S1), and 30 μL of the third elution fraction (E). Proteins were detected by silver staining.
When kifunensine was added to the cell culture medium, the production rate of rCRFR1-NT-Kif was greater than that of rCRFR1-NT by approximately two orders of magnitude and reached a maximum at a concentration of 0.5 μg/ml kifunensine as determined by analysis of the medium with immunoblotting. Under these conditions, two major species of 35 kD and 32 kD (Fig. 2B ▶, lane 3) were found by SDS-PAGE and immunoblotting in the medium. After deglycosylation of both species with PNGaseF, a single 13 kD protein (Fig. 2B ▶, lane 4) was found. Thus, the mass difference between rCRFR1-NT and rCRFR1-NT-Kif was due to altered N-glycosylation controlled by kifunensine. rCRFR1-NT-Kif was purified by batch adsorption to Ni-affinity resin for further protein chemical characterization. The two major species of the glycosylated protein were detected by SDS-PAGE with silver staining (Fig. 2C ▶, lane 7).
Ligand binding to rCRFR1-NT-Kif
Ligand binding to rCRFR1-NT and rCRFR1-NT-Kif was analyzed using [125I-Tyr0]-rUcn. Approximately 50% specific binding was detected for medium containing rCRFR1-NT-Kif. No specific binding was observed for medium containing rCRFR1-NT or medium from non-transfected HEK 293 cells. Competition of rUcn (Fig. 1C ▶) and Ast with [125I-Tyr0]-rUcn for rCRFR1-NT-Kif revealed a KD of 27 nM and a KI of 60 nM, respectively (Table 1). The CRFR2-selective antagonist antisauvagine-30 (Rühmann et al. 1998) did not compete with [125I-Tyr0]-rUcn for rCRFR1-NT-Kif.
Characterization of rCRFR1-NT and rCRFR1-NT-Kif
rCRFR1-NT-Kif was isolated from 400 mL medium by ultrafiltration and Ni-affinity purification. Subsequent SDS-PAGE followed by Western blotting and immunodetection yielded two bands which respresented 35 kD and 32 kD species of rCRFR1-NT-Kif (Fig. 2B ▶, lane 3). These bands were excised from the Western blot and subjected to Edman degradation. Both protein species contained two forms with identical sequences except that the more abundant form was N-terminally extended by a Ser residue (Fig. 3A and 3B ▶). On the basis of the initial PTH amino acid yields, the larger form was more abundant by a factor of approximately 2. Twenty nine residues of each form were sequenced with a repetitive yield of 92%. The analyzed sequences represented rCRFR1(24–52) and rCRFR1(25–53). The relative yield of PTH-Asn32 was not decreased in either sequence (Fig. 3A–B ▶), but only low levels of Asn were detected in positions 38 and 45 (Fig. 3A–B ▶), indicating a high degree of glycosylation of these two residues.
Fig. 3.
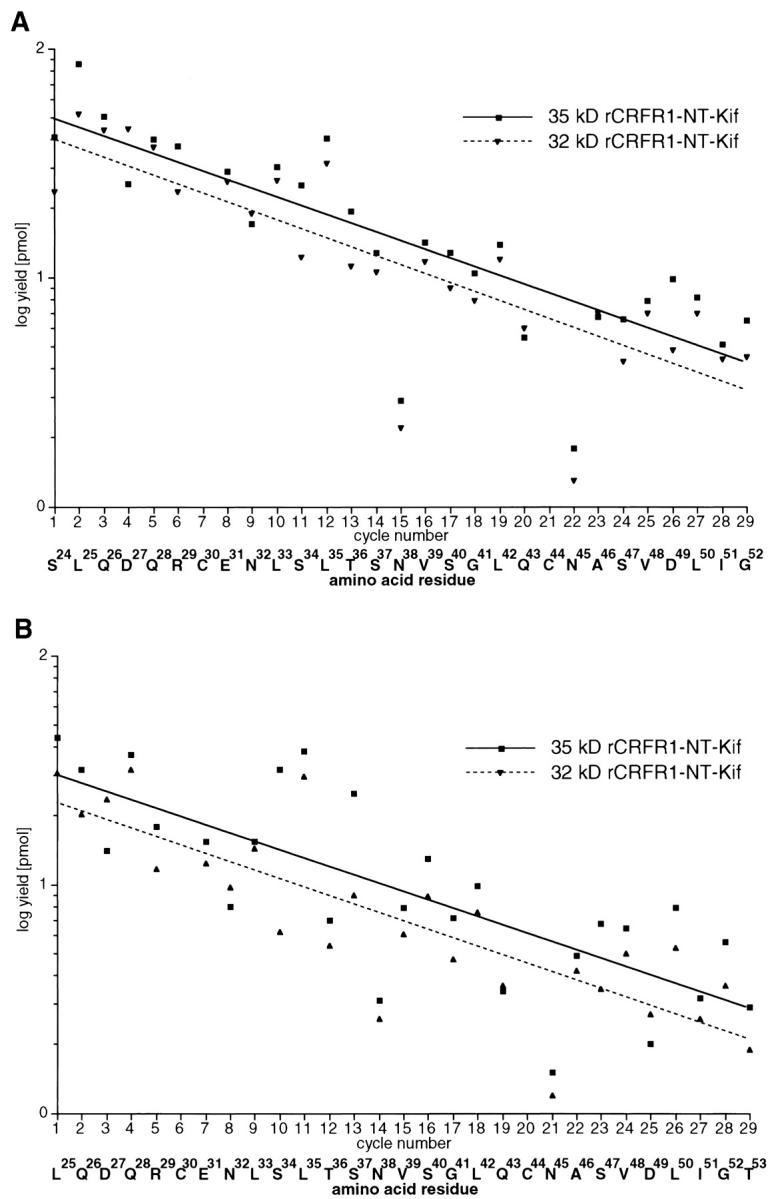
Edman degradation of 32 kD and 35 kD rCRFR1-NT-Kif. The yields of the respective amino acid residues of 29 cyles of Edman degradation of rCRFR1-NT-Kif are shown. (A) shows the data of 32 kD and 35 kD species starting with Ser24 and (B) shows the data of 32 kD and 35 kD species starting with Leu25. The initial yields of the 32 kD protein species were 47.0 pmol and 26.4 pmol for the sequences starting with Ser and Leu, respectively.
Serum free medium (400 ml) containing rCRFR1-NT was subjected also to consecutive ultrafiltration and Ni-affinity purification. Purified rCRFR1-NT was deglycosylated with PNGaseF and subjected to Western blotting. The observed 13 kD band was excised for Edman degradation (data not shown). Two protein forms with identical sequences were found except that the more abundant protein was extended N-terminally by a Ser residue. Eleven residues of each form were ideied with a repetitive yield of 91%. The analyzed sequences represented rCRFR1(24–34) and rCRFR1(25–35). Residue Asn32 followed by Leu33–Ser34 represented the first N-terminal potential glycosylation site. Edman degradation revealed no conversion of Asn32 into Asp as would have been expected if Asn32 would have been glycosylated to a significant extent.
