

Abstract
Ribonuclease A aggregates (dimers, trimers, tetramers, pentamers) can be obtained by lyophilization from 40% acetic acid solutions. Each aggregate forms two conformational isomers distinguishable by different basic net charge. The crystal structure of the two dimers has recently been determined; the structure of the higher oligomers is unknown. The results of the study of the two trimeric and tetrameric conformers can be summarized as follows: (1) RNase A trimers and tetramers form by a 3D domain-swapping mechanism. N-terminal and C-terminal types of domain swapping could coexist; (2) the secondary structures of the trimeric and tetrameric conformers do not show significant differences if compared with the secondary structure of monomeric RNase A or its two dimers; (3) a different exposure of tyrosine residues indicates that in the aggregates they have different microenvironments; (4) the two trimeric and tetrameric conformers show different susceptibility to digestion by subtilisin; (5) dimers, trimers, and tetramers of RNase A show unwinding activity on double-helical poly(dA-dT) • poly(dA-dT), that increases as a function of the size of the oligomers; (6) the less basic conformers are more stable than the more basic ones, and a low concentration in solution of trimers and tetramers favors their stability, which is definitely increased by the interaction of the aggregates with poly(dA-dT) • poly(dA-dT); (7) the products of thermal dissociation of the two trimers indicate that their structures could be remarkably different. The dissociation products of the two tetramers allow the proposal of two models for their putative structures.
Keywords: RNase A oligomers, trimers and tetramers of RNase A, properties of trimeric and tetrameric RNase A, RNase A aggregates higher than dimers
The study of the manner in which proteins aggregate in vitro can help in understanding the process of protein aggregation in vivo, and, therefore, also the origin of pathologic proteins responsible for several severe diseases.
Although it has recently been reported that native ribonuclease A can dimerize at neutral pH (Park and Raines 2000), it is known that by lyophilization from 30–50% acetic acid solutions RNase A gives rise to oligomers (Crestfield et al. 1962) ranging from dimers to pentamers and possibly higher aggregates, each oligomeric species existing in the form of at least two conformational isomers (Gotte et al. 1999).
Dimeric RNase A obtained by lyophilization consists of a minor and a major component that are in the ratio of about 1:3–1:4. Their crystal structures have been determined (Liu et al. 1998, 2001), and the two dimeric conformers are 3D domain-swapping dimers formed by the exchange of the N-terminal α-helix of each monomeric subunit in the case of the minor dimer, of the C-terminal β-strand, instead, for the major RNase A dimer. This unique behavior of RNase A, in the dimeric aggregates of which the two known mechanisms of protein aggregation by domain swapping coexist, expands the range of possibilities for the formation of oligomeric structures. It, on one hand, supports the various models involving a 3D domain-swapping mechanism (Klafki et al. 1993; Schlunegger et al. 1997; Liu et al. 1998; Cohen and Prusiner 1998; Chiti et al. 2000) already proposed to explain amyloid fibers formation; on the other hand, allows the proposal that domain swapping can occur in every protein at high local concentration under partially destabilizing conditions, and that polymerization can occur by the exchange of multiple domains (Liu et al. 2001). Moreover, by combining the 3D domain swapping and the "polar zipper" (Perutz et al. 1994) hydrogen bonding observed in the major dimer of RNase A, a 3D domain-swapping zipper model can be proposed to explain protein fiber formation (Liu et al. 2001).
Besides the definition of their crystal structures, a study of several biochemical and biophysical properties of the two RNase A dimers has also been performed (Gotte et al. 1999; Sorrentino et al. 2000). Here we present experimental results concerning a partial characterization of the two trimeric and tetrameric conformers of RNase A, and some additional properties of the dimeric forms of the enzyme.
Results and Discussion
Is the structure of the trimeric and tetrameric aggregates formed by "swapping" of the N- or C-terminals of their component subunits?
We used the procedure outlined by Ciglic et al. (1998) with divinyl sulfone as histidine cross-linking agent to estimate the amount of domain swapping, if present, in trimeric and tetrameric aggregates of RNase A, that were obtained as described (Gotte et al. 1999). Although lysine residues could also react with divinyl sulfone, the nitrogen of lysines should be protonated at pH 5.0, at which the reaction was carried out, and therefore, definitely less reactive than histidine nitrogen, that could be selectively preferred in the cross-linking reaction.
If, because of domain swapping, the active sites of the two oligomeric species were composite as they are in RNase A dimers (Liu et al. 1998, 2001) and independently of an N-terminal or C-terminal type of domain swapping, histidine residues at positions 12 of one subunit, and 119 of the other, forming an active site, would be cross-linked in a dimer. The same should be true for a third or a fourth subunit in a trimer or a tetramer, and the products of the reaction analyzed by SDS-PAGE should move as dimers, trimers, or tetramers, respectively. Otherwise, if in the absence of domain swapping two histidines of the same subunit were cross-linked, the analysis by SDS-PAGE would reveal a band migrating in any case as a monomer. Both conformers of each species have been examined. The reaction (which anyhow never went to completion) was protracted for 8 days at 20°C, a temperature at which the possibility for the aggregates to dissociate is greatly reduced. The results of one of several similar experiments appear in Figures 1A and B and 2A and B ▶ ▶. For each trimeric conformer (Fig. 1A ▶, B) three main bands were detected by SDS-PAGE after about a 24-h reaction. The upper one corresponds to the trimer, the second to the dimer, and the lower one to the monomer. It must be pointed out that the splitting of each of these bands is a side effect of the reaction (see Ciglic et al. 1998). We interpret the appearance of the cross-linked dimer as being due to a time-dependent cross-linkage of two subunits out of three as a first event, followed by the final cross-linking of the third subunit. The first step would, therefore, bring in the SDS-gel to the dissociation and appearance of the cross-linked dimer. The monomer detected at the bottom of both parts A and B of Figure 1 ▶ corresponds to the amount of trimer that did not yet react at the times chosen for the analysis. The interpretation put forth to explain the presence of a cross-linked dimer in the gel can be supported (1) by its appearence after 2 h of reaction, when no trimer yet is cross-linked, and (2) by the results of a parallel experiment (PAGE under nondenaturing conditions) performed to check the state of the trimer dissolved in the same reaction buffer at the same temperature (see legend to Fig. 1 ▶) but in the absence of divinyl sulfone, indicating that the trimer survived intact for the entire reaction time (data not shown).
Fig. 1.

Cross-linkage with divinyl sulfone of the subunits of the two conformers of trimeric RNase A. The less basic (A) and the more basic (B) trimers (T1 and T2, respectively) were incubated (final concentration, 0.8 mg prot./mL) in 0.1 M sodium acetate buffer, pH 5, with divinyl sulfone (reagent/protein ratio, 1000) at 20°C. At the times indicated on the top of the lanes (whose numbering is at the bottom of the gel), 10-μg aliquots were withdrawn and reaction stopped by adding β-mercaptoethanol (0.2 M, final concentration); 7 and 9 μg of T1 (A) and T2 (B), respectively, were then analyzed by 12% SDS-PAGE.
Fig. 2.

Cross-linkage with divinyl sulfone of the subunits of the two conformers of tetrameric RNase A. The less basic (A) and the more basic (B) tetramers (TT1 and TT2, respectively) were incubated (final concentration, 1 mg prot./mL) in 0.1 M sodium acetate, pH 5, with divinyl sulfone (reagent/protein ratio, 1000) at 20°C. At the times indicated on the top of the lanes (whose numbering is at the bottom of the gel), 10-μg aliquots were withdrawn from the incubation mixture and reaction was stopped by adding β-mercapto ethanol (0.2 M, final concentration); 7 μg of TT1 (A) or TT2 (B) were then analyzed by 12% SDS-PAGE.
