

Abstract
One of the difficulties that can impede structural work on a molecule of interest is limited solubility. Although functionally similar to the human immunodeficiency virus type-1 reverse transcriptase (HIV-1 RT), the Moloney murine leukemia virus reverse transcriptase (MMLV RT) differs both in architecture and solubility properties. Reverse transcriptase is an essential retroviral enzyme that replicates the single-stranded RNA genome of the retrovirus producing a double-stranded DNA copy, which is subsequently integrated into the host's genome. We have introduced a single amino acid substitution in the connection domain of an N-terminally truncated MMLV RT (L435K) that significantly improves the solubility of the enzyme eliminating the need for nonionic detergents in buffering storage solutions. The substituted enzyme retains near wild-type polymerase activity. An important consequence of the improved solubility of the L435K MMLV RT has been the ability to obtain diffraction quality crystals.
Keywords: Moloney murine leukemia virus, reverse transcriptase, solubility, crystallization, mutant
Reverse transcriptase (RT) is strictly a DNA polymerase, but can use either RNA or DNA as a template, and degrades the RNA genome in preparation for the second-strand DNA synthesis via a ribonuclease H activity specific for RNA : DNA duplex to replicate the retroviral genome (reviewed in Telesnitsky and Goff 1997). The enzyme includes fingers, palm, thumb, connection, and RNase H structural domains as first described in the structure of the human immunodeficiency virus type-1 (HIV-1) RT complexed with nevirapine (Kohlstaedt et al. 1992). Moloney murine leukemia virus (MMLV) RT is a monomeric ∼75 kD enzyme as isolated (Moelling 1974) in contrast to the heterodimeric HIV-1 RT, which includes 66,000 Da (p66) and 51,000 Da (p51) subunits (LeGrice 1993) (p51 results from proteolytic processing of the p66 subunit).
In addition to the architectural differences between HIV-1 RT and MMLV RT, there are significant solubility differences. MMLV RT is not soluble at concentrations suitable for structural studies in the absence of nonionic detergent and glycerol, whereas HIV-1 RT is soluble in the absence of nonionic detergent. This observation formed the basis of studies designed to improve the solubility of MMLV RT. One of the major difficulties in pursuing structural studies of MMLV RT has proven to be the limited solubility of the enzyme, which impacts the ability to purify large quantities of the enzyme as well as its behavior in crystallization experiments.
Other retroviral proteins including the catalytic core of the HIV-1 integrase (Jenkins et al. 1995) and two-domain RSV (Hyde et al. 1999) and SIV (Li et al. 1999) integrases have been modified by site-directed mutagenesis to improve the properties of the enzymes, most notably the solubility. In the initial study on HIV-1 integrase, 29 mutants were made in which hydrophobic residues were substituted with Lys or Ala residues (Jenkins et al. 1995). A single substitution of F185K was found to improve the solubility of the catalytic core of HIV-1 integrase. Single substitutions of F199K for the RSV two-domain integrase and F185H for the analogous SIV enzyme were also found to improve solubility and the ability to obtain diffraction quality crystals. In these cases, the substitutions for residues are located at a dimer interface for the catalytic core domain.
We have used a directed approach to modify MMLV RT and improve its properties, based initially on structural analysis of the fingers and palm domains of MMLV RT and on the results of sequence alignments with HIV-1 RT. The two regions of the protein that we targeted were the N-terminus and the connection domain. The amino terminus of MMLV RT includes 40 amino acids that are not present in HIV-1 RT, and was therefore an obvious target for modification in MMLV RT. In addition, alignment of MMLV RT and HIV-1 RT sequences with GCG (GCG 1991) provided a rather unsatisfactory alignment for residues in the connection domain in particular, where a large insertion of approximately 40 amino acid residues was predicted for MMLV RT. However, the overall structural similarity between the fingers and palm domains of MMLV RT and HIV-1 RT suggested to us that the structures of the remaining domains, thumb, connection, and RNase H, were likely to be similar as well. Thus, rather than a single large insertion in the connection domain, the extra approximately 40 amino acid residues found in the thumb and connection domains of MMLV RT and not in HIV-1 RT were likely to be accommodated as small insertions throughout these domains. This idea was confirmed by performing sequence alignments of the residues in the thumb and connection domains in PSI-BLAST (Altschul et al. 1997) and GenTHREADER (Jones 1999), which give more sensible alignments with no large insertions or gaps. However, because the sequence identity for the pair-wise amino acid alignments of residues from the thumb and connection domains of MMLV RT and HIV-1 RT is ∼10%, these pair-wise alignments are likely to be at best 50% accurate (Friedberg et al. 2000). Thus, an accurate sequence alignment for these residues will require a structural alignment. We also suspected, based on the structure of HIV-1 RT, that the most likely domain to include exposed hydrophobic residues was the connection domain of MMLV RT. The connection domain of p66 is involved intimately in the heterodimeric interface in HIV-1 RT, while the analogous connection domain in the monomeric MMLV RT must necessarily be involved in different interactions. This was then the starting point for ideying potentially exposed hydrophobic residues that were contributing to the low solubility of the protein.