Mass spectrometric characterization of rCRFR1-NT-Kif glycosylation
rCRFR1-NT-Kif affinity-purified was also analyzed with NanoES MS. A large degree of heterogeneity introduced by glycosylation was found (Fig. 4A ▶). rCRFR1-NT-Kif was deglycosylated with EndoHf and purified by RP-HPLC. This protein gave rise to 5 major groups of signals in the NanoES mass spectrum (Fig. 4B ▶). Each group accounted for a different charge state between + 7 and + 11. Maximum entropy deconvolution showed signals of proteins with different molecular masses (Fig. 4C ▶). Molecular masses at 12367, 12571, and 12774 accounted for the protein sequence Ser24–His127 carrying 3, 4, and 5 N-acetylglucosamine residues, respectively. In the same manner, molecular masses at 12484 and 12686 accounted for the sequence Leu25–His127 carrying 4 and 5 N-acetylglucosamine residues, respectively. EndoHf cleaved the carbohydrate moiety in a way that the N-acetylglucosamine residue linked to Asn remained on the protein chain (Maley et al. 1989). Thus, the number of glycosylated Asn residues was represented by the number of N-acetylglucosamine residues left on the protein. Furthermore, the relative abundance of the different molecular ion signals represented the abundance of the different forms of rCRFR1-NT-Kif generated by co- and posttranslational processing. On the basis of this observation the ratio of the protein sequences Ser24–His127 and Leu25–His127 was estimated to be 2 to 1, which was in agreement with the data derived from protein sequence analysis. Furthermore, the relative abundance for rCRFR1-NT-Kif containing either 5 or 4 glycosylated Asn residues was 53% and 40%, respectively.
Fig. 4.
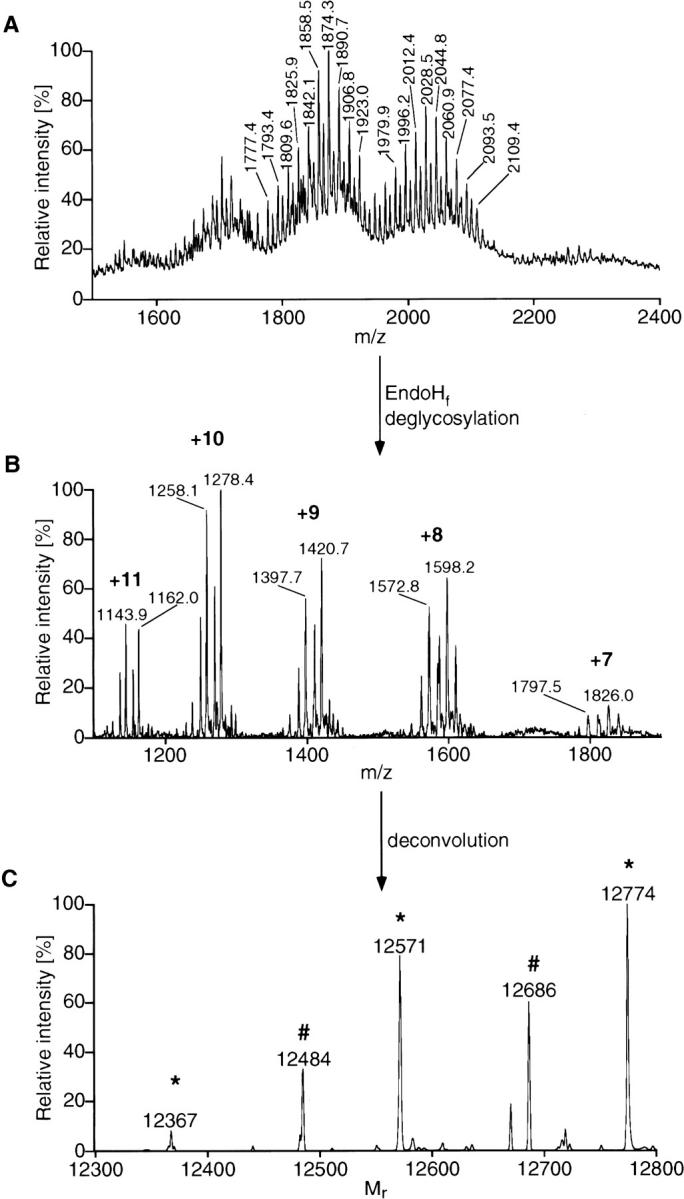
NanoES mass spectrum of rCRFR1-NT-Kif before and after EndoHf deglycosylation. rCRFR1-NT-Kif and EndoHf-deglycosylated rCRFR1-NT-Kif (c = ∼0.2 μg/μl) were dissolved in 50% methanol containing 1% acetic acid. (A) shows the ES mass spectrum of the heterogeneous glycoprotein. After deglycosylation, 5 distinct charge states (+ 11 to + 7) can be seen (B). The deconvoluted mass spectrum (C) represents the protein chain starting with amino acid Ser24 (*) and carrying three to five N-acetylhexosamine (HexNAc) residues, and the protein chain starting with amino Leu25 (#) and carrying four to five HexNAc residues.
Following affinity-purification and deglycosylation with EndoHf, rCRFR1-NT-Kif was reduced with DTT, alkylated with iodoacetamide, and purified by RP-HPLC. Approximately 2 μg of the product was digested using trypsin and subjected to RP-HPLC chromatographic separation with on-line mass spectrometric recording. This combination provided the possibility of reducing the total ion chromatographic display of the RP-HPLC separation to the display of a certain molecular mass showing exclusively the elution profile of one peptide. By using this approach, two chromatograms were generated for the tryptic peptide rCRFR1-NT-Kif(86–96). The chromatogram reconstructed by the molecular ions at m/z 1235.4 and 618.2 represented rCRFR1-NT-Kif(86–96) lacking an N-acetylglucosamine residue, whereas the chromatogram reconstructed by the molecular ions at m/z 1438.5 and 719.8 represented the same fragment carrying one N-acetylglucosamine residue. The molecular masses of these peptides differed by the mass increment 203 of the N-acetylglucosamine residue that represented one carbohydrate moiety. The relative abundances of these mass chromatograms were used to calculate directly the ratio of the non-glycosylated Asn90 (30%) and the glycosylated Asn90 (70%) in the fragment rCRFR1-NT-Kif(86–96). This approach was also used to calculate the degree of glycosylation of Asn78 (92%) in the fragment rCRFR1-NT-Kif(58–85) and of Asn98 (96%) in the fragment rCRFR1-NT-Kif(97–111).
In the same manner, three forms of the fragment rCRFR1-NT-Kif(30–57) carrying one (m/z 1678.9 and 1119.6), two (m/z 1780.5 and 1187.3), and three (m/z 1882.1 and 1255.0) N-acetylglucosamine residues, respectively, were analyzed. The relative abundances were 16%, 75%, and 9%, respectively. The degree of glycosylation of fragment rCRFR1-NT-Kif(30–57) was calculated by combining the data from RP-HPLC-MS and Edman degradation. In contrast to Edman degradation, which revealed no detectable glycosylation of Asn32, the mass spectrometric results pointed to 9% full glycosylation of rCRFR1-NT-Kif(30–57). Consequently, Asn32 must have been glycosylated to an extent of at least 9%. It was assumed that the proportion of 16% single and 75% double glycosylation accounted mainly for the residues Asn38 and Asn45. Furthermore, the data from Edman degradation suggested a similar extent of glycosylation of these residues. Therefore, glycosylation of at least 90% for either residue Asn38 and Asn45 was assumed.