As for the two tetramers of RNase A (TT1 and TT2, the less basic and more basic conformer, respectively) subjected to the same cross-linking reaction (Fig. 2A ▶, B), four types of reaction products appear in going from the top to the bottom of the gel: the cross-linked tetramer, a cross-linked trimer, a cross-linked dimer, and a monomer. Here too, to explain the presence of trimer and dimer, we think that first two out of four, then three, and finally the four subunits constituting the tetramer were cross-linked. The data supporting this view are (1) that the cross-linked trimer and dimer appear before the tetramer itself results as being cross-linked: indeed, cross-linked TT1 appears in a definite amount only after 72 h of reaction (Fig. 2A ▶), cross-linked TT2 after 48 h (Fig. 2B ▶). Moreover, (2) in a parallel experiment the two tetramers, in the absence of the cross-linking reagent, showed themselves to be remarkably stable at 20°C during the rather long reaction time (data not shown). In conclusion, the results described above support the view that also RNase A oligomers higher than dimers form by a domain-swapping mechanism.
Helix-unwinding activity of various RNase A aggregates
Many years ago bovine pancreatic RNase A was found to be an efficient "destabilizer" of double-stranded DNA. In the original experiment performed by Felsenfeld et al. (1963), the Tm of calf thymus DNA interacting with bovine pancreatic RNase was markedly lowered. This destabilizing ability of RNase A was later found to be common to several mammalian pancreatic-type ribonucleases, and to be a function of the "basicity" of the enzyme protein. In particular, RNases endowed with a higher number of specifically located positive charges than RNase A are more effective than the latter enzyme in destabilizing double-helical DNA (Carsana et al. 1983; Sorrentino and Libonati 1994; Libonati and Sorrentino 2001).
The experiments shown in Figure 3 ▶ indicate that poly(dA-dT) • poly(dA-dT) is destabilized by its interaction with various RNase A aggregates. A modest but clear shift towards hyperchromic values of the thermal transition profile of poly(dA-dT) • poly(dA-dT) interacting with monomeric RNase A, its major and more basic dimer (D2), its more basic trimer (T2), and more basic tetramer (TT2) can be observed between about 30 and 55°C. The helix-unwinding effect appears to be, in particular between 40 and 55°C, a function of the size of the oligomers, i.e., of the increasing of their basic charge density. This (1) is a further support to the hypothesis (Libonati and Sorrentino 1992; Sorrentino and Libonati 1994; Libonati and Sorrentino 2001) that the efficiency of degradation of double-stranded RNA by single-strand–preferring ribonucleases depends on the number (or density) of specifically located basic amino acid residues in the enzyme protein, through its ability to destabilize the nucleic acid secondary structure; and (2) therefore, explains why the more basic conformers of each RNase A aggregate degrade double-stranded RNA more efficiently than the less basic conformers (Gotte et al. 1999).
Fig. 3.

Effect of monomeric RNase A and its aggregates on the thermal transition profile of poly(dA-dT) • poly(dA-dT). 1.4 mL of 0.01 M imidazole-HCl/0.035 M NaCl buffer, pH 7, contained (in a stoppered cuvette) double-stranded poly(dA-dT) • poly(dA-dT) (13 μg/mL), and the various RNase A species (14 μg/mL). Absorbances at 260 nm were determined with a Beckman DU 650 spectrophotometer, equipped with a thermostatically controlled water bath, after maintaining the nucleic acid–protein mixture at the chosen temperature for 2.5 min (i.e., after hyperchromicity reached a stable value). (Open triangle) thermal transition profile of the nucleic acid in the absence of protein(s). (Filled circle) RNase A monomer; (open square) its major dimer (D2); (filled triangle) its more basic trimer (T2); (+) its more basic tetramer (TT2). Starting value of A260 was about 0.250. The contribution of protein(s) was nil.
Stability of the trimeric and tetrameric aggregates of RNase A in the absence or presence of poly(dA-dT) • poly(dA-dT), and as a function of protein concentration
The integrity of the structure of the RNase A aggregates is a crucial point for the validity of the experiment shown in Figure 3 ▶. From the experimental results presented in Figure 4 ▶, in which the more basic dimeric (D2), trimeric (T2), and tetrameric (TT2) conformers in the absence of the nucleic acid were held at the temperatures indicated for the same times (2.5 min) where they were maintained when their helix-unwinding activity was examined (Fig. 3 ▶), it could be deduced that, although the dimer was significantly more stable, a large amount of the trimer and the tetramer dissociated within the range of temperatures used.
Fig. 4.

Stability of trimer T2 and tetramer TT2 as a function of temperature. Trimer T2 (gray bar) and tetramer TT2 (empty bar), as well as the major RNase A dimer, D2 (dark bar), dissolved (final concentration, 1 mg prot./mL) in 0.01 M imidazole/0.035 M NaCl buffer, pH 7, were maintained at the various temperatures for 2.5 min, and then transferred to an ice-cold water bath; 7 μg of each sample were withdrawn and analyzed by cathodic PAGE under nondenaturing conditions. Ten percent polyacrylamide gels were run at 4°C for 100 min at 20 mA. The amounts of undissociated aggregates were estimated by measuring the band densities with an ImageMaster VDS (Pharmacia Biotech), and expressed as percentages of controls.
However, the pattern changed when the stability of the structure of T2 and TT2 was checked in the presence of poly(dA-dT) • poly(dA-dT) (Fig. 5 ▶). A mixture of the double-helical polydeoxyribonucleotide and trimer T2 was held at 45°C for 2.5 min, then rapidly brought to 25°C, and applied onto a gel filtration column in HPLC. Elution was performed with 0.2 M sodium phosphate buffer, pH 6.7. The nucleic acid eluted much before all protein species. The residual trimer and its dissociation products—dimer and monomer—could be found at their typical elution positions (details are given in the legend to the figure ▶). The quaication of each eluted species indicated that while in the absence of poly(dA-dT) • poly(dA-dT) only 31% of the trimer (empty bar) could be recovered after 2.5 min at 45°C, 72% of it (dark bar) could instead be found after maintaining the trimer under identical conditions but in the presence of the nucleic acid. Moreover, while in the absence of poly(dA-dT) • poly(dA-dT) the dissociation products of T2 were 42% of dimer and 27% of monomer (empty bars), in the presence of the nucleic acid they were reduced to 17% and 11%, respectively (dark bars). Similar results were obtained with the more basic tetrameric conformer (TT2) (data not shown): the amounts of tetramer recovered after its exposure to 45°C in the absence or presence of the nucleic acid were 28 and 44%, respectively. Accordingly, the amounts of the dissociation products—dimer and monomer—were 43 and 29%, respectively, in the absence of poly(dA-dT) • poly(dA-dT), and 33 and 23% in its presence. In conclusion, the structures of trimer T2 and tetramer TT2 were definitely preserved by the interaction with the nucleic acid, thus giving support to the significance of the experiment shown in Figure 3 ▶.
Fig. 5.

