

The expression, purification and crystallization of the full-length matrix protein from three lyssaviruses is described.
Keywords: matrix proteins, Rhabdoviridae, lyssaviruses, Lagos bat virus, SUMO tag
Abstract
The matrix (M) proteins of lyssaviruses (family Rhabdoviridae) are crucial to viral morphogenesis as well as in modulating replication and transcription of the viral genome. To date, no high-resolution structural information has been obtained for full-length rhabdovirus M. Here, the cloning, expression and purification of the matrix proteins from three lyssaviruses, Lagos bat virus (LAG), Mokola virus and Thailand dog virus, are described. Crystals have been obtained for the full-length M protein from Lagos bat virus (LAG M). Successful crystallization depended on a number of factors, in particular the addition of an N-terminal SUMO fusion tag to increase protein solubility. Diffraction data have been recorded from crystals of native and selenomethionine-labelled LAG M to 2.75 and 3.0 Å resolution, respectively. Preliminary analysis indicates that these crystals belong to space group P6122 or P6522, with unit-cell parameters a = b = 56.9–57.2, c = 187.9–188.6 Å, consistent with the presence of one molecule per asymmetric unit, and structure determination is currently in progress.
1. Introduction
Lyssaviruses, typified by the rabies virus (RV), are members of the Rhabdoviridae family of nonsegmented negative-sense single-stranded RNA viruses and cause meningoencephalitis, leading to the death of approximately 55 000 people annually (World Health Organization, 2005 ▶). At present, although infection can be prevented, rabies encephalitis remains incurable. The lyssavirus genome encodes five viral proteins: nucleoprotein (N), phosphoprotein (P), matrix (M), glycoprotein (G) and polymerase (L). M is a small (∼20–25 kDa) multifunctional protein that is essential for the budding and morphogenesis of the virus (Jayakar et al., 2004 ▶). In addition, it has been implicated in controlling the balance between transcription and replication of the viral genome and in modulation of host-cell transcription, translation and apoptosis (Kassis et al., 2004 ▶; Finke & Conzelmann, 2005 ▶; Komarova et al., 2007 ▶). During virus assembly, M interacts with the viral ribonucleoprotein particle (containing N, P, L and viral RNA), condensing it into a skeleton-like shape (Newcomb & Brown, 1981 ▶; Newcomb et al., 1982 ▶), and with the plasma membrane of the budded virion (Mebatsion et al., 1999 ▶). In addition, M interacts with G, which appears to be important for budding although the mechanism remains unclear (Nakahara et al., 1999 ▶; Mebatsion et al., 1996 ▶, 1999 ▶). The N-terminus of M is characterized by a positive charge thought to be important for membrane association (Gaudier et al., 2002 ▶; Solon et al., 2005 ▶) and contains a potential ‘late domain’, the PPXY motif, found in many RNA viruses where it has been implicated in promoting budding (Bieniasz, 2006 ▶). This PPXY motif has been shown to interact with NEDD4, part of the cellular ubiquitin–proteasome machinery; in vesicular stomatitis virus (VSV), a rhabdovirus closely related to lyssaviruses, this interaction promotes budding in an as-yet unidentified manner (Harty et al., 1999 ▶, 2001 ▶; Jayakar et al., 2000 ▶; Irie et al., 2004 ▶; Bieniasz, 2006 ▶).
To date, no high-resolution structural information is available for full-length rhabdovirus matrix proteins, although the crystal structure of a thermolysin-resistant core of the VSV matrix protein has been solved (Gaudier et al., 2002 ▶). While this structure revealed a novel fold distinct from that of the matrix proteins of other negative-sense single-stranded RNA viruses such as Ebola and influenza (Dessen et al., 2000 ▶; Harris et al., 2001 ▶; Gomis-Rüth et al., 2003 ▶), it lacked the N-terminal 57 residues (containing the late domain) and the solvent-exposed loop between residues 122 and 127 owing to proteolysis. The function of this loop remains unclear: it had previously been implicated in M self-association and/or membrane association (Gaudier et al., 2001 ▶, 2002 ▶), but recent evidence suggests that it may instead affect viral translation and induction of apoptosis (Connor et al., 2006 ▶). In short, the absence of the functionally important N-terminus and hydrophobic loop in the structure of the VSV M protein leaves many questions unanswered. In addition, no structure is available for lyssavirus M proteins, which do not share significant sequence identity with the M protein from VSV.