On the basis of the extent of glycosylation of the respective proteolytic fragments, the overall rate of glycosylation of rCRFR1-NT-Kif was calculated. Thus, it was concluded that 36% of rCRFR1-NT-Kif contained 4 glycosylated Asn residues and 50% carried 5 glycosylated Asn residues. This result did not deviate significantly from the extent of glycosylation of rCRFR1-NT-Kif obtained by the abundance of the respective molecular ions of the entire protein after deglycosylation with EndoHf (Fig. 4B ▶). In detail, Asn38, Asn45, Asn78, and Asn98 were >90% glycosylated, whereas Asn32 was glycosylated to an extent of at least 9%, and Asn90 to an extent of 70%.
rCRFR1-NT-Kif was reduced, alkylated with iodoacetamide, and purified by RP-HPLC. Approximately 3 μg protein was fragmented using trypsin and analyzed by RP-HPLC-MS in combination with cone-skimmer fragmentation in the electrospray interface (Katta et al. 1991). The method for the determination of the carbohydrate structure is demonstrated by using the results of the proteolytic fragment rCRFR1-NT-Kif(97–111). The doubly protonated molecular ion of this fragment carrying one carbohydrate moiety showed a distinct fragment ion pattern. The ten signals starting from the largest molecular ion were separated pairwise by a mass difference of 162, which was attributed to the presence of nine hexose residues; two signals separated by 203 were explained by the presence of one N-acetylhexosamine residue. The molecular ion of smallest size was compatible only with rCRFR1-NT-Kif(97–111) carrying one N-acetylhexosamine residue. From the series of these increments the oligosaccharide sequence was derived to be (N-acetylhexosamine)2(hexose)9 linked to Asn98. This pattern was compatible only with the high mannose type glycosylation (Kornfeld and Kornfeld, 1985; Settineri and Burlingame, 1996).
Assignment of disulfide bridges
Approximately 1 μg affinity-purified rCRFR1-NT-Kif was reduced with DTT. For comparison, the same amount of reduced and non-reduced protein was reacted with iodoacetamide. HPLC-MS analysis revealed a mass difference of 348 between both forms of the protein, which accounted for six sulfhydryl groups in the reduced protein modified by carboxamidomethyl residues. This result indicated the presence of three disulfide bridges in rCRFR1-NT-Kif.
Approximately 4 μg affinity-purified rCRFR1-NT-Kif was deglycosylated with PNGaseF, which was shown to convert Asn residues bound to N-linked oligosaccharides into Asp residues (Maley et al. 1989). Therefore, the glycosylated Asn residues of rCRFR1-NT-Kif were expected to be converted into proteolytic cleavage sites for AspN digestion. One half of this protein fraction was reduced with DTT and alkylated with iodoacetamide. Both fractions were purified by RP-HPLC prior to proteolytic cleavage. Comparison of the trypsin digests of both protein fractions by RP-HPLC-MS showed that only the protein fragments rCRFR1-NT-Kif(112–127) and rCRFR1-NT-Kif(114–127) were not affected upon reduction and alkylation as indicated by their elution profile (Fig. 5 ▶). Similarly, signals representing the fragments rCRFR1-NT-Kif(90–97) and rCRFR1-NT-Kif(104–127) obtained by AspN digestion were not changed by reduction and alkylation (Table 3).
Fig. 5.

HPLC chromatograms of the tryptic digests of rCRFR1-NT-Kif. After deglycosylation with PNGaseF and HPLC purification, rCRFR1-NT-Kif was digested using the endoprotease trypsin. (A) shows the chromatograms of the peptide map derived from reduced and alkylated rCRFR1-NT-Kif, whereas (B) shows the peptide map of non-reduced rCRFR1-NT-Kif. Cys-containing fragments are indicated by square brackets. The signals of the disulfide-linked peptides are marked by gray shading. Assignment of the fragments was carried out on the basis of the calculated and observed molecular masses of the peptides. Several signals were assigned to either rCRFR1-NT-Kif(30–57) or rCRFR1-NT-Kif(86–96) as a result of incomplete conversion of Asn32 and Asn90 into Asp residues caused by partial glycosylation of these residues.
Table 3.
Signal assignments of the AspN fragments of reduced and alkylated rCRFR1-NT-Kif
| Retention time [min] | Fragment | Mr (observed) | Mr (calculated) | Deviation |
| 30:56 | 38–44 | 777.9 | 777.9 | <129 ppm |
| 36:28 | 78–89 | 1413.7 | 1413.5 | 142 ppm |
| 37:55 | 27–37 | 1322.6 | 1322.4 | 151 ppm |
| 38:53 | 108–127 | 2490.2 | 2489.8 | 161 ppm |
| 42:29 | 90–97 | 860.7 | 860.9 | 232 ppm |
| 43:54 | 104–127 | 2959.8 | 2959.3 | 169 ppm |
| 50:50 | 78–97 | 2255.9 | 2255.4a | 222 ppm |
| 67:49 | 49–77 | 3384.8 | 3384.0 | 236 ppm |
a With Asn in position 90.
All major signals in the chromatograms were assigned to proteolytic peptides on the basis of their molecular mass determined by mass spectrometry. The molecular masses of the peptides were calculated on the basis of the known rCRFR1-NT-Kif sequence. The tryptic peptide fragment with the mass of 3761.3 could only be explained by the disulfide linkage connecting the fragments rCRFR1-NT-Kif(58–76) and rCRFR1-NT-Kif(97–110) (Fig. 5 ▶). Thereby, the linkage between Cys68 and Cys102 (Fig. 6A ▶) was assigned unambiguously. The fragment with the molecular mass of 2075.6 obtained by the AspN digestion matched only a species containing fragments rCRFR1-NT-Kif(38–44) and rCRFR1-NT-Kif(78–89) linked by a disulfide bridge (Table 4). Thereby, the second disulfide linkage was assigned to Cys44 and Cys87 (Fig. 6B ▶). Consequently, the third disulfide linkage must be formed between the remaining residues Cys30 and Cys54 (Fig. 6C ▶). It must be pointed out that this assignment was only possible by AspN cleavage N-terminally to residues Asp38, Asp78, and Asp90 generated by PNGaseF deglycosylation. This assignment represented >95% of the abundance of all cysteine-containing fragments.
Fig. 6.

Disulfide bridge arrangement of rCRFR1-NT-Kif. (A) and (B) show the disulfide-linked peptides of the tryptic and AspN digest, respectively. (C) represents the derived disulfide bridging. In the amino acid sequences, X represents an asparagine or aspartate residue depending on the extent of glycosylation at the corresponding position. The solid lines represent disulfide bridges directly deduced from the proteolytic digests. The dotted lines connect fragments without an unambiguous assignment of a single disulfide bridge. The dashed line represents the disulfide bridge which was concluded from the results of Cys derivatization. The 23-amino acid long signal peptide is marked by a black background and the 24 amino acid long signal peptide by a gray background.
Table 4.