Stability of trimer T2 in the absence or presence of poly(dA-dT) • poly(dA-dT). A sample of the more basic trimeric RNase A aggregate (T2), dissolved (final concentration, 200 μg prot./mL) in 0.01 M imidazole/0.035 M NaCl buffer, pH 7, was maintained at 45°C for 2.5 min in the absence or presence of poly(dA-dT) • poly(dA-dT) (final concentration, 240 μg/mL). Aliquots, containing 12 μg of protein alone or mixed with 14.4 μg of the nucleic acid, were withdrawn and loaded on a TSK gel G2000 SW HPLC column. Elution was performed with 0.2 M sodium phosphate buffer, pH 6.7, at room temperature. The areas of the peaks eluted at the positions (previously determined with standard samples) of trimeric, dimeric, or monomeric RNase A were expressed as a percentage of the input. T2, the trimer recovered after incubation at 45°C in the absence (empty bar) or presence (dark bar) of the nucleic acid. D and M, dimeric or monomeric RNase A, respectively, recovered as products of the dissociation of T2 in the absence (empty bars) or presence (dark bars) of the nucleic acid. Retention time for poly(dA-dT) • poly(dA-dT) was 50 min versus 71 min for T2, 76 min for D and 86 min for M.
The stability of the aggregates also depends on their concentration in solution. The two trimers and tetramers, dissolved at two different concentrations (0.4 or 4.0 mg/mL) in 0.03 M sodium phosphate buffer, pH 6.7, were kept at 35°C for 48 and 2 h, respectively. The aggregates were then analyzed by ion-exchange chromatography in FPLC. The results obtained in several experiments (data not shown) consistently indicated that while between 80 and 90% of the two trimeric conformers incubated at the lower concentration could be recovered intact after 48 h, only 10–15% of them retained their structure if incubated for the same time at a concentration of 4 mg/mL. The same is true for the two tetramers: about 90% of them could be recovered intact after 2 h incubation at a concentration of 0.4 mg/mL, whereas the amount of undissociated tetramers was reduced to 60–65% if they were incubated for the same time at a concentration of 4 mg/mL. These results may tentatively be interpreted in terms of an equilibrium between tetramers, trimers, dimers, and monomer: because the equilibrium constant does not change, if the concentration of any type of aggregate (left side of the reaction) increases, the concentration of its dissociation product(s) (right side of the reaction) must also increase.
Kinetics of thermal dissociation of trimeric and tetrameric aggregates of RNase A
The dissociation kinetics of the RNase A trimeric or tetrameric conformers was studied by keeping solutions (1 mg/mL) of each protein species in 0.03 M sodium phosphate buffer, pH 6.7, at 35°C for the times indicated in Figure 6A and B ▶. At each time, aliquots of the samples were withdrawn, cooled, and applied onto an ion-exchange column. Elution was performed at room temperature with a linear (2 h) 0.09–0.18 M sodium phosphate gradient, at pH 6.7. Each point appearing in Figure 6A and B ▶ (which shows one of the various experiments performed) is the amount of undissociated trimeric or tetrameric conformer recovered in the ion-exchange chromatography. The area of each peak was measured and expressed as a percentage of the area of the corresponding input (trimers or tetramers at zero time). The results were (1) that both more basic conformers (T2 and TT2) are more labile than their less basic counterparts, a property also shared by dimeric RNase A (Sorrentino et al. 2000), and (2) that both trimers are remarkably more resistant than the two tetramers. In fact, although after 2 h at 35°C about 40% of TT1 and 60% of TT2 were already dissociated, almost no dissociation could be detected for both T1 and T2. We believe that the small difference in stability between T1 and T2 (Fig. 6A ▶) might be significant on the basis of consistent indications as to the lower stability of T2 obtained in other experiments (not shown). It might also be worth mentioning that after 8 days at 35°C under higher ionic strength conditions (0.1 M sodium phosphate buffer, pH 6.7) the two trimers showed no dissociation, while the more basic tetramer (TT2) was completely dissociated, and only about 5% of the less basic one (TT1) could be recovered (data not shown).
Fig. 6.

Kinetics of the dissociation of the two trimeric or tetrameric RNase A conformers. Samples of T1 and T2 (A) or TT1 and TT2 (B), dissolved (final concentration, 1 mg prot./mL) in 0.03 M sodium phosphate buffer, pH 6.7, were incubated at 35°C. At the times indicated, aliquots (60 μg) were withdrawn and cooled to 0°C. They were then loaded onto a cation-exchange (Source 15S HR 10/10) column in FPLC. Elution was performed with a 2 h linear gradient (0.09–0.18 M sodium phosphate buffer, pH 6.7) at room temperature. The areas of the peaks were measured and expressed as percentages of the input loaded at zero time.
Products of the dissociation of the trimeric and tetrameric aggregates of RNase A
To learn how they dissociate, the two trimeric or tetrameric RNase A conformers, dissolved in 0.03 M sodium phosphate buffer, pH 6.7, were kept at 35°C for the times indicated in Figure 7A and B ▶. At each time, aliquots were withdrawn and analyzed by cathodic PAGE under nondenaturing conditions. As for the two forms of trimer (Fig. 7A ▶), lanes 6 and 7 of the gel show the bands of the remaining, not yet dissociated trimers T1 and T2, and of the products of their partial dissociation. The less basic trimer (T1) produces major dimer (D2), a small amount of minor dimer (D1) and monomer (M). No traces of minor dimer can instead be found as dissociation product of trimer T2, which only dissociates to a rather large amount of major dimer and proportionally less monomer.
Fig. 7.
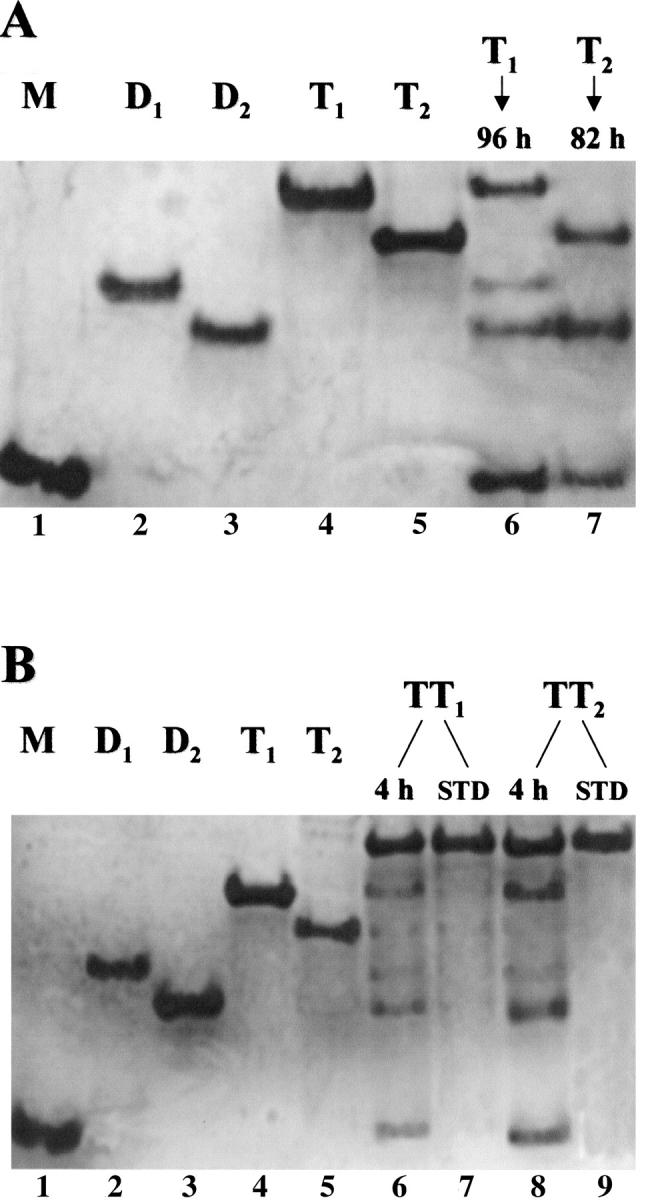
Analysis of the products of thermal dissociation of RNase A trimers and tetramers by cathodic PAGE under nondenaturing conditions. Trimers and tetramers (1 mg/mL of 0.03 M sodium phosphate buffer, pH 6.7) were maintained at 35°C for the times indicated. (A) Lanes 1–5: "standard" (undissociated) aggregates of RNase A: 1. Monomeric RNase A (M), 8 μg. 2. Minor dimer (D1), 5 μg. 3. Major dimer (D2), 5 μg. 4. Trimer T1, 6 μg. 5. Trimer T2, 6 μg. Lane 6: trimer T1, 6 μg, after 96 h incubation. Lane 7: trimer T2, 5 μg, after 82 h incubation. (B) Lanes 1–5: "standard" (undissociated) aggregates of RNase A, as under (A). Lane 6: tetramer TT1, 6 μg, after 4-h incubation. Lane 7: (STD) standard TT1, 5 μg. Lane 8: Tetramer TT2, 6 μg, after 4-h incubation. Lane 9: (STD) standard TT2, 5 μg.