Comparative structural studies of MMLV RT and HIV-1 RT to date have been limited to the fingers and palm domains. Structural information on MMLV RT includes high-resolution structures reported for the fingers and palm domains of MMLV RT alone (Georgiadis et al. 1995) and in complexes with DNA (Coté et al. 2000; Najmudin et al. 2000). As the only other member of the reverse transcriptase family that has proven amenable to structural studies, we expect the structure of the full-length MMLV RT to provide the basis for further understanding of structure–function relationships in RT and ultimately aiding the design of useful inhibitors of this enzyme. We describe here our directed experimental approach to improving the solubility of MMLV RT to obtain crystals of the full-length MMLV RT that are suitable for structural analysis. We expect that this approach will prove useful for other problems in which functionally related proteins have proven intractable to structural studies.
Results
Improved solubility of MMLV RT
Efforts to improve the solubility of the protein have included N-terminal truncation of the enzyme and substitution of hydrophobic residues that might be solvent exposed. Based on the crystal structure of the N-terminal fragment of MMLV RT and limited proteolysis experiments (Georgiadis et al. 1995), we knew that the N-terminal 24 amino acid residues were disordered. We, therefore, constructed an enzyme including residues 24–671, which we refer to as RT24 (Najmudin et al. 2000; Gu et al. 2001). We have previously shown that RT24 has enzymatic properties that are indistinguishable from the wild-type enzyme (Gu et al. 2001). In addition, we constructed an enzyme including residues 45–671, which we refer to as RT45. This enzyme was designed to resemble the HIV-1 RT, which lacks the N-terminal 40 amino acid residues found in MMLV RT. In contrast to MMLV RT in which the N-terminal residues form part of the palm domain, the fold of HIV-1 RT begins in the fingers domain. Of the N-terminally truncated enzymes, we found that RT24 was indeed more soluble, and could be concentrated to over 20 mg/mL in the presence of detergent and glycerol. We were not able to purify the RT45 enzyme because it was quite susceptible to proteolysis in the initial steps of the purification, as judged by initial expression levels and recovery of the enzyme following the initial chromatographic separation.
In our search for solvent-exposed hydrophobic residues, we ideied a stretch of five hydrophobic residues LVILA (432–436) bounded by Pro residues within the connection domain of MMLV RT by inspection of the sequence and substituted each of the five residues with Lys. The substitutions were introduced within the RT24 construct, expressed, and purified as described in Materials and Methods. The N-terminal hexa-His affinity tag used for the initial step of purification was removed by digestion with thrombin. Of the substituted enzymes, V433K and L435K were substantially more soluble in the absence of detergent than the other enzymes and the parent RT24 enzyme. We have pursued subsequent enzymatic characterizations and crystallization studies with the L435K enzyme for which expression levels were slightly better than those of V433K.
Enzymatic characterization of L435K
Enzyme assays were performed to determine whether the L435K substitution affected the enzymatic properties of the enzyme. The L435K enzyme was compared to RT24 in a standard homopolymeric assay in addition to an assay using mRNA as the template, which more closely resembles retroviral RNA with numerous stem-loop structures that must be resolved by the enzyme during active synthesis. We have previously shown that RT24 retains wild-type activity in a number of different polymerase assays (Najmudin et al. 2000; Gu et al. 2001). The specific activity of L435K was determined using poly rA-oligo dT as the template primer. In this assay, the enzyme synthesizes poly dT directed by a poly rA template and primed with oligo dT. The L435K mutant retains approximately 78% of wild-type activity. In addition, the lengths of the products synthesized on poly rA-oligo dT are also comparable to RT24, as shown in Figure 1 ▶. In the mRNA template assay, we used oligo dT as the primer and provided radioactively labeled dATP to be incorporated by the enzyme to ensure that the products visualized resulted from synthesis directed by the mRNA and not just from its poly A tail. The products synthesized by RT24 and L435K enzymes were again very similar, as shown in Figure 2 ▶, indicating that the L435K enzyme has no difficulty resolving stem-loop structures and synthesizes full-length products corresponding to 600 or 650 base pairs.