In an attempt to resolve these issues, we report here the cloning, expression, purification and crystallization of the full-length M protein from three lyssaviruses, Lagos bat virus, Mokola virus and Thailand dog virus, the first of which has been crystallized in space group P6122 or P6522.
2. Experimental procedures and results
2.1. Construction of a novel In-Fusion expression vector, pOPINS, for expressing proteins with an N-terminal His6-SUMO fusion tag
pOPINS was generated by amplification of the SUMO protein tag (Malakhov et al., 2004 ▶) from a synthetic (codon-optimized) SUMO template using the primers 5′-GAGATATACCATGGGTAGCAGCCATCACCATCATCATCACGGGAGCGATAGCGAAGTGAACCAG-3′ (forward) and 5′-CGGGGTACCACCGATCTGTTCGCGATG-3′ (reverse). The PCR product was gel-purified, digested with NcoI and KpnI and ligated into NcoI/KpnI-cut pOPINB (Berrow et al., 2007 ▶). This vector encodes the 12.4 kDa N-terminal His6-SUMO fusion tag, MGSSHHHHHHGSDSEVNQEAKPEVKPEVKPETHINLKVSDGSSEIFFKIKKTTPLRRLMEAFAKRQGKEMDSLRFLYDGIRIQADQTPEDLDMEDNDIIEAHREQIGG↓, where ↓ denotes the cleavage point for SUMO protease, which is structure-dependent rather than sequence-dependent (Muller et al., 2001 ▶). The sequence of the protein of interest starts directly after the cleavage point. Hence, treatment with SUMO protease yields the native target without additional amino acids. The target protein can be separated from the His6-tagged SUMO protease (see below) by Ni–nitrilotriacetic (Ni–NTA) acid metal-affinity chromatography.
2.2. Cloning of the SUMO protease from Saccharomyces cerevisiae and expression of the N-terminally His6-tagged protease
The SUMO protease was PCR-amplified from S. cerevisiae genomic DNA (Invitrogen) using the primers 5′-ATCATCACAGCAGCGGCCTTGTTCCTGAATTAAATGAAAAAGACGA-3′ (forward) and 5′-ATGGTCTAGAAAGCTTTACTATTTTAAAGCGTCGGTTAAAATCAAATGG-3′ (reverse). The PCR product was gel-purified and cloned into KpnI/HindIII-cut pOPINE using the In-Fusion protocol (Berrow et al., 2007 ▶). The vector encodes the S. cerevisiae SUMO protease MAHHHHHHSSGLVPELNEKDDDQVQKALASRENTQLMNRDNIEITVRDFKTLAPRRWLNDTIIEFFMKYIEKSTPNTVAFNSFFYTNLSERGYQGVRRWMKRKKTQIDKLDKIFTPINLNQSHWALGIIDLKKKTIGYVDSLSNGPNAMSFAILTDLQKYVMEESKHTIGEDFDLIHLDCPQQPNGYDCGIYVCMNTLYGSADAPLDFDYKDARMRRFIAHLIILTDALK.
The protease was expressed and purified following the standardized OPPF purification methods as described by Ren et al. (2005 ▶) and its identity was verified by mass spectrometry (data not shown).