By taking into account the linear model proposed for an RNase A trimer (Liu et al. 2001), in which two monomers are associated with each other through the C-terminal type of domain swapping, and a third monomer is associated with one of them through the N-terminal type of domain swapping (the central subunit having, therefore, both the N- and C-terminal type of exchange), the dissociation products of T1 and T2 suggest that the structures of the two trimers could differ remarkably. Indeed, from the products of the dissociation of trimer T1—that we assume to be identical to the model proposed (Liu et al. 2001; see also Fig. 8 ▶)—it appears that both the domain swapping involving the two N-terminal ends (forming the "minor dimer" contained in the trimer) and the one involving the C-terminal ends (forming the "major dimer" contained in the trimer) dissociate. Trimer T2 appears instead to dissociate exclusively in the forms of major dimer and monomer.
Fig. 8.
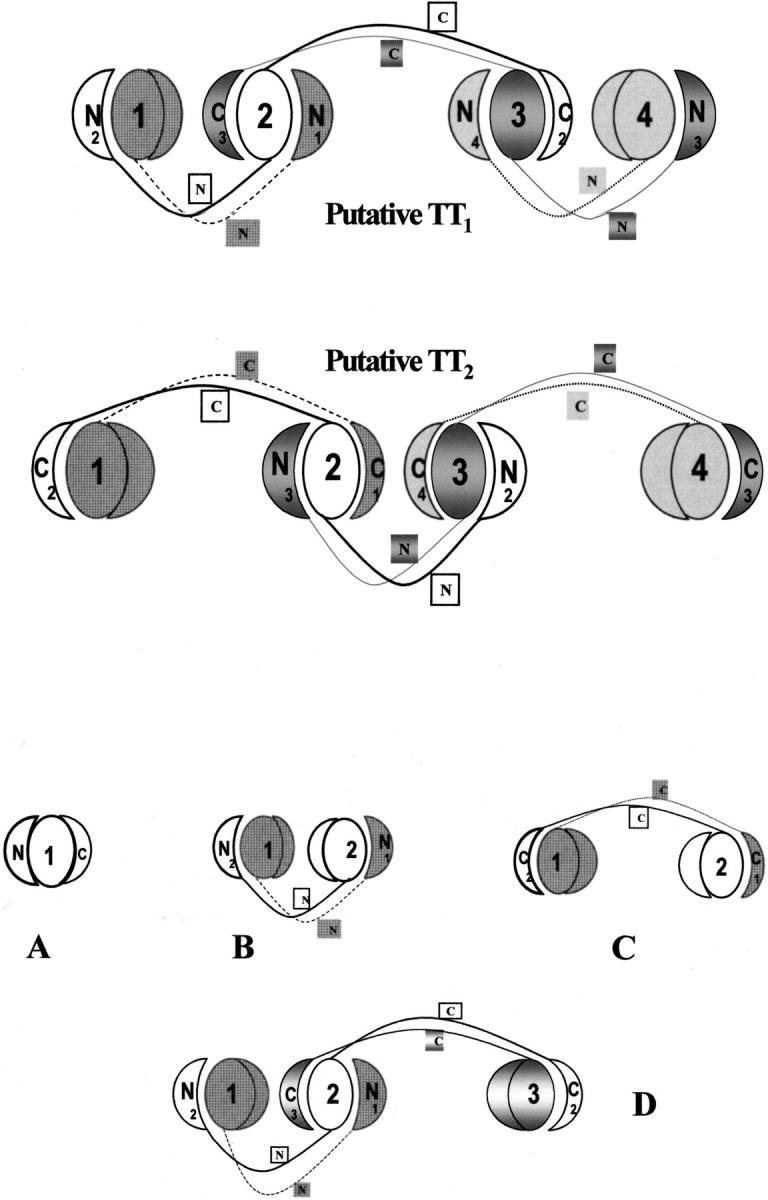
Models of the putative structures of the two tetrameric conformers of RNase A. In the subunits 1, 2, 3, 4 of both models the N- and C-terminal ends are shown as half circles and quarter circles. The N- and C-termini of each monomer involved in the domain-swapping mechanism are labeled and slightly removed from the labeled (1, 2, 3, 4) "core"-domain (full circles) of each subunit. A, model of RNase A; B, model of RNase A minor dimer (Liu et al. 1998); C, model of RNase A major dimer, and D, putative model of an RNase A trimer (Liu et al. 2001).
As for the two RNase A tetramers, Figure 7B ▶ shows the results obtained. The less basic tetramer, TT1 (lane 6), dissociates mainly in the form of less basic trimer (T1), major dimer (D2), and monomer; lower amounts of the more basic trimer (T2) and minor dimer (D1) are also present. The tetramer TT2 (lane 8), which is less stable than TT1 (see Fig. 6 ▶), dissociates to remarkable amounts of the less basic trimer (T1), major dimer (D2), and monomer, but traces of minor dimer (D1) and T2 can also be seen. On the basis of these results, one might postulate that, among other possible models (Bennet et al. 1995), the less basic tetramer (TT1) could be a linear structure composed of one central major dimer, each monomer of which would be linked through an N-terminal type of domain swapping to another monomer. The opposite structure, i.e., a central minor dimer, each monomer of which would exchange its C-terminal end with that of another monomer, could be that of tetramer TT2 (see Fig. 8 ▶). In favor of these ideas are: (1) the higher basicity of TT2 (Gotte et al. 1999), ascribable to the presence of two C-terminal swapping conformations versus one N-terminal type of domain swapping (the opposite would occur in tetramer TT1); (2) the higher apparent molecular weight of TT2, as detected in gel filtration experiments (Gotte et al. 1999), and, in turn, attributable to a greater extension, and therefore, to a higher steric hindrance, of the major dimer if compared to the minor dimer (Liu et al. 2001); (3) its higher dissociability, which could be due to the lower stability of the major dimer compared to that of the minor dimer (Sorrentino et al. 2000).