Fig. 1.
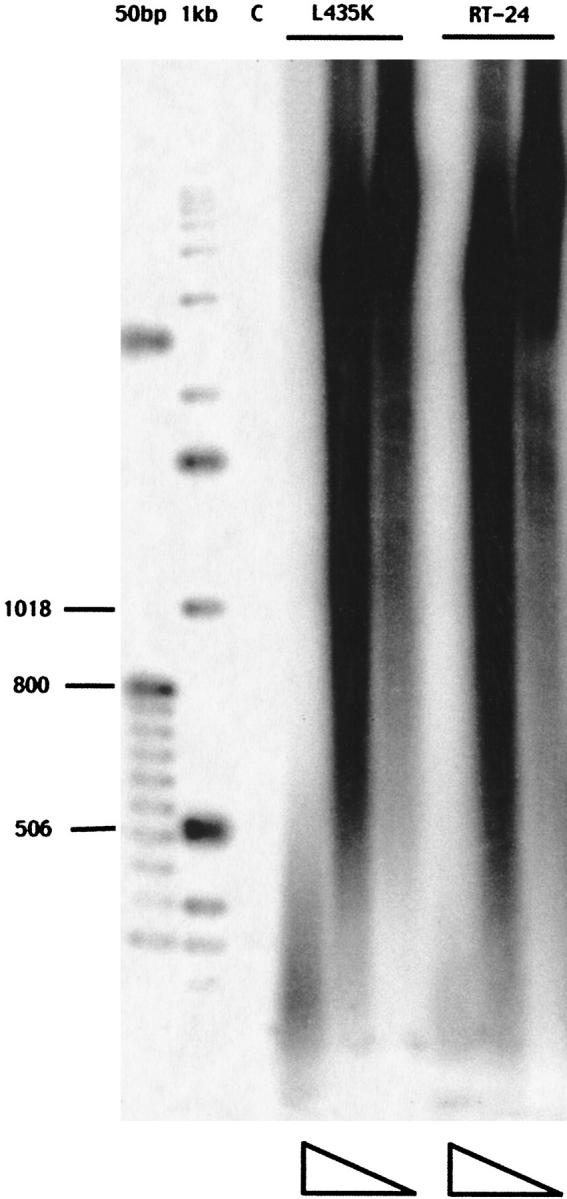
Analysis of products for synthesis directed by poly rA as the template. The reaction products were separated on a 1.4% (w/v) agarose gel and visualized on a Molecular Dynamics Phosphorimager plate. The size markers are 50 bp and 1 kb-labeled DNA ladders. The column labeled "C" is the control reaction without enzyme. For the L435K mutant and RT24, the columns from left to right indicate decreasing concentration of enzyme, with each enzyme at 40 nm, 4 nm, and 0.8 nm. The products synthesized by RT24 and L435K are very similar.
Fig. 2.

Analysis of products for synthesis directed by a mRNA template. The reaction products were separated on a 2.0% (w/v) agarose gel and visualized on a Molecular Dynamics Phosphorimager plate. The size markers are 50 bp and 1 kb-labeled DNA ladders. The column labeled "C" is the control reaction without enzyme. For the L435K mutant and RT24, the columns from left to right indicate decreasing concentration of enzyme, with each enzyme at 40 nm, 13.3 nm, and 4.4 nm. A similar distribution of full-length products, 600 and 650 bases long, can be seen for both enzymes.
Crystallization of MMLV RT
In our efforts to obtain diffraction quality crystals of MMLV RT, we have screened crystallization conditions using the wild-type MMLV RT, ΔRH (1–497), RT24, R116A, and L435K enzymes alone and in complexes with DNA. The R116A substitution was introduced for functional studies of the fingers domain binding site as previously described (Gu et al. 2001). Of the enzymes screened, only ΔRH was successfully crystallized in the absence of DNA. However, the ΔRH crystals diffracted very weakly, and were not suitable for a structure determination. Very small crystals were obtained for all of the other enzymes complexed initially with a template-primer (25 and 20 nucleotides, respectively) that were not sufficiently large for X-ray diffraction studies. The most promising crystallization results were obtained for complexes with the R116A and L435K enzymes.