Signal assignments of the AspN fragments of non-reduced rCRFR1-NT-Kif
| Retention time [min] | Fragment | Mr (observed) | Mr (calculated) | Deviation |
| 43:01 | 90–97 | 860.9 | 860.9 | <116 ppm |
| 44:24 | 104–127 | 2959.6 | 2959.3 | 101 ppm |
| 45:20 | 38–44-S-S-78–89 | 2075.6 | 2075.3 | 145 ppm |
| 47:23 | 38–48-S-S-78–89 | 2447.2 | 2447.6 | 163 ppm |
| 53:51 | 38–44-S-S-78–97 | 2917.5 | 2917.2a | 103 ppm |
| 62:35 | 27–31-S-S-49–77-S-S-98–127 | 7601.5 | 7600.6 | 118 ppm |
| 63:26 | 27–37-S-S-49–77-S-S-98–127 | 8217.7 | 8216.3 | 170 ppm |
| 64:47 | 27–37-S-S-49–77-S-S-90–127 | 9061.1 | 9059.2 | 210 ppm |
| 65:54 | 27–37-S-S-49–77-S-S-98–103 | 5275.6 | 5275.0 | 114 ppm |
a With Asn in position 90.
Secondary structure prediction of rCRFR1-NT
By using the consensus method for protein secondary structure prediction Jpred2 (Cuff and Barton 1999), an α-helical domain was predicted for residues 25–35 of rCRFR1-NT. β-structures were proposed for the stretches of residues 62–66, 83–86, and 116–119. Cys30 was found to be located in the α-helical domain, whereas the remaining Cys residues were not part of the secondary structure domains. In the same manner, only Asn32, which represented the first potential glycosylation site, was located in a region with secondary structure. In comparison, the N-terminal domain of rCRFR2α and rCRFR2β exhibited a similar pattern of secondary structure domains with a replacement of the C-terminal β-strand of rCRFR1-NT by an α-helical domain.
Discussion
Mannosidase I of HEK 293 cells was inhibited by kifunensine (Elbein et al. 1990) to prevent the formation of hybrid or complex oligosaccharide structures. Deglycosylation experiments revealed a complex glycosylation type for rCRFR1 and, as expected, a high mannose glycosylation type for rCRFR1-Kif. The binding data obtained with the SPA for rCRFR1 agreed with earlier observations for rCRFR1 (Perrin et al. 1993,1998,1999). Both receptors bound rUcn, h/rCRF, and Ast with high affinity, demonstrating that kifunensine treatment did not prevent the correct folding of the receptor and that the investigated glycosylation types of rCRFR1 did not influence the binding of the tested ligands. Furthermore, the presented cAMP data indicated that rCRFR1-Kif was fully functional. Alteration of the glycosylation type impaired neither the targeting of the receptor to the cell surface nor the intracellular coupling to G-proteins. Thus, the kifunensine treatment did not prevent the correct insertion of the receptor into the membranes.
Recently, it was shown that the molecular size of native CRFR1 varies not only between mouse and rat brain, but also between different brain regions (Radulovic et al. 1998). These differences are probably caused by alterations in the glycosylation of CRFR1. The presented binding and cAMP data for rCRFR1 and rCRFR1-Kif suggested that the different glycosylation of CRFR1 did not influence the binding affinities or the coupling to adenylate cyclase in vivo.
Although rCRFR1-NT was detected as a soluble glycoprotein in the medium, the extremely low production level prevented the pharmacological and protein chemical characterization of rCRFR1-NT. The addition of kifunensine to the serum-free medium increased the rCRFR1-NT yield by approximately two orders of magnitude and changed the glycosylation type of this protein. This increased yield may be a result of impaired cytosolic proteasomal degradation of rCRFR1-NT-Kif. Recently, the fate of terminally misfolded α1-antitrypsin was studied in the presence of different glycosidase inhibitors (Liu et al. 1999). It was demonstrated that inhibition of mannosidase I prolonged the retention phase of misfolded α1-antitrypsin in the endoplasmic reticulum and impaired proteasomal degradation, but did not affect the secretion of misfolded α1-antitrypsin. In analogy, it can be speculated that kifunensine extended the retention phase of rCRFR1-NT in the endoplasmatic reticulum and thus enhanced the folding process to generate a CRFR1-like spatial structure that might be more resistant to proteasomal degradation.
The binding constants obtained for rCRFR1-NT-Kif were probably similar to those of rCRFR1-NT in view of the observation that for the full length receptor the glycosylation type altered by kifunensine did not change the binding affinities to rUcn, h/rCRF, and Ast significantly. rCRFR1-NT-Kif bound rUcn and Ast specifically with relatively high affinity, whereas the CRFR2-selective antagonist antisauvagine-30 (Rühmann et al. 1998) did not compete with radiolabeled rUcn. These findings showed that the membrane interaction of the full length receptor was not required for specific interactions of rUcn and Ast with the soluble N-terminal domain of rCRFR1.
The observation that rCRFR1-NT-Kif did not bind radiolabeled h/rCRF, in contrast to rUcn and Ast, indicated that h/rCRF required more than the N-terminal domain of rCRFR1 for specific binding. Thus, CRF in comparison to Ucn and Ast interacted in a different manner with the full length receptor. This observation was supported by the finding that binding of Ucn and Ast was independent of the G protein-coupling state of CRFR1, whereas binding of h/rCRF and oCRF was impaired by uncoupling of CRFR1 from G proteins (Spiess et al. 1998; Perrin et al. 1999). In agreement with this observation, the importance of the fourth extracellular domain (EC4) for binding of oCRF to rCRFR1 was demonstrated recently (Sydow et al. 1999). The specific binding of rUcn and Ast to rCRFR1-NT-Kif indicated that this soluble protein was a valuable model for the corresponding domain of the full length receptor.
rCRFR1-NT and rCRFR1-NT-Kif were found to have identical start sequences, demonstrating that the kifunensine treatment did not influence the signal peptide processing in HEK 293 cells. The major form starting with Ser24 and the minor form starting with Leu25 were predicted with the highest probability using an established algorithm for the ideication of signal peptides and their cleavage sites (Nielsen et al. 1997). In view of these results, it was suggested that both isolated forms of rCRFR1-NT and rCRFR1-NT-Kif which started either with Ser24 or Leu25 were products of the precursor protein cleaved by signal peptidase which removed the first 23 or 24 amino acids. Alternatively, the possibility has to be considered that the smaller species was derived from the larger species by action of an amino peptidase. By using the above algorithm, we found for human, mouse, and sheep CRFR1 the same signal peptides of 23 and 24 amino acids as most probable. It is proposed that the full length rCRFR1 which was expressed in HEK 293 cells was N-terminally processed in a similar manner as rCRFR1-NT-Kif.