2.3. Cloning and expression of three lyssavirus matrix proteins
In order to improve the chances of successful crystallization, a set of constructs was designed for three lyssaviruses: Mokola virus (MOK; accession No. AY540347), Thailand dog virus (THA; accession No. AY540348) and Lagos bat virus (LAG; accession No. AY540349) (Kassis et al., 2004 ▶). cDNA encoding the three matrix proteins was prepared from viral RNA using random hexamer-mediated reverse transcription, followed by PCR with gene-specific primers. Genes encoding full-length M were cloned into pOPINF [adding an N-terminal His6-3C (NH3C) tag], pOPINE [adding a C-terminal Lys-His6 (CKH) tag] or pOPINS [adding an N-terminal His6-SUMO (NHS) tag] using In-Fusion cloning (Berrow et al., 2007 ▶). Small-scale expression screening in Escherichia coli Rosetta(DE3)pLysS was performed as described by Berrow et al. (2007 ▶). Strikingly, high yields of soluble full-length M were observed for all three matrix proteins, but only when fused to the SUMO tag. Using the NH3C tag resulted in the production of insoluble full-length protein only and none of the C-terminally tagged constructs showed any soluble expression, underscoring the solubilizing effect of the SUMO tag.
2.4. Purification of full-length SUMO-tagged matrix proteins
The full-length NHS constructs were scaled up in native or selenomethionine (SeMet) form as described by Sutton et al. (2004 ▶) and Ren et al. (2005 ▶), yielding between 1 and 5 mg soluble protein per litre of culture. The initial lysis and Ni–NTA affinity purification used our standard protocol (Ren et al., 2005 ▶). Briefly, the pellets were resuspended in lysis buffer [25 mM Tris–HCl pH 7.5, 500 mM NaCl, 40 mM imidazole, 2 mM DTT, 0.2% Triton X-100, DNase (Sigma–Aldrich) and protease inhibitors (Roche)] and passed through a cell disruptor at 207 MPa (Basic Z model cell disruptor, Constant Systems). Following centrifugation, binding to Ni–NTA Sepharose (GE Healthcare), washing and elution, the protein solution was loaded directly onto a Superdex 200 column (HiLoad 16/60, GE Healthcare) equilibrated in gel-filtration buffer (25 mM HEPES pH 8.0, 100 mM NaCl, 5 mM DTT). The peak fractions were pooled and 250 µg SUMO protease added and incubated for 2 h at room temperature with gentle agitation. 2 ml fresh Ni-Sepharose was added to the suspension and incubated for an additional 30 min at room temperature. Following transfer to a disposable chromatography column (Econopak, Bio-Rad) the flowthrough was collected, concentrated and applied onto a Superdex 75 column (HiLoad 16/60, GE Healthcare) equilibrated in gel-filtration buffer. The peak fractions were pooled and analysed by SDS–PAGE (Fig. 1 ▶) and mass spectroscopy (Nettleship et al., 2005 ▶), confirming the identity of the purified proteins and 100% selenium incorporation for the SeMet-labelled samples (data not shown).
Figure 1.
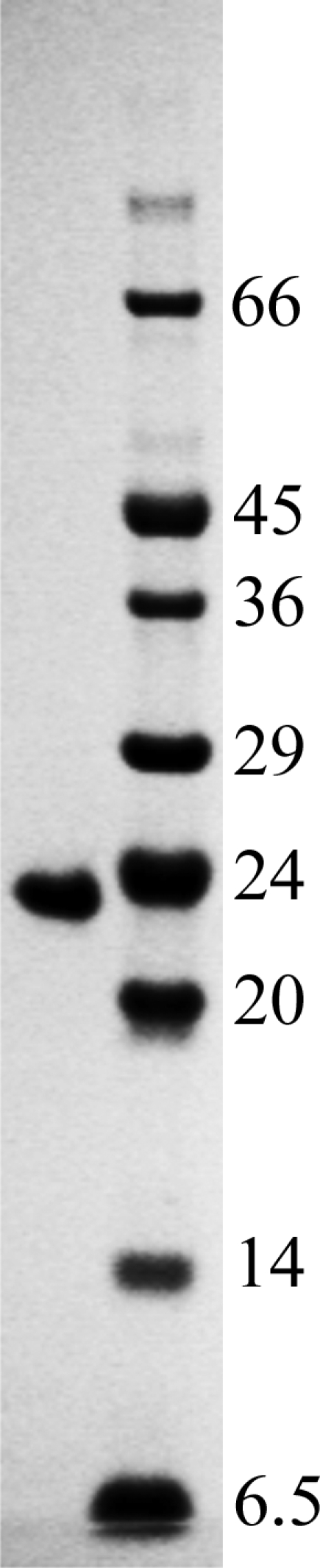
SDS–PAGE of purified LAG M (molecular weights are indicated in kDa).