We then screened complexes of R116A and L435K enzymes with approximately 15 different DNA molecules including duplex lengths of 14 to 21 base pairs with varying sequences. Due to the nature of the biological activity of RT, the binding of the enzyme to template primer DNA may have length dependence but not sequence dependence. We obtained crystals suitable for a structure determination of L435K complexed with a duplex with the following sequence, 5′-ATTGATATATTAAATT-3′; complemented by 3′-ACTATATAATTTAAAT-5′, resulting in a 14 base pair duplex with two base pair overhang on either end, shown in Figure 3a ▶. Crystals of R116A complexed with the same DNA duplex had been obtained under similar conditions, but grew to a maximum thickness of approximately 5–10 μ. It should be noted that R116A was purified in the presence of nonionic detergent. To directly compare its behavior in crystallization experiments with that of the L435K enzyme, RT24 was purified in the absence of detergent and complexed with the same DNA duplex. The crystals obtained of the RT24 complex were not sufficiently large for X-ray diffraction analysis, as shown in Figure 3b ▶.
Fig. 3.
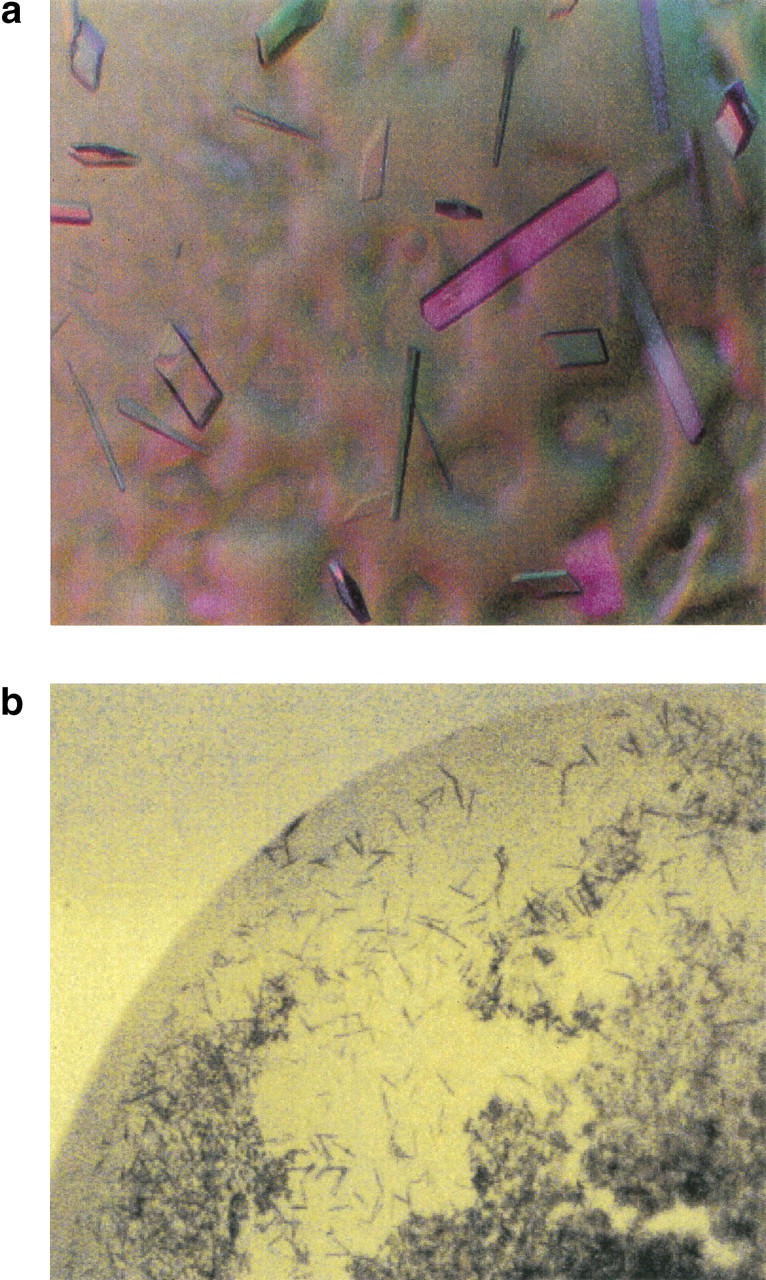
Comparison of crystals obtained with RT24 (wild-type sequence) and the L435K substituted MMLV RT. (a) Crystals of the L435K MMLV RT complexed with DNA are shown. (b) Crystals of RT24 complexed with the same DNA duplex as in (a) and grown under similar conditions are shown for comparison and photographed at the same magnification.