Disulfide bridges are important determinants for protein conformations by stabilizing tertiary structures. Since we could demonstrate that rCRFR1-NT-Kif interacted specifically with rUcn and Ast, it was concluded that it probably possessed the tertiary structure of the respective domain of the full length receptor. Therefore, the disulfide linkages were established using protein chemical methods. It was demonstrated that rCRFR1-NT-Kif did not contain free cysteine residues. Thus, the six cysteine residues of rCRFR1-NT-Kif formed three disulfide bonds. It was concluded that neither of the cysteine residues Cys188 and Cys258 of rCRFR1 located in the extracellular domains 2 (EC2) and 3 (EC3), respectively, formed a disulfide bond with a Cys residue in the N-terminal domain. In almost all known GPCRs, the two Cys residues located in EC2 and EC3 are highly conserved. It has been proposed that they form a disulfide bridge stabilizing the tertiary structure of the receptor (Strader et al. 1994). This proposal agrees with our findings. Site-directed mutagenesis was performed on several GPCRs (Karnik and Khorana 1990; Savarese et al. 1992; Ohyama et al. 1995; Perlman et al. 1995; Cook and Eidne 1997) including the secretin receptor (Vilardaga et al. 1997) and suggestive evidence was found for the linkage between these two conserved Cys residues. This disulfide bond is also proposed for mCRFR1 (Qi et al. 1997).
Two disulfide bridges connecting residue Cys44 with Cys102 and residue Cys68 with Cys87 of the mCRFR1 precursor protein were proposed on the basis of mutations of single and paired Cys residues to Ser residues (Qi et al. 1997). In addition, it was found that mutating residue Cys30 did not affect the function of mCRFR1. These results contrasted our finding for rCRFR1 showing three disulfide bridges connecting residues Cys30 and Cys54, Cys44 and Cys87, and Cys68 and Cys102 of rCRFR1. However, site-directed mutagenesis provides only indirect evidence for protein structure. In addition to local changes, point mutations may influence the protein structure even in remote regions. In contrast, the disulfide structure of the functional rCRFR1-NT-Kif was elucidated by analyzing the protein structure. The disulfide bridge arrangement determined for rCRFR1-NT-Kif may represent the pattern of disulfide linkages of the full length CRFR1. CRFR belongs to the secretin-like GPCR family which is characterized by at least five conserved Cys residues in the N-terminal domain of its members. rCRFR1 contains an additional Cys residue located N-terminally to the conserved Cys residues. It is conceivable that these residues form a pattern of disulfide bridges which is also conserved within this receptor family. Thus, the receptors of the secretin-like GPCR family may contain two of the three disulfide linkages shown for rCRFR1-NT-Kif.
It is noteworthy that Cys30, which is missing in rCRFR2α but not in rCRFR2β, was located in the predicted α-helical domain of rCRFR1. Therefore, it is concluded that the tertiary structure of the N-terminal domain of rCRFRs is stable without the formation of a disulfide linkage of the Cys residue located in the α-helical part. This conclusion agrees with the site-directed mutagenesis of the first Cys residue of mCRFR1, which led to a functional receptor (Qi et al. 1997).
Asn98, which is part of the most C-terminally-located glycosylation site, was almost fully glycosylated, whereas Asn90 was glycosylated to an extent of only 70%. This lower glycosylation may be explained by the neighborhood of Trp93 to the glycosylation site Asn90-Gly91-Ser92. It has been demonstrated that tryptophan residues following glycosylation sequons impair the glycosylation efficiency (Mellquist et al. 1998), probably due to poor accessibility of these residues to oligosaccharyl transferase. It was probable that truncation of rCRFR1 did not influence the degree of glycosylation in view of the observation that the more terminally located residue Asn98 was almost fully glycosylated. The first potential glycosylation site (Asn32) was barely glycosylated. This site is located in the predicted α-helical structure. The remaining glycosylation sites are located in regions where no specific secondary structure was found by the prediction method used. Only CRFR1 of the rat contains in this position a potential glycosylation m. Therefore, we propose that the glycosylation of rCRFR1 in position 32 is not important for ligand binding and receptor function. Since it has been reported that kifunensine does not influence protein glycosylation even at concentrations leading to full inhibition of mannosidase I (Elbein et al. 1990), we concluded that glycosylation of rCRFR1-NT-Kif was not affected by kifunensine and thus probably resembled the glycosylation of full length CRFR1.
Materials and methods
Generation of the rCRFR1-NT cDNA
cDNA coding for the first 121 amino acids of rCRFR1 was extended at the 3` end by a sequence coding for a His6 tag. The clone was amplified by PCR and ligated into the mammalian expression vector pcDNA3 (Invitrogen, San Diego, CA) using the restriction enzymes KpnI and EcoR1 (Sydow et al. 1997).
Production of rCRFR1 and rCRFR1-NT
rCRFR1 was produced in HEK 293 cells (Rühmann et al. 1996). Transfection and culturing of HEK 293 cells and membrane preparations were carried out as described earlier (Rühmann et al. 1996; Sydow et al. 1997). The soluble proteins rCRFR1-NT and rCRFR1-NT-Kif were produced by using serum-free cell culture conditions (Jahn et al. 2001). For the production of high mannose type-glycosylated proteins the mannosidase I inhibitor kifunensine (ICN Biomedicals, Eschwege, Germany) (Elbein et al. 1990) was added to the media at a final concentration of 0.5 μg/ml for at least 4 days. Twenty days after transfection individual clones were isolated and screened for highest protein expression.
SDS-PAGE, Western blotting, and immunodetection
Serum-free medium was used directly as source for rCRFR1-NT. rCRFR1 was obtained from cell membrane preparations. Samples of rCRFR1-NT (medium) and rCRFR1 (cell membranes) treated with 2% SDS were run on 10% and 7.5% polyacrylamide gels, respectively (Fling and Gregerson 1986). Immunodetection of rCRFR1-NT and rCRFR1 was accomplished with 0.4 μg/ml polyclonal antibody anti-rCRFR1-NT using a secondary antibody coupled to alkaline phosphatase (Sydow et al. 1997). Protein detection by silver staining was performed using a standard protocol (Merril et al. 1981).
Enzymatic deglycosylation
For N-deglycosylation of rCRFR1, 19 μg of membrane protein was incubated for 60 min at 37°C with 500 units PNGaseF or with 1000 units EndoHf (New England BioLabs, Schwalbach, Germany) in the presence of trasylol, bacitracin, and PMSF as suggested by the supplier. For deglycosylation of rCRFR1-NT, 30 μg affinity-purified rCRFR1-NT was incubated for 6 h at 37°C with 1000 units PNGaseF or 2000 units EndoHf in 50 mM phosphate buffer pH 7.4 or 5.5, respectively, containing 2 M urea and 2 mM PMSF.