2.5. Crystallization
Crystallization trials were attempted for all three lyssavirus matrix proteins under identical conditions. The matrix proteins could only by concentrated to 1.1 mg ml−1 as estimated by A 280 using predicted extinction coefficients; further concentration led to heavy protein precipitation. Protein samples were centrifuged for 5 min at 20 000g and 288 K immediately prior to crystallization to ensure that samples were free of particulate matter. Unless otherwise stated, all crystallization experiments were performed at room temperature in 96-well nanolitre sitting drops (100 nl protein solution plus 100 nl reservoir solution) equilibrated against 95 µl reservoir solution as described by Walter et al. (2003 ▶, 2005 ▶). A total of 768 crystallization conditions were tested for each M protein. Despite similar behaviour throughout purification, crystals were only obtained for LAG M. Small needle-like crystals (Fig. 2 ▶ a) grew against a reservoir of 100 mM sodium acetate pH 5.0, 10%(v/v) 2-methyl-2,4-pentanediol, but were found to be recalcitrant to optimization by altering the reservoir pH, concentration of precipitant or protein:reservoir ratio using the procedure described in Walter et al. (2005 ▶). The presence of zinc has previously been shown to promote self-association and aggregation of the M protein from VSV (Gaudin et al., 1997 ▶). To ascertain whether zinc could enhance the crystallization of M, 0.1 mM ZnCl2 was added to LAG M prior to crystallization. Re-screening all 768 conditions with the Zn-supplemented M protein yielded larger hexagonal crystals, which were grown against a reservoir consisting of 100 mM citrate pH 4.0 and 10%(w/v) polyethylene glycol (PEG) 6000 (Fig. 2 ▶ b). These diffraction-quality crystals (Fig. 2 ▶ b) grew within a fortnight to approximate dimensions of 80 × 40 × 40 µm. Reproduction of these crystals with selenomethionine-labelled (SeMet) protein proved difficult, crystallization being critically dependent upon the starting concentrations of protein and PEG 6000, the presence of 0.1 mM ZnCl2 in the protein solution and the protein:reservoir ratio in the drops (200 nl protein solution plus 100 nl reservoir being optimal). Crystallization trials against reservoirs containing 100 mM citrate pH 4.0 and a range of PEG 6000 concentrations [0.25–5%(w/v)] successfully yielded SeMet crystals that were suitable for diffraction analysis (Fig. 2 ▶ c). Crystals of native and SeMet LAG M also consistently appeared in 100 mM citrate pH 4.0 with either 0.5–1 M NaCl or 0.5–1 M LiCl2 (Fig. 2 ▶ d), but these gave poor diffraction. Subsequent attempts to crystallize MOK and THA M supplemented with 0.1 mM ZnCl2 did not yield crystals either in standard sparse-matrix screens or in the PEG 6000/citrate optimization screens used for LAG M.
Figure 2.

Crystals obtained for native and SeMet-substituted LAG M with or without ZnCl2 added to the protein. The scale bar corresponds to 50 µm. (a) Native LAG M, no ZnCl2 added, crystallization conditions 10%(v/v) 2-methyl-2,4-pentanediol, 0.1 M sodium acetate pH 5.0. (b) Native LAG M, ZnCl2 added, crystallization conditions 10%(w/v) PEG 6000, 0.1 M citrate pH 4.0. (c) SeMet-substituted LAG M, ZnCl2 added, crystallization conditions 0.75%(w/v) PEG 6000, 0.1 M citrate pH 4.0. (d) Native LAG M, ZnCl2 added, crystallization conditions 0.1 M citrate pH 4.0, 1.0 M sodium chloride.