The optimized crystallization condition for the L435K : DNA complex included 0.1 M KCl, 0.1 M Mg(OAc)2, 0.05 M sodium cacodylate (pH 6.5), and 9%–11% PEG 8000 in the reservoir solution of vapor diffusion hanging drops. Crystals have been reproducibly obtained at temperatures of 20°C in experiments from several different preparations of L435K enzyme complexed with the 14-mer DNA duplex under the conditions previously noted, and are highly dependent on the concentration of the enzyme and temperature. We found that incubation of the crystallization trays at a slightly elevated temperature (∼23°C) overnight improved the overall thickness of the crystals from approximately 5 to 30 μ in the smallest dimension. From crystals with approximate dimensions of 100 × 50 × 30 microns shown in Figure 3a ▶, it was possible to collect data to 2.9 Å resolution on cryocooled crystals at the Advanced Photon Source, IMCA CAT 17-ID beamline. The crystals belong to space group P21212 with cell dimensions of a = 244.90 Å, b = 94.66 Å, and c = 52.26 Å.
Discussion
The goal of our studies with MMLV RT is to determine the structure of the full-length enzyme. To achieve this goal, it was necessary to obtain diffraction quality crystals of the enzyme. Initial attempts to obtain crystals of the wild-type MMLV RT were not successful potentially due in part to its limited solubility. We have, therefore, sought to improve the solubility of the enzyme without significantly affecting its catalytic properties. The use of a directed approach involving N-terminal truncation and substitution of Lys for potential solvent-exposed residues in MMLV RT has resulted in an enzyme with improved solubility without resorting to a labor-intensive screening process.
Specifically, truncation of the N-terminal 24 amino acid residues and substitution of Lys for Leu 435 has improved the solubility of the enzyme and allowed us to purify significant quantities of L435K MMLV RT in the absence of detergent. We find that the L435K enzyme retains near wild-type polymerase activity in that it does not differ significantly in processivity (as judged by product analysis for synthesis directed by poly rA or mRNA) or specific activity from RT24. Presumably, this substitution has not affected the overall structure of the enzyme. In addition, the improved solubility of the L435K enzyme in the absence of detergent has enabled us to obtain diffraction quality crystals, which are suitable for a structure determination. We propose that a directed approach similar in strategy to that used in this study may be successful in improving solubility for other problems.
Materials and methods
Site-directed mutagenesis of MMLV RT
The gene encoding the full-length wild-type MMLV RT from residues 24–671(wt-RT24) was cloned into the pET15b vector (Novagen) at the NdeI/XhoI restriction sites. This recombinant vector expresses the protein with a cleavable N-terminal hexa-histidine tag for affinity purification. The point mutations L432K, V433K, I434K, L435K, and A436K were made by site-directed mutagenesis using the QuikChange mutagenesis kit (Stratagene). The plasmid containing the RT24 insert was used as the template in the initial PCR step. The primers used to make these mutations were as follows: for L432K 5′-G GGA CAG CCG AAA GTA ATA TTG GCC C-3′, for V433K 5′-G GGA CAG CCA CTT AAG ATT CTG GCC C-3′, for I434K 5′-CAG CCA CTA GTC AAG CTT GCC CCC CAT G-3′, for L435K 5′-CA CTA GTC ATT AAG GCC CCC CAT GCA G-3′, and for A436K 5′-CTA GTC ATT CTG AAG CCC CAT GCA GTA G-3′. The underlined bases represent the mutated codon for lysine. All the primers necessary for making the substitutions were purchased from Integrated DNA Technologies in a 0.1 μm scale, purified by gel electrophoresis, gel extracted (Qiagen kit) and quantitated prior to use. After PCR, the parent plasmids were digested with DpnI(NEB). The mutant plasmids were then transformed into supercompetent XL-1 cells and then into DH5α for plasmid amplification. After this step, the plasmids were sequenced to verify the point mutations using the T7 Sequenase kit (Amersham Life Science).