Radioligand binding assay
A new method, based on a SPA (Udenfriend et al. 1985), was established that allowed the binding analysis of membrane-bound rCRFR1 and soluble rCRFR1-NT. The competition binding assay was performed in 96-well microtiter plates and consisted of unlabeled peptide (300 nM as highest concentration), radiolabeled peptide ([125I-Tyr0]-rat Ucn or [125I-Tyr0]-human/rat CRF, NEN Life Science, Dreieich, Germany, 0.07 nM), and membrane suspension (2 μg total protein) in a total volume of 150 μl assay buffer (50 mM Tris-HCl, 5 mM MgCl2, 2 mM EGTA, 100 KIU trasylol, 1 mM DTT and 1% BSA). After a two hour incubation at room temperature, 50 μl wheat germ agglutinin (WGA) bead suspension (250 μg beads/well; neuropeptide Y receptor SPA binding assay, RPNQ 0085, Amersham Pharmacia Biotech, Uppsala, Sweden) in assay buffer was added to the wells. The microtiter plate was sealed and shaken vigorously. The beads were allowed to settle down overnight at 4°C and the bound radioactivity was detected with a Wallac 1450 Microbeta scintillation counter. The same method was used for the binding analysis of rCRFR1-NT, with the exception that DTT in the assay buffer was omitted, the WGA bead mass was 500 μg beads/well, and the unlabeled peptides were added in a final concentration up to a maximum of 3 μM. Binding data were analyzed using the Prism computer program (GraphPad Software, San Diego, CA). The KD values for rUcn and h/rCRF were calculated on the basis of the assumption that the affinity of the radioligand and the respective unlabeled peptide were identical. The inhibition constant (KI) of Ast was determined from IC50 values using the Cheng-Prusoff equation (Cheng and Prusoff, 1973).
Measurement of intracellular cAMP accumulation
The cells were stimulated as described (Sydow et al. 1997) using increasing concentrations of rUcn or h/rCRF. Intracellular cAMP was measured with the Biotrak™ cAMP [125I] SPA system (Amersham Pharmacia Biotech, Uppsala, Sweden) according to the manufacturer's product manual.
Protein purification and analysis
The methods for protein purification, Cys alkylation, HPLC-MS analysis, and Edman degradation were described recently (Jahn et al. 2001). Prior to purification, the medium containing rCRFR1-NT was concentrated by ultrafiltration using a membrane with a molecular weight cut-off of 8,000 (Millipore, Eschborn, Germany). Nickel-affinity purification was performed as a batch procedure at 4°C under native conditions as suggested by the supplier except for the elution step, which was performed at pH 4. Digestion with TPCK-treated trypsin (Sigma, Deisenhofen, Germany) or endoprotease AspN (Boehringer Mannheim, Mannheim, Germany) was performed at 37°C for 2 to 8 hours as described (Jahn et al. 2001). An enzyme to substrate ratio of 1:10 (w/w) and 1:20 was applied with AspN and trypsin, respectively. The amino acid residues of all forms of rCRFR1-NT were counted on the basis of the amino acid sequence of the pre-form of the rCRFR1 precursor.
Mass spectra were recorded using a Micromass AutoSpec-T tandem mass spectrometer. For nano-electrospray mass spectrometry (NanoES MS), 2 μL sample solution in a mixture of 49.5% methanol, 49.5% H2O, and 1% acetic acid was loaded into gold/palladium-coated NanoES spray capillaries pulled from boro-silicate glass (Protana, Odense, Denmark). Deconvolution of the protein ES mass spectra was carried out by employing the MaxEnt algorithm implemented into the OPUS (Micromass, Manchester, UK) data system. Fragmentation in the sampling cone-skimmer region (Katta et al. 1991) was induced by doubling the potential difference between sampling cone and skimmer.
Secondary structure prediction
The secondary structures were calculated using the consensus method for protein secondary structure prediction Jpred2 (Cuff and Barton, 1999) available on the internet (http://jura.ebi.ac.uk:8888/).
Acknowledgments
Gudrun Fricke-Bode, Bernd Hesse-Niessen, and Sandra Wille are gratefully thanked for their excellent technical assistance. We thank Dr. Olaf Brauns, Dr. Frank M. Dautzenberg, and Dr. Claus O. Markert for helpful discussions.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CRF, corticotropin-releasing factor
Ucn, urocortin
Svg, sauvagine
CRFR, CRF receptor
rCRFR, rat CRFR
hCRFR, human CRFR
mCRFR, mouse CRFR
xCRFR, Xenopus laevis CRFR
rCRFBP, rat CRF binding protein
NT, amino-terminal domain
Ast, astressin
EC, extracellular domain
HEK, human embryonic kidney
SPA, scintillation proximity assay
SDS-PAGE, SDS-polyacrylamide gel electrophoresis
EndoHf, endo-β-acetylglucosaminidase H
PNGaseF, peptide-N-glycosidase F
WGA, wheat germ agglutinin
RP-HPLC, reversed phase-high performance liquid chromatography
NanoESMS, nano-electrospray mass spectrometry
rCRFR1-NT-Kif, rCRFR1-NT expressed in presence of kifunensine
rCRFR1-Kif, rCRFR1 expressed in presence of kifunensine
GPCR, G protein-coupled receptor
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.12101.
Parts of this work were presented at the meetings of the Society for Neuroscience in Miami (October 23–28 1999) and New Orleans (November 4–9, 2000), and the meeting of the Endocrine Society in Toronto (June 21–24 2000).
References
- Behan, D.P., Grigoriadis, D.E., Lovenberg, T., Chalmers, D., Heinrichs, S., Liaw, C., and De Souza, E.B. 1996. Neurobiology of corticotropin releasing factor (CRF) receptors and CRF-binding protein: Implications for the treatment of CNS disorders. Mol. Psychiatry 1: 265–277. [PubMed] [Google Scholar]
- Chalmers, D.T., Lovenberg, T.W., and De Souza, E.B. 1995. Localization of novel corticotropin-releasing factor receptor (CRF-2) mRNA expression to specific subcortical nuclei in rat brain: Comparison with CRF-1 receptor mRNA expression. J. Neurosci. 15 6340–6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, C.P., Pearse, R.V., O'Connell, S., and Rosenfeld, M.G. 1993. Ideication of a seven transmembrane helix receptor for corticotropin-releasing factor and sauvagine in mammalian brain. Neuron 11 1187–1195. [DOI] [PubMed] [Google Scholar]
- Chen, R., Lewis, K.A., Perrin, M.H., and Vale, W.W. 1993 Expression cloning of a human corticotropin-releasing-factor receptor. Proc. Natl. Acad. Sci. USA 908967–8971. [DOI] [PMC free article] [PubMed]
- Cheng, Y. and Prusoff, W.H. 1973. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22 3099–3108. [DOI] [PubMed] [Google Scholar]
- Cook, J.V.F. and Eidne, K.A. 1997. An intramolecular disulfide bond between conserved extracellular cysteines in the gonadotropin-releasing hormone receptor is essential for binding and activation. Endocrinology 138 2800–2806. [DOI] [PubMed] [Google Scholar]