2.6. Data collection
Prior to data collection, crystals were transferred to fresh drops containing reservoir solution supplemented with 25%(v/v) glycerol and flash-cryocooled by transfer directly into a cold stream of nitrogen gas (100 K). Crystals thus cryocooled diffracted quite poorly (Fig. 3 ▶ a), but re-annealing by means of transferring the crystals into the cryoprotecting solution then re-cryocooling them in the cold nitrogen-gas stream yielded a significant improvement in diffraction quality (Fig. 3 ▶ b). Diffraction data were recorded from a single frozen (100 K) crystal of native LAG M on a MAR225 CCD detector at ESRF beamline ID23-2 and from a single frozen (100 K) crystal of SeMet LAG M at three wavelengths around the Se K edge on a MAR225 CCD detector at ESRF BM14. Data were processed using XDS via the xia2 automated processing pipeline (native LAG M; Kabsch, 1993 ▶; Winter et al., manuscript in preparation) or using the HKL-2000 processing suite (SeMet LAG M; Otwinowski & Minor, 1997 ▶).
Figure 3.

Re-annealing of cryocooled crystals greatly enhanced diffraction quality. Identical diffraction images (Δϕ = 0.5°) were collected from a single crystal of SeMet LAG M (a) before and (b) after re-annealing the crystal by removing it from the cold nitrogen-gas cryostream, quickly sweeping it through the cryoprotectant solution and then re-cryocooling in the cryostream.
3. Discussion
We have successfully cloned, expressed and purified M proteins from three lyssaviruses: Lagos bat virus, Mokola virus and Thailand dog virus. This represents the first successful recombinant expression and purification of a full-length lyssavirus M protein. The use of a SUMO fusion tag at the N-terminus of the protein was essential for obtaining soluble protein, highlighting the utility of SUMO fusion tags in enhancing solubility of ‘difficult’ proteins.
Despite sharing >79% sequence identity, crystals were obtained for only one of the three M proteins, that from Lagos bat virus. Extensive screening around the condition used to crystallize Lagos bat virus M in the presence or absence of ZnCl2 failed to yield crystals of MOK or THA M. Note that the crystals were obtained at an unusually low protein concentration (1.1 mg ml−1), explaining why they were rather small (maximum dimension 80 µm).
Diffraction data were recorded from crystals of both native and SeMet LAG M (Table 1 ▶). The unit-cell parameters (a = b = 56.9–57.2, c = 187.9–188.6 Å) and point group (622) are consistent with the presence of 36% solvent and one molecule per asymmetric unit. Systematic absences in the diffraction data are consistent with the presence of a 61 or 65 screw axis along c. The mass-spectrometric analysis demonstrated that the level of SeMet incorporation was ∼100% and the presence of selenium is consistent with the anomalous scattering signal observed in the peak, inflection and remote-wavelength diffraction data (see Table 1 ▶). Space-group and structure determination are currently under way.
Table 1. Data-collection and processing statistics.
Values in parentheses are for the highest resolution shell.
| SeMet | ||||
|---|---|---|---|---|
| Native | Peak | Remote | Inflection | |
| X-ray source | ESRF ID23-2 | ESRF BM14 | ||
| Detector | MAR225 CCD | MAR225 CCD | ||
| Space group | P6122 or P6522 | P6122 or P6522 | ||
| Unit-cell parameters (Å) | a = b = 56.9, c = 187.9 | a = b = 57.2, c = 188.6 | ||
| Wavelength (Å) | 0.873 | 0.978 | 0.900 | 0.979 |
| Energy (eV) | 14208 | 12679 | 13776 | 12670 |
| Oscillation angle (°) | 1 | 0.5 | 0.5 | 0.5 |
| No. of frames | 60 | 720 | 420 | 360 |
| Resolution (Å) | 40–2.75 (2.82–2.75) | 25–3.0 (3.16–3.00) | 25–3.2 (3.37–3.20) | 25–3.2 (3.36–3.19) |
| Unique reflections | 5164 (367) | 4089 (567) | 3427 (478) | 3464 (475) |
| Completeness (%) | 99.6 (99.4) | 99.7 (100.0) | 99.7 (99.9) | 99.7 (100.0) |
| Redundancy | 6.2 (5.4) | 39.3 (41.1) | 22.9 (24.2) | 19.5 (20.5) |
| Rmerge† | 0.128 (0.769) | 0.188 (1.158) | 0.185 (0.940) | 0.187 (0.966) |
| Rmeas‡ | 0.140 (0.853) | 0.196 (1.181) | 0.193 (0.972) | 0.197 (1.002) |
| Rp.i.m.§ | 0.055 (0.360) | 0.031 (0.183) | 0.040 (0.196) | 0.044 (0.220) |
| Average I/σ(I) | 10.2 (2.2) | 26.3 (4.9) | 20.3 (4.4) | 19.1 (3.8) |
| Reflections with average I/σ(I) ≥ 2 (%) | 79.3 (44.7) | 80.7 (52.8) | 78.1 (48.1) | 75.1 (46.6) |
| d′′/σ¶ | — | 6.1/1.4 | 2.7/1.0 | 3.1/1.1 |
R
merge = 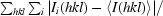
 .