Circular dichroism analyses
The near-UV spectra of monomeric RNase A and its aggregates (Fig. 9 ▶) are characterized by two dichroic bands, one negative, centered at 278 nm, the other positive, centered at 242 nm. These signals can reasonably be attributed to the phenolic rings of tyrosine residues (Simons and Blout 1968; Grandi et al. 1979). It has already been reported (Sorrentino et al. 2000) that in going from the monomer to the minor and to the major dimer, the CD signals become more positive at 242 nm, and less negative at 278 nm. This finding has been reported as being the evidence of a progressively higher exposure of the phenolic rings of tyrosine residues (Grandi et al. 1979). Although the minor RNase A dimer is formed by the swapping of the N-terminal helixes (residues 1–15) of each subunit (Liu et al. 1998), the structure of the major dimer is due to the domain swapping of the C-terminal β-strands (residues 116–124) of each monomer (Liu et al. 2001). One tyrosine (Tyr-25) is located rather close to the loop (residues 16–22), termed the hinge loop (Bennet et al. 1994), linking the two domains of the minor dimer. Another tyrosine residue (Tyr-115) is located in the hinge loop of the major dimer (residues 112–115). These two tyrosines could reasonably be responsible for most of the differences observed in the CD spectra being located rather or very close to the interchangable portions of the two monomers constituting the minor or major dimer. Therefore, it could be possible that the changes observed in the CD signals are mainly due to those residues.
Fig. 9.

Near UV-CD spectra of monomeric RNase A and its oligomeric aggregates. Each protein species was dissolved in 0.1 M sodium phosphate buffer, pH 6.7. Details are given in the Materials and Methods section. (—) monomer, M; (• • • •) minor dimer, D1; (- - - -) major dimer, D2; (-•-•-) less basic trimer, T1; (– –) more basic trimer, T2; (– - – -) less basic tetramer, TT1; (-••-••-) more basic tetramer, TT2.
The near-UV CD spectra of the two trimeric and the two tetrameric conformers are similar to each other and lie between the spectra of the minor and major dimers. Thus, here again, the data obtained allow us to postulate that trimers and tetramers of RNase A could combine both types of domain swapping, involving the N-terminal and the C-terminal ends of their monomeric subunits.
A quantitative analysis of the CD data in terms of secondary structure content of the various oligomers was performed by using the deconvolution of Boehm et al. (1992). The results obtained, summarized in Table 1, indicate that the percentage values calculated for α-helix, β-structure, and random coil are very similar for the various oligomeric species, suggesting that no significant changes in their secondary structures occur. The percentage values for the RNase A monomer, and its minor and major dimer are rather close to those determined by X-ray crystallographic analyses of the crystal structures of the two dimeric conformers of RNase A (Y. Liu, pers. comm.).
Table 1.
Secondary structure content of the various oligomeric species evaluated from CD measurements
| α-Helix | β-Structure | β-turns | Random coil | Total | |
| Monomer | 16.7 | 33.7 | 19.8 | 29.8 | 100 |
| Minor dimer | 16.2 | 29.1 | 21.6 | 33.1 | 100 |
| Major dimer | 15.8 | 31.8 | 21.3 | 32.9 | 100 |
| Trimer 1 | 14.9 | 33.0 | 20.6 | 31.5 | 100 |
| Trimer 2 | 15.0 | 33.5 | 20.3 | 31.2 | 100 |
| Tetramer 1 | 16.7 | 27.2 | 21.0 | 35.1 | 100 |
| Tetramer 2 | 16.9 | 32.3 | 19.9 | 30.9 | 100 |
CD spectra were recorded with three different preparations of the various aggregates; variations in secondary structure estimates were less than 2%.
Susceptibility of the two forms of RNase A trimers or tetramers to digestion by subtilisin
SDS-PAGE analyses of aliquots of each trimeric or tetrameric conformer digested with subtilisin for 2, 4, and 8 h at 5°C are shown in Figure 10A and B ▶. It is worth pointing out that in SDS-PAGE each type of RNase A aggregate dissociates into monomer. After incubation with the protease two close bands can be detected for each RNase A species analyzed, as it was found with the two RNase A dimers (Nenci et al. 2001). The slower moving band, migrating as the control (untreated RNase A), represents residual, not digested RNase A. The faster moving band is the S-protein. Figure 10C ▶, in which the quaication of the results presented in Figure 10A and B ▶ is shown, summarizes the data obtained, on the basis of which it can be advanced that the sensitivity of the various aggregates to the action of subtilisin increases in the order T2, T1, M, TT1, TT2.
Fig. 10.
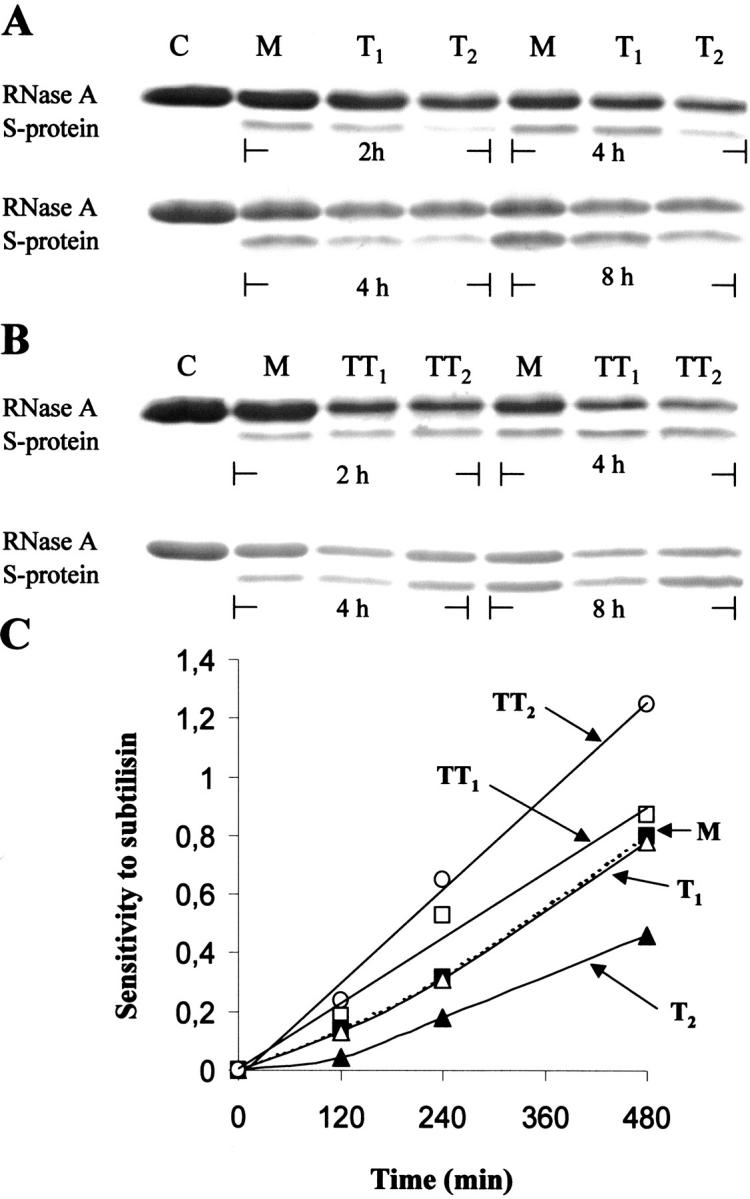
Susceptibility of the trimeric and tetrameric RNase A aggregates to digestion by subtilisin. (A) C, purified RNase A (4 μg), used as a control; M, purified RNase A, its less (T1) and more (T2) basic trimers (1 mg/mL). (B) C, purified RNase A (4 μg), used as a control; M, purified RNase A, its less basic (TT1) and more (TT2) basic tetramers. All samples in (A) and (B), except the controls, were incubated with subtilisin (concentration of each species, 0.8 mg/mL; substrate/protease ratio, 1000) in 0.33 M Tris-HCl/0.13 M sodium phosphate buffer, pH 8, at 5°C, for 2, 4, and 8 h, as indicated. Reaction was interrupted by adding PMSF (1.5 mM, final concentration); 3 μg of each RNase A species were analyzed by 18% SDS-PAGE. (C) Quaication of the digestion reaction. The densities of the remaining, undigested RNase A bands, and the densities of the S-protein bands, appearing in (A) and (B), were measured with an ImageMaster VDS (Pharmacia Biotech). The sensitivity of each protein species (RNase A monomer, filled square; T1, open triangle; T2, filled triangle; TT1, open square; TT2, open circle) to subtilisin was expressed as the ratio between the density of each S-protein band and the density of the corresponding band of the remaining, undigested RNase A.