- Cuff, J.A. and Barton, G.J. 1999. Evaluation and improvement of multiple sequence methods for protein secondary structure prediction. Proteins 34 508–519. [DOI] [PubMed] [Google Scholar]
- Dautzenberg, F.M., Dietrich, K., Palchaudhuri, M.R., and Spiess, J. 1997. Ideication of two corticotropin-releasing factor receptors from Xenopus laevis with high ligand selectivity: unusual pharmacology of the type 1 receptor. J. Neurochem. 69 1640–1649. [DOI] [PubMed] [Google Scholar]
- Dautzenberg, F.M., Wille, S., Lohmann, R., and Spiess, J. 1998. Mapping of the ligand-selective domain of the xenopus-laevis corticotropin-releasing factor-receptor-1 - implications for the ligand-binding site. Proc. Natl. Acad. Sci. USA 95 4941–4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson, C.J., Sutton, S.W., Perrin, M.H., Corrigan, A.Z., Lewis, K.A., Rivier, J., Vaughan, J.M., and Vale, W.W. 1996. Cloning and characterization of human urocortin. Endocrinology 137 2167–2170. [DOI] [PubMed] [Google Scholar]
- Eckart, K., Radulovic, J., Radulovic, M., Jahn, O., Blank, T., Stiedl, O., and Spiess, J. 1999. Actions of CRF and its analogs. Curr. Med. Chem. 6 1035–1053. [PubMed] [Google Scholar]
- Elbein, A.D., Tropea, J.E., Mitchell, M., and Kaushal, G.P. 1990. Kifunensine, a potent inhibitor of the glycoprotein processing mannosidase I. J. Biol. Chem. 265 15599–15605. [PubMed] [Google Scholar]
- Fling, S. and Gregerson, D. 1986. Peptide and protein molecular weight determination by electrophoresis using a high-molar tris buffer system without urea. Anal. Biochem. 155 83–86. [DOI] [PubMed] [Google Scholar]
- Gulyas, J., Rivier, C., Perrin, M., Koerber, S.C., Sutton, S., Corrigan, A., Lahrichi, S.L., Craig, A.G., Vale, W.W., et al. 1995. Potent, structurally constrained agonists and competitive antagonists of corticotropin-releasing factor. Proc. Natl. Acad. Sci. USA 92 10575–10579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn, O., Eckart, K., Sydow, S., Hofmann, B.A., and Spiess, J. 2001. Pharmacological characterization of recombinant rat corticotropin-releasing factor binding protein using different sauvagine analogs. Peptides 22 47–56. [DOI] [PubMed] [Google Scholar]
- Karnik, S.S. and Khorana, H.G. 1990. Assembly of functional rhodopsin requires a disulfide bond between cysteine residues 110 and 187. J. Biol. Chem. 265 17520–17524. [PubMed] [Google Scholar]
- Katta, V., Chowdhury, S.K., and Chait, B.T. 1991. Use of a single-quadrupole mass spectrometer for collision-induced dissociation studies of multipy charged peptide ions produced by electrospray ionization. Anal. Chem. 63 174–178. [DOI] [PubMed] [Google Scholar]
- Kishimoto, T., Pearse, R.V., Lin, C.R., and Rosenfeld, M.G. 1995. A sauvagine/corticotropin-releasing factor receptor expressed in heart and skeletal muscle [published erratum appears in Proc. Natl. Acad. Sci. USA 1995 92: 4074]. Proc. Natl. Acad. Sci. USA 92 1108–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld, R. and Kornfeld, S. 1985. Assembly of asparagine-linked oligosaccharides. Ann. Rev. Biochem. 54 631–664. [DOI] [PubMed] [Google Scholar]
- Kostich, W.A., Chen, A., Sperle, K., and Largent, B.L. 1998. Molecular ideication and analysis of a novel human corticotropin-releasing factor (CRF) receptor: the CRF2gamma receptor. Mol. Endocrinol. 12 1077–1085. [DOI] [PubMed] [Google Scholar]
- Liaw, C.W., Lovenberg, T.W., Barry, G., Oltersdorf, T., Grigoriadis, D.E., and de Souza, E.B. 1996. Cloning and characterization of the human corticotropin-releasing factor-2 receptor complementary deoxyribonucleic acid. Endocrinology 137 72–77. [DOI] [PubMed] [Google Scholar]
- Liu, Y., Choudhury, P., Cabral, C.M., and Sifers, R.N. 1999. Oligosaccharide modification in the early secretory pathway directs the selection of a misfolded glycoprotein for degradation by the proteasome. J. Biol. Chem. 274 5861–5867. [DOI] [PubMed] [Google Scholar]
- Lovenberg, T.W., Liaw, C.W., Grigoriadis, D.E., Clevenger, W., Chalmers, D.T., De Souza, E.B., and Oltersdorf, T. 1995. Cloning and characterization of a functionally distinct corticotropin-releasing factor receptor subtype from rat brain [published erratum appears in Proc. Natl. Acad. Sci. USA 1995. 92: 5759]. Proc. Natl. Acad. Sci. USA 92 836–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maley, F., Trimble, R.B., Tarentino, A.L., and Plummer, T.H., Jr. 1989. Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal. Biochem. 180 195–204. [DOI] [PubMed] [Google Scholar]
- Mellquist, J.L., Kasturi, L., Spitalnik, S.L., and Shakin-Eshleman, S.H. 1998. The amino acid following an Asn-X-Ser/Thr sequon is an important determinant of N-linked core glycosylation efficiency. Biochemistry 37 6833–6837. [DOI] [PubMed] [Google Scholar]
- Merril, C.R., Goldman, D., Sedman, S.A., and Ebert, M.H. 1981. Ultrasensitive stain for proteins in polyacrylamide gels shows regional variation in cerebrospinal fluid proteins. Science 211 1437–1438. [DOI] [PubMed] [Google Scholar]
- Montecucchi, P. and Henschen, A. 1981. Amino acid composition and sequence analysis of sauvagine, a new active peptide from the skin of Phyllomedusa sauvagei. Int. J. Peptide Protein Res. 18 113–120. [DOI] [PubMed] [Google Scholar]
- Myers, D.A., Trinh, J.V., and Myers, T.R. 1998. Structure and function of the ovine type 1 corticotropin releasing factor receptor (CRF1) and a carboxyl-terminal variant. Mol. Cell. Endocrinol. 144 21–35. [DOI] [PubMed] [Google Scholar]
- Nielsen, H., Engelbrecht, J., Brunak, S., and Heijne, G.v. 1997. Ideication of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 10 1–6. [DOI] [PubMed] [Google Scholar]
- Ohyama, K., Yamano, Y., Sano, T., Nakagomi, Y., Hamakubo, T., Morishima, I., and Inagami, T. 1995. Disulfide bridges in extracellular domains of angiotensin II receptor type IA. Regul. Pept. 57 141–147. [DOI] [PubMed] [Google Scholar]
- Palchaudhuri, M.R., Hauger, R.L., Wille, S., Fuchs, E., and Dautzenberg, F.M. 1999. Isolation and pharmacological characterization of two functional splice variants of corticotropin-releasing factor type 2 receptor from Tupaia belangeri. J. Neuroendocrinol. 11 419–428. [DOI] [PubMed] [Google Scholar]
- Palchaudhuri, M.R., Wille, S., Mevenkamp, G., Spiess, J., Fuchs, E., and Dautzenberg, F.M. 1998. Corticotropin-releasing factor receptor type 1 from Tupaia belangeri- cloning, functional expression and tissue distribution. Eur. J. Biochem. 258 78–84. [DOI] [PubMed] [Google Scholar]