Expression and purification of the mutant enzymes
Following sequencing, the plasmids were transformed into BL21 (DE3) for overexpression in E. coli. For trial expression and purification, the enzymes were expressed by growing 1L bacterial cultures at 37°C until the OD at 600 nm was ∼1.0. IPTG ∼0.7 mM was then added, and induction was carried out at 37°C for 3 h. Cell pellets were obtained by centrifuging the cells at 4200 rpm for 35 min and stored at −80°C. Before purification, the pellets were suspended in 50 mM NaH2PO4/Na2HPO4 (pH 7.8), 5% glycerol, 0.3 M NaCl, and 10 mM imidazole. The resuspended cells were lysed using two passes through a French pressure cell at 1000 lb/in2. Lysates were then centrifuged at 35,000 rpm for 40 min, and crude extracts were applied to 1 mL Ni-NTA (Qiagen) columns, washed with 20 mM imidazole, and eluted with 250 mM imidazole contained in buffer of the same composition as used for lysis. Based on SDS-PAGE analysis following these trial purifications, the L435K mutant had a higher yield than the other mutants, and was chosen for subsequent large-scale purification and crystallization.
In the scaleup, the L435K mutant was expressed in 6L bacterial cultures in the conditions described above and lysed in the same manner. A number of trials were required to obtain optimum purification conditions. The final purification protocol involved three chromatographic separations performed using an FPLC. After lysis, the crude extracts were applied to an 8-mL Ni-NTA (Qiagen) affinity column. The buffer in this step was the same as for the small-scale Ni-NTA column with the protein now eluted in an imidazole gradient from 20 mM to 250 mM. This was followed by an 8-mL S-Sepharose (Pharmacia) ion-exchange column after dilution in the purification buffer containing 50 mM Tris HCl (pH 8.5), 1 mM EDTA, 1 mM DTT, 5% glycerol. The N-terminal hexa-histidine tag was cleaved by digestion at 4°C overnight using thrombin protease (3.0 units/μL). The sample was then passed through a Mono-S (Pharmacia) ion-exchange column after dilution in purification buffer composed of 50 mM HEPES (pH 7.5), 1 mM EDTA, 1 mM DTT, and 5% glycerol. In the ion-exchange purification steps, the protein sample was diluted to make the initial salt concentration 75 mM NaCl and eluted by NaCl gradient fractionation. Cleavage of the histidine tag was verified by observing size variation in a SDS-PAGE gel and also by the altered elution profile in the second ion-exchange step. The fractions were then concentrated in Amicon 30 centricons, with the final salt concentration changed to 0.2 M NaCl by buffer exchange during concentration and stored at −80°C. The purified enzymes were at least 95% pure as assessed by SDS-PAGE. The protein concentration was determined using the MicroBCA assay (Pierce).
Enzyme assays
Homopolymeric assay
We have determined specific activities of the enzymes and analyzed the products synthesized using the standard poly(rA)-oligo(dT) primer assay as previously described (Roth et al. 1985). The purified enzymes were diluted in 50 mM HEPES (pH 7.5), 1 mM DTT, 1 mM EDTA, 0.2 M NaCl, 5% glycerol, and 2 mM n-decyl-β-d-maltopyranoside. Concentrations of 40 nM, 4 nM, and 0.8 nM were used for the analysis of products as visualized on a 1.4% (w/v) agarose gel. Specific activities were determined for enzyme concentrations of 0.8 nM, 0.4 nM, 0.13 nM, and 0.04 nM, which are within the linear range of the assay. The specific activities of RT24 and L435K enzymes were 200,925 nmol dTMP incorporated/mg and 156,777 nmol dTMP incorporated/mg in 15 min at 37°C, respectively. The reactions included 50 mM Tris HCl pH 8.3), 0.05% (v/v) NP40, 20 μM dTTP, 20 μg/mL poly(rA), 0.6 mM MnCl2, 10 μg/mL poly(dT), and 10 μCi of [α32P] dTTP (NEN Dupont) per mL.
Heteropolymeric assay
The reactions included 50 mM Tris HCl (pH 8.3), 4 mM HEPES (pH 7.5), 21 mM DTT, 20 μM dNTPs, 2 μg/mL mRNA, 75 mM NaCl, 0.05% (v/v) NP40, 0.16 mM n-decyl-β-d-maltopyranoside, 0.4% glycerol, 80 nm EDTA, 1.0 mM MgCl2, 1 μg/mL oligo dT used as primer, 2 μCi of [α32P] dATP (NEN Dupont) and RT enzyme concentrations of 40 nM, 13.3 nM, and 4.4 nM. The rabbit globin mRNA (GIBCO BRL) template includes transcripts that are 600 and 650 bases long. The reactions were carried out at 37°C for 60 min and then stopped by placing the reactions on ice. The products were separated on 2% (w/v) agarose gel and visualized using a Molecular Dynamics Phosphorimager. The markers used for gel analysis in both assay were 1 kb and 50 bp (GIBCO BRL), and were end-labeled using 0.04 units of T4 polynucleotide kinase and 0.02 mCi of [γ32P] ATP (NEN Dupont) per 1.5 μg of markers.