By observing Figure 10A and C ▶, it appears rather clearly that at all incubation times the more basic form of the RNase A trimer (T2) is more resistant to the action of subtilisin than both monomeric RNase A (M) and the less basic trimer (T1), which actually appears to be almost as sensitive as the RNase A monomer. The resistance of T2, higher than that of monomeric RNase A, is reminiscent of the similar property already described for the major and more basic RNase A dimer, D2, which was, in fact, found to be more resistant to subtilisin than monomeric RNase A (Nenci et al. 2001), a property that, incidentally, is now explainable on the basis of its structure, defined by Liu et al. (2001). On the contrary, the sensitivity to subtilisin of the less basic trimer (T1), which is only slightly more resistant than monomeric RNase A (Fig. 10A ▶, C), appears to be quite different from that of the minor, less basic, RNase A dimer (D1), which was found to be remarkably more sensitive to subtilisin than both monomeric RNase A and its major dimer (Nenci et al. 2001). Because the resistance to subtilisin of a mixture of minor and major dimer (in a 1:3 ratio, similar to that characterizing their formation by lyophilization of RNase A from acetic acid solutions) has indeed to be ascribed to the prevalent presence of the major dimer (Nenci et al. 2001), the results obtained with the two trimers indicate again notable structural differences between them.
As for the tetrameric conformers of RNase A, both of them show a lower resistance to subtilisin than do all other species (Fig. 10B ▶, C), although, between the two, the less basic tetramer (TT1) is more resistant than the more basic one (TT2), this being—in terms of molecule basicity—just the contrary of what was found with the two trimers. Here too, because dimers are among the dissociation products of both tetrameric conformers, it seems that the way in which the two dimeric components associate in the tetramer could make both dimers more and differently sensitive to the protease than they are when not structured in form of tetramer.
It might be worth pointing out that the minor RNase A dimer appears to be unique in two respects, namely in its remarkable susceptibility to the action of subtilisin (Nenci et al. 2001), and in its amount relative to that of the major dimer (Gotte et al. 1999). Although in fact the minor and major dimeric conformers are always in the ratio of at least 1:3–1:4, so that the formation of the major RNase A dimer appears to be definitely favored if compared to that of the minor dimer, no such great difference can be observed in the relative amounts of the two trimeric or tetrameric conformers. The relative proportions of monomeric RNase A and its various aggregates (average values calculated from 15 experiments; see also Gotte et al. 1999) are, in fact, the following: monomer, 73.39 ± 2.60%; minor (less basic) dimer, 3.56 ± 0.59%; major (more basic) dimer, 16.16 ± 1.82%; less basic (major) trimer, 3.26 ± 0.55%; more basic (minor) trimer, 2.08 ± 0.43%; less basic tetramer, 0.49 ± 0.13%; more basic tetramer, 0.81 ± 0.18%; higher order aggregates, 0.22 ± 0.08%.
Conclusions
It could be reasonably proposed that the two forms of RNase A trimers and tetramers form by a domain-swapping mechanism, as was found for the two dimeric conformers (Liu et al. 1998, 2001). Possibly, the two types of domain swapping, involving the N-terminal and C-terminal ends of each monomeric subunits, coexist in trimers and tetramers. From the products of the dissociation of the two trimers it can be deduced that the structure of one of them (T1) can fit the model already proposed (Liu et al. 2001), while the structure of the other (T2) might be substantially different. On the basis of the dissociation products of the two tetramers, we might advance, among other equally possible structures (Bennet et al. 1995), two linear models of tetrameric RNase A in which the two types of domain swapping coexist. The two putative structures could account for some biophysical properties of the two forms of tetramers, namely their different basic net charges and apparent molecular weights (Gotte et al. 1999), and their different dissociability. Moreover, substantial structural differences between the two trimers and tetramers are undoubtedly revealed by their different sensitivities to the action of subtilisin.
The secondary structure of the RNase A oligomers appears not to change significantly in going from monomer to dimers, trimers, or tetramers. Instead, tyrosine residues show themselves as being differently exposed, so that they appear to have different microenvironments in trimers and tetramers. This could be in line with the properties shown by the oligomers. The different structures of each couple of conformers confer to their components, as repeatedly noticed, different charge characteristics. In other words, the different exposure of some positively charged or, alternatively, negatively charged amino acid residues can explain the nucleic acid helix-unwinding ability of trimers and tetramers, as well as the ion-exchange properties of each couple of conformers (Gotte et al. 1999). Moreover, it is quite reasonable that the interaction of trimers and tetramers with a (double-stranded) polydeoxyribonucleotide makes both protein species remarkably more stable than they are in the absence of the nucleic acid. Although in fact the electrostatic interactions between the polycationic aggregates and the polyanionic quasi-substrate can, on one hand, be responsible for the partial "unwinding" of the double-helical nucleic acid due to the binding by the protein(s) of the polydeoxyribonucleotide sequences made single-stranded by the thermal fluctuation of the nucleic acid secondary structure (Jensen and von Hippel 1976), they can, on the other hand, unspecifically stabilize the structure of each RNase A oligomer.
A final comment could be that the various forms in which RNase A can aggregate, and the possibility that its aggregation can occur through two types of 3D domain swapping simultaneously serve to highlight the peculiarity of this enzyme protein, and once more the general importance of the 3D domain-swapping mechanism (Bennet et al. 1994, 1995) as a tool by which a protein can form aggregates, in particular amyloid fibers. The various RNase A oligomers form under rather drastic environmental conditions, i.e., by lyophilization from acetic acid solutions. But, besides the possibility, recently reported (Park and Raines 2000), that native RNase A could form dimers at neutral pH, dimers and higher oligomers could also form as a consequence of the change of other, possibly less drastic, environmental variables. This could particularly be true in the light of the data and ideas already discussed (Liu et al. 2001), and of the results recently obtained by Faendrich, Fletcher, and Dobson (2001) with a prototype of globular proteins like muscle myoglobin, which under conditions where its native structure is destabilized can form amyloid-like fibrils. Therefore, the idea that every protein under proper conditions could form fibrils similar to amyloid is appealing, and the study and possible definition of the structure of the higher oligomers of RNase A, like trimers and tetramers, could contribute to the understanding of the basic mechanisms of protein aggregation.