.
R
meas is the redundancy-independent merging R factor, also known as R
r.i.m. (Diederichs & Karplus, 1997 ▶; Weiss & Hilgenfeld, 1997 ▶; Weiss et al., 1998 ▶). R
meas = 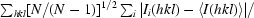 , where N is the number of times a given reflection has been observed.
, where N is the number of times a given reflection has been observed.
R
p.i.m. is the precision-indicating merging R factor (Weiss & Hilgenfeld, 1997 ▶; Weiss et al., 1998 ▶). R
p.i.m. =  .
.
As calculated using SHELXC (Sheldrick, 2008 ▶). The values shown were calculated using data in the resolution ranges 25–8.0 and 4.0–3.8 Å, respectively.
Acknowledgments
We thank Joanne Nettleship for mass spectrometry, Florence Larrous and the staff at BM14 and ID23-2 at the ESRF, Grenoble, France for technical assistance. JMG is supported by the Royal Society and DIS and the OPPF are supported by the UK MRC and European Commission grant Nos. QLG2-CT-2002-00988 (SPINE) and LSHG-CT-2004-511960 (VIZIER). SCG is a Nuffield Medical Fellow. BM14 is supported by the UK research councils. OD, HB and UPRE LDHA are supported by European Commission grant No. LSHG-CT-2004-511960 (VIZIER).
References
- Berrow, N. S., Alderton, D., Sainsbury, S., Nettleship, J., Assenberg, R., Rahman, N., Stuart, D. I. & Owens, R. J. (2007). Nucleic Acids Res.35, e45. [DOI] [PMC free article] [PubMed]
- Bieniasz, P. D. (2006). Virology, 344, 55–63. [DOI] [PubMed]
- Connor, J. H., McKenzie, M. O. & Lyles, D. S. (2006). J. Virol.80, 3701–3711. [DOI] [PMC free article] [PubMed]
- Dessen, A., Volchkov, V., Dolnik, O., Klenk, H. D. & Weissenhorn, W. (2000). EMBO J.19, 4228–4236. [DOI] [PMC free article] [PubMed]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol.4, 269–275. [DOI] [PubMed]
- Finke, S. & Conzelmann, K. K. (2005). Virus Res.111, 120–131. [DOI] [PubMed]
- Gaudier, M., Gaudin, Y. & Knossow, M. (2001). Virology, 288, 308–314. [DOI] [PubMed]
- Gaudier, M., Gaudin, Y. & Knossow, M. (2002). EMBO J.21, 2886–2892. [DOI] [PMC free article] [PubMed]
- Gaudin, Y., Sturgis, J., Doumith, M., Barge, A., Robert, B. & Ruigrok, R. W. (1997). J. Mol. Biol.274, 816–825. [DOI] [PubMed]
- Gomis-Rüth, F. X., Dessen, A., Timmins, J., Bracher, A., Kolesnikowa, L., Becker, S., Klenk, H. D. & Weissenhorn, W. (2003). Structure, 11, 423–433. [DOI] [PMC free article] [PubMed]
- Harris, A., Forouhar, F., Qiu, S., Sha, B. & Luo, M. (2001). Virology, 289, 34–44. [DOI] [PubMed]
- Harty, R. N., Brown, M. E., McGettigan, J. P., Wang, G., Jayakar, H. R., Huibregtse, J. M., Whitt, M. A. & Schnell, M. J. (2001). J. Virol.75, 10623–10629. [DOI] [PMC free article] [PubMed]
- Harty, R. N., Paragas, J., Sudol, M. & Palese, P. (1999). J. Virol.73, 2921–2929. [DOI] [PMC free article] [PubMed]
- Irie, T., Licata, J. M., Jayakar, H. R., Whitt, M. A., Bell, P. & Harty, R. N. (2004). J. Virol.78, 7823–7827. [DOI] [PMC free article] [PubMed]
- Jayakar, H. R., Jeetendra, E. & Whitt, M. A. (2004). Virus Res.106, 117–132. [DOI] [PubMed]
- Jayakar, H. R., Murti, K. G. & Whitt, M. A. (2000). J. Virol.74, 9818–9827. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (1993). J. Appl. Cryst.26, 795–800.