Crystallization and data collection
After obtaining pure and concentrated protein, the Hampton Crystal Screen was used in an attempt to obtain crystals of the protein alone. However, no crystalline growth was observed. DNA duplexes of various lengths and sequences were screened next in the Natrix Screen (Hampton Research). The most promising results were obtained with a 14-mer duplex with a two base overhang 5′-ATTGATATATTAAATT-3′ and complementary strand 3′-AC TATATAATTTAAAT-5′. These two single strands of DNA were synthesized in a 1-μmole scale (Integrated DNA Technologies) and purified using standard reverse phase HPLC methods. The purified samples were stored as 5-mM stock solutions. The ds-DNA was annealed at 80°C followed by slow cooling to room temperature resulting in 2.5 mM solutions for use in crystallization screens.
The protein : DNA complex was made by mixing the components in a 1 : 2 ratio, respectively, with a protein concentration of 18–20 mg/mL (∼0.25 mM). The Hampton Natrix screen was used for initial trials, and crystalline growth was observed in condition 26 (0.2 M KCl, 0.1 M Mg(OAc)2, 0.05 M sodium cacodylate (pH 6.5), 10% PEG 8000). To optimize growth conditions, salt, buffer, and PEG concentrations were varied to arrive at the final condition of 0.1 M KCl, 0.1 M Mg (OAc)2, 0.05 M sodium cacodylate (pH 6.5), and 9%–11% PEG 8000. Different protein concentrations ranging from 10–50 mg/mL were used, but the concentration of 18–20 mg/mL gave the best crystals. The complex was incubated on ice for about 45 min, and 2.5 μL of the complex was mixed with 2.5 μL of drop solution and crystallized by the hanging drop vapor diffusion method over 500 μL of reservoir solution. The reservoir solution differed from the drop solution by the presence of 5% glycerol in it. The crystal trays were kept in the bottom shelf of a 20°C incubator overnight. The bottom shelf of this incubator shows a temperature variation between 23°C and 25°C. During this stage, the drops expanded in size, presumably from absorption of water from the reservoir. After this time period, the trays were relocated to the other shelves of the incubator, which are at 20°C, and the larger crystals grew in about a week with dimensions of approximately 100 × 50 × 30 microns. This particular crystal growth process is reproducible, but shows sensitivity to temperature and concentration. Native data sets were collected on crystals grown in this manner. Nucleation of smaller plate-like crystals approximately 5–10 μ thick resulted from placement of the trays with the same crystallization conditions on upper shelves, which remain at 20°C.
Crystals were serially soaked in cryosolutions including 0.2 M NaCl, 0.1 M KCl, 0.1 M Mg(OAc)2, 0.05 M sodium cacodylate (pH 6.5), 5% glycerol, 11% PEG 8000 and incremental ethylene glycol concentrations of 0%–16% prior to flash freezing. The crystals diffracted X-rays to Bragg spacings of ∼5.0Å as measured with an R-Axis IV image plate detector mounted on a Rigaku RU200 generator in a 4-h exposure. Data were collected on prefrozen crystals at NSLS X25 complete to 3.5-Å resolution and at APS IMCA CAT 17-ID to 2.9-Å resolution at 100 K.
Acknowledgments
We thank Mike Becker, Lonnie Berman, and Bill Nolan from NSLS X25, Andy Howard and Jorge Rios from IMCA CAT; the members of the Georgiadis group for helpful discussions, in particular Xiuping Liu for technical assistance; Richard Leidich and Tom Weaver for maintenance of home-sourced X-ray equipment; and Attilio Defalco and Greg Listner for systems support. Data were collected at the National Synchrotron Light Source, Brookhaven National Laboratory, which is supported by the U.S. Department of Energy, Division of Materials sciences and Division of Chemical Sciences, and at beamline 17-ID in the facilites of the Industrial Macromolecular Crystallography Association Collaborative Access Team (IMCA CAT) at the Advanced Photon Source. The IMCA CAT facilities are supported by the companies of the Industrial Macromolecular Crystallography Center for Synchrotron Radiation Research and Instrumentation. This work was funded by a grant from the National Institutes of Health, GM 55026 (M.M.G.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.16301.