Materials and methods
Bovine ribonuclease A (type XII-A), Subtilisin Carlsberg, type VIII (from Bacillus licheniformis), Trypsin from bovine pancreas, TPCK treated, yeast RNA, and the double-stranded poly(A) • poly(U) and poly(dA-dT) • poly(dA-dT) complexes were purchased from Sigma Chemical Co. Divinyl sulfone was a Sigma-Aldrich product. Acetic acid was from Merck. Other chemicals were of the highest purity available. Source 15S HR 10/10 or 16/10 columns were from Pharmacia, TSK gel 2000SW column was from Tosohaas. PMSF (Phenylmethane-sulphonyl fluoride) was purchased from Boehringer. The concentration of RNase A and its aggregates was estimated spectrophotometrically on the basis of an ɛ280 of 7.3 for a 1% solution (Gotte et al. 1999). Alternatively, it was determined by the Bradford method (1976). If necessary, samples were concentrated with Centricon 3 or 10 against 0.13 M sodium phosphate/0.33 M Tris-HCl buffer, final pH 8. Samples were desalted by using Centricon 3 or 10 against distilled water.
Purification of bovine ribonuclease A
RNase A was dissolved in 0.1 M sodium phosphate buffer, pH 6.7, and subjected to ion-exchange chromatography on a Pharmacia Source 15S column in a Pharmacia FPLC system, as described (Gotte et al. 1999). The large peak of material, with the highest specific activity (Kunitz units), emerging as the last protein species (Gotte et al. 1999), was collected and used for all following procedures.
Preparation of RNase A aggregates
Aggregation of RNase A was performed as described (Crestfield et al. 1962; Gotte et al. 1999) by lyophilizing solutions of the purified enzyme protein (15–20 mg/mL) in 40% acetic acid. The lyophilized material was dissolved in 0.1 M sodium phosphate buffer, pH 6.7, diluted to 0.065 M immediately before applying it onto cationic exchange column.
Purification and separation of the RNase A aggregates
The ribonuclease A aggregates dissolved as described above were loaded on a Source 15S HR 16/10 column in an FPLC system. About 35 mg protein (in 2 mL) were applied and eluted at room temperature with a linear gradient of sodium phosphate buffer, pH 6.7 (0.09–0.18 M, 120 min), at a flow rate of 1.2 mL/min. Under these experimental conditions the various oligomers elute with a pattern identical to that already described (Gotte et al. 1999). The peaks of interest were finally collected and used immediately or kept frozen until use.
Enzyme assays
Ribonuclease activity was assayed by the procedure described by Kunitz (1946). The activity of the various aggregates and of purified monomeric RNase A towards poly(A) • poly(U) was assayed spectrophotometrically at 260 nm, as described (Libonati and Floridi 1969; Gotte et al. 1999).
Gel electrophoresis
(a) SDS-PAGE was carried out with 12 or 18% polyacrylamide gel. Gels were run at 100 volts for 2–3 h at 22°C. Staining was performed with 0.025% Coomassie brilliant blue R-250. (b) Cathodic gel electrophoresis under nondenaturing conditions was performed according to Goldenberg (1989) with slight modifications, using a β-alanine/acetic acid buffer, pH 4.0. Ten or 12.5% polyacrylamide gels were run at 20 mA for 100–120 min, at 4°C. Fixing and staining were performed with 12.5% trichloroacetic acid and 0.1% Coomassie brilliant blue.
Assessing the extent of peptide swapping
Cross-linking of the various RNase A aggregates were carried out, according to the procedure outlined by Ciglic et al. (1998), by using divinyl sulfone as cross-linker of histidine residues. Each protein species, dissolved in 0.1 M sodium acetate buffer, pH 5, and the reagent, as a 10% solution in ethanol, were mixed in a 1:1000 ratio and incubated at 20°C up to 220 h. At the times indicated, aliquots were withdrawn from each incubation mixture, and the reaction quenched by adding 2-mercaptoethanol (0.2 M, final concentration). The samples were then analyzed by 12% SDS-PAGE, and the extent of the cross-linked conformers estimated by Comassie blue staining.
Measurement of the DNA helix-unwinding activity
Thermal transition profile analyses of the synthetic double-helical polydeoxyribonucleotide poly(dA-dT) • poly(dA-dT) in the absence or presence of the various RNase A species were determined by following the hyperchromicity at 260 nm of the nucleic acid/protein mixture as a function of temperature between 20 and 60°C. A typical mixture in stoppered cuvette was composed of 13 μg of the double-stranded polydeoxyribonucleotide and 14 μg of the various RNase A aggregates in 1.4 mL of 0.01 M imidazole/0.035 M NaCl buffer, pH 7 (buffer A). Determinations of the absorbances of the mixtures maintained at the chosen temperature until hyperchromicity reached a stable value (about 2.5 min) were performed with a Beckman DU 650 spectrophotometer equipped with a thermostatically controlled water bath.
Determination of the stability of the various RNase A oligomers
Stability of the more basic RNase A trimer (T2) and tetramer (TT2) was analyzed and compared with that of the major (and more basic) dimeric form in a range of temperature between 20 and 50°C. In one type of experiment 10 μg of the sample, contained in 10 μL of buffer A (see above) (see legend to Fig. 4 ▶), were maintained for exactly 2.5 min at the chosen temperature, then rapidly transferred to an ice-cold bath, and analyzed by PAGE (10% polyacrylammide) under nondenaturing conditions. The amounts of each undissociate RNase A aggregate, as well as of its dissociation products were determined with an ImageMaster VDS (Pharmacia Biotech). A different procedure was used to assay the stability of trimer T2 and tetramer TT2 in the absence or presence of poly(dA-dT) • poly(dA-dT) (details are given in the legend to Fig. 5 ▶).
In a second type of experiment, both trimers (T1 and T2) and tetramers (TT1 and TT2) were incubated in 0.03 M sodium phosphate buffer, pH 6.7, at 35°C. At times indicated, 60 μg of each sample were withdrawn, cooled down, and analyzed by ion-exchange chromatography with a Pharmacia Source 15S HR 10/10 column in a Pharmacia FPLC system. The various protein species were eluted with a linear 2-h gradient (0.09–0.18 M sodium phosphate, pH 6.7), and the area of each peak was calculated and expressed as a percentage of the input. For some additional verification, 10 μg of each sample were analyzed by 12.5% PAGE under nondenaturing conditions.
Circular dichroism
CD spectra of the various RNase A aggregates were obtained with a JASCO 710 Spectropolarimeter equipped with a thermostatically controlled cell, at 25°C. Spectra were recorded at a scan speed of 50 nm/min with a bandwidth of 2 nm, and averaged automatically. For the near-UV wavelengths (240–320 nm) the protein concentration of the sample was between 0.95 and 1.3 mg/mL with a 1-cm path length. For the far-UV region (190–240 nm) the protein concentration was 0.13 mg/mL with a 0.1-cm path length. All samples were dissolved in 0.1 M sodium phosphate buffer, pH 6.7. Molar ellipticities were calculated by taking into account that the mean residue molecular weight for pancreatic RNase A is 110.5 (Puett 1972).
Treatment with subtilisin under controlled conditions
Digestion of the two trimeric and tetrameric RNase A conformers with subtilisin was performed at 5°C according to Doscher (1967), and essentially as already described for the RNase A dimers (Nenci et al. 2001). Details can be found in the legends to the figures. The action of subtilisin was interrupted by adding PMSF (1.5 mM, final concentration). Samples to be analyzed by SDS-PAGE were treated with PMSF for 15 min and boiled for 5 min. The amounts of digested and undigested species were measured with a Pharmacia Biotech ImageMaster VDS.