- Perlman, J.H., Wang, W., Nussenzveig, D.R., and Gershengorn, M.C. 1995. A disulfide bond between conserved extracellular cysteines in the thyrotropin-releasing hormone receptor is critical for binding. J. Biol. Chem. 270 24682–24685. [DOI] [PubMed] [Google Scholar]
- Perrin, M., Donaldson, C., Chen, R., Blount, A., Berggren, T., Bilezikjian, L., Sawchenko, P., and Vale, W. 1995. Ideication of a second corticotropin-releasing factor receptor gene and characterization of a cDNA expressed in heart. Proc. Natl. Acad. Sci. USA 92 2969–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin, M.H., Donaldson, C.J., Chen, R., Lewis, K.A., and Vale, W.W. 1993. Cloning and functional expression of a rat brain corticotropin releasing factor (CRF) receptor. Endocrinology 133 3058–3061. [DOI] [PubMed] [Google Scholar]
- Perrin, M.H., Sutton, S., Bain, D.L., Berggren, W.T., and Vale, W.W. 1998. The first extracellular domain of corticotropin releasing factor-R1 contains major binding determinants for urocortin and astressin. Endocrinology 139 566–570. [DOI] [PubMed] [Google Scholar]
- Perrin, M.H., Sutton, S.W., Cervini, L.A., Rivier, J.E., and Vale, W.W. 1999. Comparison of an agonist, urocortin, and an antagonist, astressin, as radioligands for characterization of corticotropin-releasing factor receptors. J. Pharmacol. Exp. Ther. 288 729–734. [PubMed] [Google Scholar]
- Potter, E., Sutton, S., Donaldson, C., Chen, R., Perrin, M., Lewis, K., Sawchenko, P.E., and Vale, W.W. 1994. Distribution of corticotropin-releasing factor receptor mRNA expression in the rat brain and pituitary. Proc. Natl. Acad. Sci. USA 91 8777–8781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, L.J., Leung, A.T., Xiong, Y.T., Marx, K.A., and Abou-Samra, A.B. 1997. Extracellular cysteines of the corticotropin-releasing factor-receptor are critical for ligand interaction. Biochemistry 36 12442–12448. [DOI] [PubMed] [Google Scholar]
- Radulovic, J., Blank, T., Eckart, K., Radulovic, M., Stiedl, O., and Spiess, J. 1999. CRF and CRF receptors. In Regulatory peptides and cognate receptors (Richter, D., ed) Vol. 26, pp. 67–90. Springer, Heidelberg. [DOI] [PubMed]
- Radulovic, J., Rühmann, A., Liepold, T., and Spiess, J. 1999. Modulation of learning and anxiety by corticotropin-releasing factor (CRF) and stress: differential roles of CRF receptors 1 and 2. J. Neurosci. 19 5016–5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulovic, J., Sydow, S., and Spiess, J. 1998. Characterization of native corticotropin-releasing factor-receptor type 1 (crfr1) in the rat and mouse central nervous system. J. Neurosci. Res. 54 507–521. [DOI] [PubMed] [Google Scholar]
- Rühmann, A., Bonk, I., Lin, C.J.R., Rosenfeld, M.G., and Spiess, J. 1998. Structural requirements for peptidic antagonists of the corticotropin-releasing factor receptor (CRFR): Development of CRFR2 beta-selective antisauvagine-30. Proc. Natl. Acad. Sci. USA 95 15264–15269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rühmann, A., Köpke, A.K.E., Dautzenberg, F.M., and Spiess, J. 1996. Synthesis and characterization of a photoactivatable analog of corticotropin-releasing factor for specific receptor labeling. Proc. Natl. Acad. Sci. USA 93 10609–10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarese, T.M., Wang, C.D., and Fraser, C.M. 1992. Site-directed mutagenesis of the rat m1 muscarinic acetylcholine receptor. Role of conserved cysteines in receptor function. J. Biol. Chem. 267 11439–11448. [PubMed] [Google Scholar]
- Settineri, C.A. and Burlingame, A.L. 1996. Structural characterization of protein glycosylation using HPLC/electrospray ionization mass spectrometry and glycosidase digestion. In Protein and peptide analysis by mass spectrometry (Chapmann, J.R., ed) Vol. 61, pp. 255–278. Humana Press, Totowa, NJ. [DOI] [PubMed]
- Spiess, J., Dautzenberg, F.M., Sydow, S., Hauger, R.L., Ruhmann, A., Blank, T., and Radulovic, J. 1998. Molecular-properties of the CRF receptor. Trends Endocrinol. Metab. 9 140–145. [DOI] [PubMed] [Google Scholar]
- Spiess, J., Rivier, J., Rivier, C., and Vale, W. 1981. Primary structure of corticotropin-releasing factor from ovine hypothalamus. Proc. Natl. Acad. Sci. USA 78 6517–6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenzel, P., Kesterson, R., Yeung, W., Cone, R.D., Rittenberg, M.B., and Stenzel-Poore, M.P. 1995. Ideication of a novel murine receptor for corticotropin-releasing hormone expressed in the heart. Mol. Endocrinol. 9 637–645. [DOI] [PubMed] [Google Scholar]
- Strader, C.D., Fong, T.M., Tota, M.R., Underwood, D., and Dixon, R.A. 1994. Structure and function of G protein-coupled receptors. Ann. Rev. Biochem. 63 101–132. [DOI] [PubMed] [Google Scholar]
- Sydow, S., Flaccus, A., Fischer, A., and Spiess, J. 1999. The role of the fourth extracellular domain of the rat corticotropin-releasing factor receptor type 1 in ligand binding. Eur. J. Biochem. 259 55–62. [DOI] [PubMed] [Google Scholar]
- Sydow, S., Radulovic, J., Dautzenberg, F.M., and Spiess, J. 1997. Structure-function relationship of different domains of the rat corticotropin-releasing factor-receptor. Mol. Brain Res. 52 182–193. [DOI] [PubMed] [Google Scholar]
- Udenfriend, S., Gerber, L.D., Brink, L., and Spector, S. 1985. Scintillation proximity radioimmunoassay utilizing 125I-labeled ligands. Proc. Natl. Acad. Sci. USA 82 8672–8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale, W., Spiess, J., Rivier, C., and Rivier, J. 1981. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science 213 1394–1397. [DOI] [PubMed] [Google Scholar]
- Vaughan, J., Donaldson, C., Bittencourt, J., Perrin, M.H., Lewis, K., Sutton, S., Chan, R., Turnbull, A.V., Lovejoy, D., Rivier, C., et al. 1995. Urocortin, a mammalian neuropeptide related to fish urotensin I and to corticotropin-releasing factor. Nature 378 287–292. [DOI] [PubMed] [Google Scholar]
- Vilardaga, J.P., Di Paolo, E., Bialek, C., De Neef, P., Waelbroeck, M., Bollen, A., and Robberecht, P. 1997. Mutational analysis of extracellular cysteine residues of rat secretin receptor shows that disulfide bridges are essential for receptor function. Eur. J. Biochem. 246 173–180. [DOI] [PubMed] [Google Scholar]
- Vita, N., Laurent, P., Lefort, S., Chalon, P., Lelias, J.M., Kaghad, M., Le Fur, G., Caput, D., and Ferrara, P. 1993. Primary structure and functional expression of mouse pituitary and human brain corticotrophin releasing factor receptors. FEBS Lett. 335 1–5. [DOI] [PubMed] [Google Scholar]
- Yu, J., Xie, L.Y., and Abou-Samra, A.B. 1996. Molecular cloning of a type A chicken corticotropin-releasing factor receptor with high affinity for urotensin I. Endocrinology 137 192–197. [DOI] [PubMed] [Google Scholar]