- Kassis, R., Larrous, F., Estaquier, J. & Bourhy, H. (2004). J. Virol.78, 6543–6555. [DOI] [PMC free article] [PubMed]
- Komarova, A. V., Real, E., Borman, A. M., Brocard, M., England, P., Tordo, N., Hershey, J. W., Kean, K. M. & Jacob, Y. (2007). Nucleic Acids Res.35, 1522–1532. [DOI] [PMC free article] [PubMed]
- Malakhov, M. P., Mattern, M. R., Malakhova, O. A., Drinker, M., Weeks, S. D. & Butt, T. R. (2004). J. Struct. Funct. Genomics, 5, 75–86. [DOI] [PubMed]
- Mebatsion, T., Konig, M. & Conzelmann, K.-K. (1996). Cell, 84, 941–951. [DOI] [PubMed]
- Mebatsion, T., Weiland, F. & Conzelmann, K.-K. (1999). J. Virol.73, 242–250. [DOI] [PMC free article] [PubMed]
- Muller, S., Hoege, C., Pyrowolakis, G. & Jentsch, S. (2001). Nature Rev. Mol. Cell Biol.2, 202–210. [DOI] [PubMed]
- Nakahara, K., Ohnuma, H., Sugita, S., Yasuoka, K., Nakahara, T., Tochikura, T. S. & Kawai, A. (1999). Microbiol. Immunol.43, 259–270. [DOI] [PubMed]
- Nettleship, J. E., Walter, T. S., Aplin, R., Stammers, D. K. & Owens, R. J. (2005). Acta Cryst. D61, 643–645. [DOI] [PubMed]
- Newcomb, W. W. & Brown, J. C. (1981). J. Virol.39, 295–299. [DOI] [PMC free article] [PubMed]
- Newcomb, W. W., Tobin, G. J., McGowan, J. J. & Brown, J. C. (1982). J. Virol.41, 1055–1062. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Ren, J., Sainsbury, S., Berrow, N. S., Alderton, D., Nettleship, J. E., Stammers, D. K., Saunders, N. J. & Owens, R. J. (2005). BMC Struct. Biol.5, 13. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Solon, J., Gareil, O., Bassereau, P. & Gaudin, Y. (2005). J. Gen. Virol.86, 3357–3363. [DOI] [PubMed]
- Sutton, G. et al. (2004). Structure, 12, 341–353. [DOI] [PMC free article] [PubMed]
- Walter, T. S., Diprose, J., Brown, J., Pickford, M., Owens, R. J., Stuart, D. I. & Harlos, K. (2003). J. Appl. Cryst.36, 308–314.
- Walter, T. S. et al. (2005). Acta Cryst. D61, 651–657. [DOI] [PMC free article] [PubMed]
- Weiss, M. S. & Hilgenfeld, R. (1997). J. Appl. Cryst.30, 203–205.
- Weiss, M. S., Metzner, H. J. & Hilgenfeld, R. (1998). FEBS Lett.243, 291–296. [DOI] [PubMed]
- World Health Organization. (2005). WHO Technical Report Series 931. Geneva, Switzerland: World Health Organization. http://www.who.int/rabies/trs931_%2006_05.pdf.