Acknowledgments
We are grateful to Yanshun Liu for comments and suggestions. This work was supported by the Italian MURST-PRIN 1999 and 2000.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.14101.
References
- Bennett, M.J., Choe, S., and Eisenberg, D. 1994. Domain swapping: Entangling alliances between proteins. Proc. Natl. Acad. Sci. 91 3127–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennet, M.J., Schlunegger, M.P., and Eisenberg, D. 1995. 3D domain swapping: A mechanism for oligomer assembly. Protein Sci. 4 2455–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm, G., Muhr, R., and Jaenicke, R. 1992. Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 5 191–195. [DOI] [PubMed] [Google Scholar]
- Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72 248–254. [DOI] [PubMed] [Google Scholar]
- Carsana, A., Furia, A., and Libonati, M. 1983. Influence of protein net charge on the nucleic acid helix-destabilizing activity of various pancreatic ribonucleases. Mol. Cell. Biochem. 56 89–92. [DOI] [PubMed] [Google Scholar]
- Chiti, F., Taddei, N., Bucciantini, M., White, P., Ramponi, G., and Dobson, C.M. 2000. Mutational analysis of the propensity for amyloid formation by a globular protein. EMBO J. 19 1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciglic, M.I., Jackson, P.J., Raillard, S.A., Haugg, M., Jermann, T.M., Opitz, J.G., Trabesinger-Rauf, N., and Benner, S.A. 1998. Origin of dimeric structure in the ribonuclease superfamily. Biochemistry 37 4008–4022. [DOI] [PubMed] [Google Scholar]
- Cohen, F.E. and Prusiner, S.B. 1998. Pathologic conformations of prion proteins. Annu. Rev. Biochem. 67 793–819. [DOI] [PubMed] [Google Scholar]
- Crestfield, A.M., Stein, W.H., and Moore, S. 1962. On the aggregation of bovine pancreatic ribonuclease. Arch. Biochem. Biophys. Suppl. 1: 217–222. [PubMed]
- Doscher, M.S. 1967. Preparation of ribonuclease S and its component pats, S-peptide and S-protein. Methods Enzymol. 11 640–648. [Google Scholar]
- Faendrich, M., Fletcher, M.A., and Dobson, C.M. 2001. Amyloid fibrils from muscle myoglobin. Nature 410 165–166. [DOI] [PubMed] [Google Scholar]
- Felsenfeld, G., Sandeen, G., and von Hippel, P.H. 1963. The destabilizing effect of ribonuclease on the helical DNA structure. Proc. Natl. Acad. Sci. 50 644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg, D.P. 1989. Analysis of protein conformation by gel electrophoresis. In Protein structure. A practical approach (ed. T.E. Creighton), pp. 225–250. Oxford University Press, IRL Press, Oxford, UK.
- Gotte, G., Bertoldi, M., and Libonati, M. 1999. Structural versatility of bovine ribonuclease A. Distinct conformers of trimeric and tetrameric aggregates of the enzyme. Eur. J. Biochem. 265 680–687. [DOI] [PubMed] [Google Scholar]
- Grandi, C., D'Alessio, G., and Fontana, A. 1979. Comparative study on the structure and stability of bovine seminal ribonuclease, its monomeric bis (s-carboxymethylated-31,32) derivative, and bovine pancreatic ribonuclease. Biochemistry 18 3413–3420. [DOI] [PubMed] [Google Scholar]
- Jensen, D.E. and von Hippel, P.H. 1976. DNA `melting' proteins: I. Effects of bovine pancreatic ribonuclease binding on the conformartion and stability of DNA. J. Biol. Chem. 251 7198–7214. [PubMed] [Google Scholar]
- Klafki, H.W., Pick A.I., Pardowitz, I., Cole, T., Awni, L.A., Barnikol, .U., Mayer, F., Kratzin, H.D., and Hilschmann, N. 1993. Reduction of disulfide bonds in an amyloidogenic Bence Jones protein leads to formation of "amyloid-like" fibrils in vitro. Biol. Chem. Hoppe-Seyler 374 1117–1122. [DOI] [PubMed] [Google Scholar]
- Kunitz, M. 1946. A spectrophotometric method for measurement of ribonuclease activity. J. Biol. Chem. 164 563–568. [PubMed] [Google Scholar]
- Libonati, M. and Floridi, A. 1969. Breakdown of double-stranded RNA by bull semen ribonuclease. Eur. J. Biochem. 8 81–87. [DOI] [PubMed] [Google Scholar]
- Libonati, M. and Sorrentino, S. 1992. Revisiting the action of bovine ribonuclease A and pancreatic-type ribonucleases on double-stranded RNA. Mol. Cell. Biochem. 117139–151. [DOI] [PubMed] [Google Scholar]
- Libonati, M. and Sorrentino, S. 2001. Degradation of double-stranded RNA by mammalian pancreatic-type ribonucleases. Methods Enzymol. 341 234–248. [DOI] [PubMed] [Google Scholar]
- Liu, Y., Gotte, G., Libonati, M., and Eisenberg, D. 2001. A domain-swapped RNase A dimer with implications for amyloid formation. Nat. Struct. Biol. 8 211–214. [DOI] [PubMed] [Google Scholar]
- Liu, Y., Hart, P.J., Schlunegger, M.P., and Eisenberg, D.S. 1998. The crystal structure of a 3D domain-swapped dimer of RNase A at 2.1 Å resolution. Proc. Natl. Acad. Sci. 95 3437–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nenci, A., Gotte, G., Maras, B., and Libonati, M. 2001. Different susceptibility of the two dimers of ribonuclease A to subtilisin. Implications for their structure. Biochim. Biophys. Acta 1545 255–262. [DOI] [PubMed] [Google Scholar]
- Park, C. and Raines, R.T. 2000. Dimer formation by a "monomeric" protein. Protein Sci. 9 2026–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perutz, M.F., Johnson, T., Suzuki, M., and Finch, J.T. 1994. Glutamine repeats as polar zippers: Their possible role in neurodegenerative diseases. Proc. Natl. Acad. Sci. 91 5355–5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puett, D. 1972. A study on the conformation and conformational stability of ribonuclease A and its peptic derivative, des(121–124)-ribonuclease. Biochemistry 111980–1990. [DOI] [PubMed] [Google Scholar]
- Schlunegger, M.P., Bennet, M.J., and Eisenberg, D.S. 1997. Oligomer formation by 3D domain swapping: A model for protein assembly and misassembly. Adv. Protein Chem. (eds. F.M. Richards, D.S. Eisenberg, and P.S. Kim), vol. 50, pp. 61–122. Academic Press, New York. [DOI] [PubMed]
- Simons, E.R. and Blout, E.R. 1968. Circular dichroism of ribonuclease A, ribonuclease S, and some fragments. J. Biol. Chem. 243 218–221. [PubMed] [Google Scholar]
- Sorrentino, S., Barone, R., Bucci, E., Gotte, G., Russo, N., Libonati, M., and D'Alessio, G. 2000. The two dimeric forms of RNase A. FEBS Lett. 466 35–39. [DOI] [PubMed] [Google Scholar]
- Sorrentino, S. and Libonati, M. 1994. Human pancreatic-type and nonpancreatic-type ribonucleases: A direct side-by-side comparison of their catalytic properties. Arch. Biochem. Biophys. 312 340–348. [DOI] [PubMed] [Google Scholar]