References
- Altschul, S.F., Madden, T.L., Schäffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coté, M.L., Yohannan, S., and Georgiadis, M.M. 2000. Use of an N-terminal fragment from Moloney murine leukemia virus reverse transcriptase to facilitate crystallization and analysis of pseudo-16-mer DNA molecule containing G-A mispairs. Acta Crystallogr. D56 1120–1131. [DOI] [PubMed] [Google Scholar]
- Friedberg, I., Kaplan, T., and Margalit, H. 2000. Evaluation of PSI-BLAST alignment accuracy in comparison to structural alignments. Protein Sci. 9 2278–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GCG. 1991. Program manual for the GCG package, version 7. GCG, Madison, WI .
- Georgiadis, M.M., Jessen, S.M., Ogata, C.M., Telesnitsky, A., Goff, S.P., and Hendrickson, W.A. 1995. Mechanistic implications from the structure of a catalytic fragment of Moloney murine leukemia virus reverse transcriptase. Structure 3 879–892. [DOI] [PubMed] [Google Scholar]
- Gu, J., Villanueva, R.A., Smith-Snyder, C., Roth, M.J., and Georgiadis, M.M. 2001. Substitution of Asp 114 or Arg 116 in the fingers domain of Moloney murine leukemia virus reverse transcriptase affects interactions with the template-primer resulting in decreased processivity. J. Mol. Biol. 305 341–359. [DOI] [PubMed] [Google Scholar]
- Hyde, C.C., Bushman, F.D., Mueser, T.C., and Yang, Z.-N. 1999. Crystal structure of an active two-domain derivative of rous sarcoma virus integrase. J. Mol. Biol. 296 535–548. [DOI] [PubMed] [Google Scholar]
- Jenkins, T.M., Hickman, A.B., Dyda, F., Ghirlando, R., Davies, D.R., and Craigie, R. 1995. Catalytic domain of human immunodeficiency virus type 1 integrase: Ideication of a soluble mutant by systematic replacement of hydrophobic residues. Proc. Natl. Acad. Sci. USA 92 6057–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, D.T. 1999. GenTHREADER: An efficient and reliable protein fold recognition method for genomic sequences. J. Mol. Biol. 287 797–815. [DOI] [PubMed] [Google Scholar]
- Kohlstaedt, L.A., Wang, J., Friedman, J.M., Rice, P.A., and Steitz, T.A. 1992. Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 256 1783–1790. [DOI] [PubMed] [Google Scholar]
- LeGrice, S.F.J. 1993. Human immunodeficiency virus reverse transcriptase. In Reverse transcriptase (eds. A.M. Skalka and S.P. Goff), pp. 156–191. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Li, Y., Yan, Y., Zugay-Mruphy, J., Xu, B., Cole, J.L., Witmer, M., Felock, P., Wolfe, A., Hazuda, D., Sardana, M. K., et al. 1999. Purification, solution properties and crystallization of SIV integrase containing a continous core and C-terminal domain. Acta Crystallogr. D55 1906–1910. [DOI] [PubMed] [Google Scholar]
- Moelling, K. 1974. Characterization of reverse transcriptase and RNaseH from friend-murine leukemia virus. Virology 146 46–59. [DOI] [PubMed] [Google Scholar]
- Najmudin, S., Cote, M.L., Sun, D., Yohannan, S., Montano, S.P., Gu, J., and Georgiadis, M.M. 2000. Crystal structures of an N-terminal fragment from Moloney murine leukemia virus reverse transcriptase complexed with nucleic acid: Functional implications for template-primer binding to the fingers domain. J. Mol. Biol. 296 613–632. [DOI] [PubMed] [Google Scholar]
- Roth, M.J., Tanese, N., and Goff, S.P. 1985. Purification and characterization of murine retroviral reverse transcriptase expressed in Escherichia coli. J. Biol. Chem. 260 9326–9335. [PubMed] [Google Scholar]
- Telesnitsky, A. and Goff, S.P. 1997. Reverse transcriptase and the generation of retroviral DNA. In Retroviruses (eds. J.M. Coffin, S.H. Hughes, and H.E. Varmus). Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed]