

The crystal structure of the complex of mouse Keap1-DC with a fragment of the nuclear protein prothymosin α was determined and refined to 1.9 Å resolution and revealed that the peptide binds to the bottom region of the β-propeller domain of Keap1-DC.
Keywords: oxidative stress, Nrf2 transcription factor, prothymosin α, Keap1, β-propeller domain
Abstract
The Nrf2 transcription factor, which plays important roles in oxidative and xenobiotic stress, is negatively regulated by the cytoplasmic repressor Keap1. The β-propeller/Kelch domain of Keap1, which is formed by the double-glycine repeat and C-terminal region domains (Keap1-DC), interacts directly with the Neh2 domain of Nrf2. The nuclear oncoprotein prothymosin α (ProTα) also interacts directly with Keap1 and may play a role in the dissociation of the Keap1–Nrf2 complex. The structure of Keap1-DC complexed with a ProTα peptide (amino acids 39–54) has been determined at 1.9 Å resolution. The Keap1-bound ProTα peptide possesses a hairpin conformation and binds to the Keap1 protein at the bottom region of the β-propeller domain. Complex formation occurs as a consequence of their complementary electrostatic interactions. A comparison of the present structure with recently reported Keap1-DC complex structures revealed that the DLG and ETGE motifs of the Neh2 domain of Nrf2 and the ProTα peptide bind to Keap1 in a similar manner but with different binding potencies.
1. Introduction
In higher animals, the cellular response arising from environmental stress is controlled by several coordinated functions of cellular factors. The events can be divided into three steps: (i) a cellular protein acts as a sensor and detects signals from the environmental changes, (ii) the sensor transduces the signal to the gene-expression machinery and lastly (iii) the transduced signals then activate transcription factors, which induce the expression of a set of stress-responsive genes involved in cellular protection. These processes must be tightly regulated and precisely controlled in order to sustain cellular homeostasis (reviewed in Kobayashi & Yamamoto, 2006 ▶). Environmental stress factors, including reactive oxygen species, electrophilic chemicals and heavy metals, damage biological macromolecules and disrupt normal cellular functions. Oxidative and xenobiotic stress are responsible for the development of many diseases, including cancer, cardiovascular disease, diabetes and neurodegeneration. The defence against oxidative stress and electrophilic attack is controlled by the activation of an array of genes encoding detoxifying and anti-oxidative stress enzymes/proteins.
Nrf2 is a basic region leucine zipper (bZIP) transcription factor for cytoprotective enzymes that counteract oxidative and electrophilic attacks (Itoh et al., 1997 ▶). Nrf2 is composed of a conserved N-terminal regulatory domain, termed the Neh2 domain, two transactivation domains and a C-terminal bZIP domain. Under homeostatic/unstressed conditions, the cellular concentration of Nrf2 remains low and is modulated by Keap1 (Kelch-like ECH-associated protein 1) and proteasomal degradation (Itoh et al., 2004 ▶).
The Keap1 protein interacts directly with Nrf2 and is the major negative regulator of cytoprotective gene expression (Itoh et al., 1999 ▶). Keap1 contains an N-terminal BTB domain, an intervening region (IVR), a double glycine repeat or Kelch repeat (DGR) domain and a C-terminal region (CTR) domain. As the structurally homologous BTB domains form homodimers (Stogios et al., 2005 ▶), dimerization of Keap1 probably occurs through its BTB domain (Zipper & Mulcahy, 2002 ▶).
Under homeostatic/unstressed conditions, Keap1 represses the transcription functions of Nrf2 by preventing its translocation into the nucleus and directs Nrf2 for ubiquitin-dependent degradation (Wakabayashi et al., 2003 ▶; Zhang & Hannink, 2003 ▶; Motohashi & Yamamoto, 2004 ▶) by associating with the Neh2 domain of Nrf2 (Itoh et al., 1999 ▶). However, under stressed conditions, such as exposure to oxidative stress or electrophilic attack, Keap1 loses its ability to repress Nrf2, allowing Nrf2 to escape proteasomal degradation (Kobayashi et al., 2006 ▶). Nrf2 then translocates to the nucleus and transcriptionally activates a battery of cytoprotective genes to protect the cell.
A recent study showed that Keap1 also interacts directly with a nuclear oncoprotein, prothymosin α (ProTα; Karapetian et al., 2005 ▶). The ubiquitious ProTα protein is involved in the proliferation of mammalian cells (Gomez-Marquez et al., 1989 ▶) and in their protection against apotopsis (Evstafieva et al., 2003 ▶; Jiang et al., 2003 ▶). ProTα also plays important roles in transcriptional regulation (Karetsou et al., 2002 ▶; Martini et al., 2000 ▶). Karapetian et al. (2005 ▶) proposed that ProTα associates with Keap1 through its β-propeller domain and may participate in dissociating Nrf2 from the Keap1–Nrf2 complex, thus allowing transcription initiation.
As the molecular interaction of ProTα with Keap1 has not been clarified, we have undertaken the structural determination of mouse Keap1 (containing the DGR and CTR domains; amino acids 309–624; hereafter referred to as Keap1-DC) in complex with a peptide corresponding to the ProTα-interacting region (amino acids 39–54). The structure of the complex revealed that the ProTα peptide binds firmly to Keap1-DC at the bottom region of the propeller domain. Intriguingly, the mode of ProTα-peptide binding is essentially similar to those of the ETGE and DLG motifs of Nrf2 in their respective Keap1-DC complexes (Padmanabhan et al., 2006 ▶; Tong et al., 2007 ▶).
2. Materials and methods
2.1. Crystallization and data collection
The mouse Keap1-DC protein was expressed in Escherichia coli and purified and crystallized as described elsewhere (Padmanabhan et al., 2005 ▶). The best crystals were obtained at 293 K in a drop containing 0.8 M lithium sulfate, 0.5 M ammonium sulfate and 0.1 M sodium citrate pH 5.2. For cocrystallization of the protein with the ProTα peptide (amino acids 39–54: AQNEENGEQEADNEVD), which was purchased from Promega K.K., the peptide and protein solutions were mixed (in a 10:1 molar ratio) and incubated at 277 K for about 1 d before setting up the crystallization experiment. Diffraction data were collected under cryogenic conditions on beamline BL5A at the Photon Factory, Tsukuba, Japan. The data were integrated and scaled with HKL-2000 (Otwinowski & Minor, 1997 ▶). The results of the data-reduction statistics are summarized in Table 1 ▶.
Table 1. Summary of data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Source | BL5A, Photon Factory, Tsukuba |
| Wavelength () | 1.00 |
| Space group | P61 |
| Unit-cell parameters (, ) | a = b = 103.52, c = 56.16, = 120 |
| Resolution () | 50.02.0 |
| No. of unique reflections (all) | 23352 |
| No. of unique reflections (observed) | 21733 |
| Completeness (%) | 93.1 (99.0) |
| I/(I)† | 50.9 (10.0) |
| Redundancy | 10.6 (10.3) |
| R merge ‡ (%) | 7.4 (22.8) |
| Refinement statistics | |
| No. of complex molecules in ASU | 1 |
| Resolution limit () | 20.02.0 |
| cutoff | 0 |
| No. of reflections | 20530 |
| R factor§/R free ¶ (%) | 16.6/22.3 |
| No. of protein residues | 290 |
| No. of peptide residues | 9 |
| No. of sulfate ions | 5 |
| No. of water molecules | 252 |
| R.m.s. deviations | |
| Bond lengths () | 0.016 |
| Bond angles () | 1.62 |
I/(I) is the average intensity divided by its standard deviation.
R
merge = 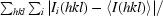
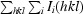 .
.
R
cryst = 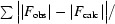
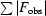 , where F
obs and F
calc are observed and calculated structure factors, respectively.
, where F
obs and F
calc are observed and calculated structure factors, respectively.
R free was calculated with 5% of data omitted from refinement.
2.2. Structure determination and refinement
The structure of the Keap1-DC–ProTα peptide complex was determined by the molecular-replacement method, employing the native Keap1 structure (PDB code 1x2j; Padmanabhan et al., 2006 ▶) as a search model, using the program MOLREP from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶). It gave a distinct peak with an R factor and a correlation coefficient of 0.363 and 0.690, respectively, for the resolution range 20–3.0 Å. The model was refined with REFMAC5 (Murshudov et al., 1997 ▶) in CCP4 and several rounds of manual fitting and re-fitting were carried out using the program Coot (Emsley & Cowtan, 2004 ▶) with careful inspection of the 2F o − F c, F o − F c and OMIT electron-density maps. The refined current model consists of 299 residues, five sulfate ions and 252 water molecules, with a final R work and R free of 16.6% and 22.3%, respectively, at 2.0 Å resolution. The electron densities for 15 residues at the N-terminus and 11 residues at the C-terminus were absent. The stereochemistry of the Keap1-DC complex was good as assessed by PROCHECK (Laskowski et al., 1993 ▶). The refinement statistics are summarized in Table 1 ▶.
3. Results and discussion
3.1. Overall structure of the Keap1-DC–ProTα peptide complex
The structure of Keap1-DC complexed with the prothymosin α peptide fragment corresponding to amino acids 39–54 was solved by the molecular-replacement method using the apo form of the Keap1-DC structure (Padmanabhan et al., 2006 ▶) as a model. The overall tertiary structure of Keap1-DC is composed of a six-bladed β-propeller motif in which each blade of the propeller is composed of four antiparallel β-strands (β1–β4; Fig. 1 ▶; Padmanabhan et al., 2006 ▶; Li et al., 2004 ▶). The β-propeller domain (also known as the Kelch domain) of the Keap1-DC protein resembles a small cylindrical/disc-shaped structure with dimensions of about 49 × 39 Å. The β-propeller ring closure is achieved by a strand in the CTR, which completes a β-sheet with three strands from blade 1 at the N-terminus of the protein.
Figure 1.

Overall tertiary structure of the mouse Keap1-DC–ProTα complex. (a) Structure-based sequence alignment of the C-terminal region of Keap1, containing six DGR repeats and the CTR domain. Conserved amino-acid residues are shown in red. (b), (c) Ribbon models of the tertiary structure of the Keap1-DC β-propeller domain (blue to red) and the ProTα peptide (sticks) are shown in a side view and from a bottom view, respectively. All structure figures were generated with PyMOL (http://www.pymol.org).
The shorter loops that connect either β-strands β1 and β2 (β1–β2) or β-strands β3 and β4 (β3–β4) define the top face of the propeller domain, whereas the longer loops that connect either β-strands β4 and β1 (β4–β1) or β-strands β2 and β3 (β2–β3) define the bottom face of the propeller domain (Fig. 1 ▶). The ‘velcro’ closure or molecular clasp (Neer & Smith, 1996 ▶) configuration is achieved in the present Keap1-DC–ProTα complex structure by the closing of the β-strands of both termini, as observed in other β-propeller domain structures.
The ProTα peptide (amino acids 39–54) binds to the Keap1-DC protein at the bottom side of the β-propeller domain (Figs. 1 ▶ and 2 ▶ a). In the structure, the peptide residues corresponding to the region from residue 39 to 48 are visible; however, the side chains of Gln47 and Glu48 were truncated to Ala since the electron density corresponding to this region is absent. As shown in Fig. 2 ▶(b), the electrostatic surface potential at the bottom region of the β-propeller domain is highly basic. The ProTα peptide binds to Keap1-DC by means of charge-complementary interactions. The peptide possesses a tight β-turn conformation formed by residues Glu42, Glu43, Asn44 and Gly45. The β-turn is stabilized by two intramolecular main chain–main chain hydrogen bonds between the carbonyl group of Asn41 and the amide group of Gly45 and between the amide groups of Asn41 and the carbonyl group of Glu46. In addition, an intramolecular hydrogen-bond interaction between Asn44 OD1 and the amide group of Glu46 also contributes to stabilize the peptide main-chain conformation (Fig. 2 ▶ a).
Figure 2.

The structure of the ProTα peptide in the Keap1-DC protein-bound form. (a) The refined structure of the ProTα peptide is shown, with its 2m|F o| − D|F c| (final) and m|F o| − D|F c| (initial) electron-density maps contoured at 1.0σ (in green) and 2.0σ (in red), respectively. (b) The peptide (stick model) binds to the highly basic bottom region of Keap1-DC. Surface acidic, basic and neutral residues are shown in red, blue and white, respectively.
In the structure of the complex, about 17 intermolecular electrostatic interactions are formed between the Keap1-DC protein and the ProTα peptide (Table 2 ▶). Intriguingly, almost all of the Kelch motifs are involved in intermolecular interactions with the peptide. Glu42 OE1 forms a salt link with Gln530 NE2 as well as with two bound water molecules. The carbonyl group of Glu42 hydrogen bonds to the side-chain atom (NE2) of Gln530. The side chain of Glu43 is surrounded by Arg415, Ser508, Arg483 and Phe470 (Fig. 3 ▶). The carboxylate group contributes two salt links each to Arg415 and Arg483, respectively, and forms a potential hydrogen-bonding interaction with Ser508. The main-chain carbonyl group of Glu43 makes a potential hydrogen-bonding interaction with the hydroxyl group of Ser555. The carbonyl group of Asn44 also forms a potential hydrogen bond with the side chain of Ser602. The side-chain carboxyl group of Glu46 makes substantial electrostatic interactions with Ser363, Asn382 and Arg380.
Table 2. Potential hydrogen-bonding interactions of ProT with Keap1-DC and water molecules.
| ProT | Keap1-DC | Distance () |
|---|---|---|
| Gln40NE2 | Tyr572OH | 3.38 |
| Wat148O | 2.77 | |
| Glu42N | Wat91O | 2.95 |
| Gln42OE1 | Gln530NE2 | 2.99 |
| Wat135O | 3.19 | |
| Wat142O | 3.02 | |
| Gln42O | Gln530NE2 | 2.77 |
| Glu43OE1 | Arg415NH1 | 2.88 |
| Arg415NH2 | 3.05 | |
| Ser508OG | 2.62 | |
| Glu43OE2 | Arg483NH1 | 2.57 |
| Ser508OG | 3.28 | |
| Glu43O | Ser555OG | 2.36 |
| Asn44ND2 | Wat32O | 3.01 |
| Asn44O | Ser602OG | 2.66 |
| Gly45O | Wat134O | 3.04 |
| Glu46OE1 | Asn382ND2 | 2.95 |
| Arg380NH2 | 3.00 | |
| Wat18O | 2.62 | |
| Glu46OE2 | Ser363OG | 2.70 |
| Wat13O | 2.59 |
Figure 3.

Close-up views of the peptide-binding regions. The interacting residues are shown as sticks and hydrogen bonds are depicted by dashed lines. The Keap1-DC and the ProTα peptide are shown in salmon and dark slate, respectively. N and O atoms in the interacting residues are shown in blue and red, respectively.
In addition to these interactions, some buried water molecules also contribute to mediate interactions between the peptide and the Keap1-DC protein (not shown). W135 bridges Tyr525 and Gln528 of Keap1-DC and Glu42 of the peptide, while W142 bridges Glu530 and Glu42. Another water molecule, W13, hydrogen bonds to Ser602 and Glu46. The W24 water molecule bridges Asn414 and Glu46, while W18 bridges Arg380 and Ser363 of Keap1-DC and Glu46 of the peptide.
3.2. Comparison with the complex of Keap1-DC and the Nrf2 ETGE motif peptide
The Keap1 protein negatively regulates the transcription function of Nrf2 (Itoh et al., 1999 ▶). Structures of Keap1-DC in complex with Nrf2 interacting peptides containing the ETGE motif have recently been reported and the alteration of any one of these interacting residues severely affected complex formation (Padmanabhan et al., 2006 ▶; Lo et al., 2006 ▶).
Intriguingly, the ProTα peptide binds to Keap1-DC in a similar fashion to that observed in the structure of the complex of Keap1-DC and the Nrf2 ETGE motif peptide (PDB code 1x2r). The ETGE motif of Nrf2 and the ProTα peptide bind to Keap1-DC at the same bottom region of the β-propeller domain (Fig. 4 ▶ a). A superposition of these two complexes for all main-chain atoms of Keap1-DC gave an r.m.s.d. value of 0.19 Å, suggesting that the tertiary structure of the Kelch domain maintains its rigidity. As shown in Fig. 4 ▶(a), the overall conformations of these two peptides from two different proteins are nearly the same. The conformations and orientations of the interacting residues in the peptides are essentially comparable and maintain similar interactions with the protein in both cases. The structural similarity observed in these two peptide fragments is reflected in their sequence similarity within the protein-interacting region. The sequence EETGE (78–82) in Nrf2 is equivalent to EENGE (42–46) in ProTα. The Thr80 residue in Nrf2, which is replaced with Asn42 in ProTα, does not contribute to the interaction with the Keap1-DC protein; however, it stabilizes the backbone conformation of the peptide by interacting with the backbone amide group. In the ProTα complex, a similar intramolecular interaction occurs through the side chain of Asn42.
Figure 4.

Comparison of the Nrf2 complexes with the ETGE and DLG motif peptides. Close-up views of superpositions of the Keap1-DC–ProTα complex over the main-chain atoms of Keap1-DC (a) with the Keap1-DC–ETGE motif complex and (b) with the Keap1-DC–DLG motif complex. The colouring scheme for the ProTα complex is the same as in Fig. 3 ▶. For the ETGE and DLG complexes, the Keap1-DC interacting residues are shown in cyan and green, respectively, and the ETGE and DLG peptides in the complexes are shown in yellow and pink, respectively.
3.3. Comparison with the Keap1-DC–DLG motif Nrf2 peptide complex
Our previous functional studies showed that the DLG motif in Nrf2 is also essential for the Keap1 interaction (Kobayashi et al., 2002 ▶). To understand the molecular mechanism of the Keap1-DC–DLG motif interaction, we have recently solved the structure of the Keap1-DC–DLG motif complex and proposed a hinge-and-latch mechanism for the interaction between Keap1 and Nrf2 (Tong et al., 2007 ▶). The DLG complex structure revealed that the DLG motif of Nrf2 also interacts with Keap1-DC at the bottom region of the β-propeller domain. The recognition modes of these two ETGE and DLG motifs for Keap1 interaction are essentially similar (Tong et al., 2007 ▶) and they also resemble that found in the present ProTα complex structure.
A superposition of the DLG motif complex structure (PDB code 2dyh) over the main-chain atoms of the Keap1-DC structure revealed that the overall structures of Keap1-DC in these two complexes are nearly the same (0.24 Å r.m.s. deviation). However, a small variation was observed in the loop connecting strands β2 and β3 of the second Kelch motif in the Keap1-DC protein (not shown). At the binding-interface region, the side-chain conformations of the Keap1-DC interacting residues are quite similar in these two complexes, except for Arg415, Arg483 and Asn382. As noted in Fig. 4 ▶(b), although these two peptides adopt similar tight β-turn conformations they did not superimpose well on each other. However, these two peptides are oriented and positioned in essentially the same manner with respect to the Keap1-DC structure. This study further suggests that the loop conformations are quite flexible in the peptide fragments to facilitate adjustments to form similar interactions with their respective Keap1-DC interacting residues.
3.4. Functional implications
The Keap1 protein exists as a homodimer in solution and dimerization occurs through its BTB domain (Tong, Katoh et al., 2006 ▶). In this context, one Nrf2 molecule interacts through its ETGE and DLG motifs with two Keap1 monomers of a homodimer. An extensive point-mutation analysis of the interactions of the ETGE and DLG motifs of Nrf2 with Keap1 revealed that the ETGE-motif region possesses high binding affinity for Keap1 compared with the DLG-motif region (Tong, Katoh et al., 2006 ▶; Tong, Kobayashi et al., 2006 ▶). Based on this study, we have proposed a lock-and-latch mechanism for regulation of the Nrf2 protein in the Keap1–Cul3–Nrf2–ROC1 degradation pathway (Tong et al., 2007 ▶).
A recent study showed that Keap1 is a nuclear-cytoplasm shuttling protein equipped with a nuclear-export signal that is important for its inhibitory function (Karapetian et al., 2005 ▶). The study also revealed that prothymosin α was able to release Nrf2 from the Nrf2–Keap1 inhibitory complex in vitro through direct competition with Nrf2 binding to the same domain: the β-propeller domain of the Keap1 protein. Our ProTα complex structure results support the findings for the association between ProTα and the C-terminal region of Keap1, which contains the Kelch domain. Nevertheless, our results unexpectedly revealed that ProTα interacts with Keap1 in the same region where Nrf2 protein fragments containing the ETGE and DLG motifs bind to the Keap1 protein. The interaction modes in these three complexes are essentially the same, suggesting that the nuclear oncoprotein prothymosin α competes with Nrf2 for Keap1 interaction in the nucleus and that particular biological events in the nucleus may allow ProTα to dissociate Nrf2 from Keap1. We speculate from our structural results that ProTα competes either with the ETGE-motif region and/or the DLG-motif region of Nrf2 for Keap1 interaction. Our results further suggest that the ProTα interacting region is substantially buried within the Keap1-DC interacting region, as observed in the ETGE complex, which indicates that the binding properties of the ProTα interacting fragment may be very similar to those of the ETGE region of Nrf2, as evidenced from the recent biochemical studies of Nrf2 peptides with Keap1 (Padmanabhan et al., 2006 ▶; Tong, Katoh et al., 2006 ▶; Tong, Kobayashi et al., 2006 ▶; Tong et al., 2007 ▶). Although our studies suggest that the ProTα interacting region may compete with the ETGE/DLG motifs of the Nrf2 transcription factor for the dissociation of Nrf2, which subsequently commences its transcription function, further functional and biochemical studies on ProTα with Keap1 in the presence and absence of Nrf2 are required in order to understand the molecular mechanism of the interaction of ProTα with Keap1.
Supplementary Material
PDB reference: α peptide complex, 2z32
Acknowledgments
We are grateful to the staff of beamline BL5A, Photon Factory, Japan and to Dr Chengwei Shang for crystallization. We thank Drs Masayuki Yamamoto and Kit Tong for providing the Keap1-DC protein sample. We are grateful to Ms T. Nakayama and Ms A. Ishii for their clerical assistance. This work was supported in part by the RIKEN Structural Genomics/Proteomics Initiative (RSGI), the National Project on Protein Structural and Functional Analyses and the Integrated Database Project of the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evstafieva, A. G., Belov, G. A., Rubtsov, Y. P., Kalkum, M., Joseph, B., Chichkova, N. V., Sukhacheva, E. A., Bogdanov, A. A., Pettersson, R. F., Agol, V. I. & Vartapetian, A. B. (2003). Exp. Cell Res. 284, 211–223. [DOI] [PubMed]
- Gomez-Marquez, J., Segade, F., Dosil, M., Pichel, J. G., Bustelo, X. R. & Freire, M. (1989). J. Biol. Chem. 264, 8451–8454. [PubMed]
- Itoh, K., Chiba, T., Takahashi, S., Ishii, T., Igarashi, K., Katoh, Y., Oyake, T., Hayashi, N., Satoh, K., Hatayama, I., Yamamoto, M. & Nabeshima, Y. (1997). Biochem. Biophys. Res. Commun. 236, 313–322. [DOI] [PubMed]
- Itoh, K., Tong, K. I. & Yamamoto, M. (2004). Free Radic. Biol. Med. 36, 1208–1213. [DOI] [PubMed]
- Itoh, K., Wakabayashi, N., Katoh, Y., Ishii, K., Igarashi, K., Engel, J. D. & Yamamoto, M. (1999). Genes Dev. 13, 76–86. [DOI] [PMC free article] [PubMed]
- Jiang, X., Kim, H. E., Shu, H., Zhao, Y., Zhang, H., Kofron, J., Donnelly, J., Burns, D., Ng, S. C., Rosenberg, S. & Wang, X. (2003). Science, 299, 223–226. [DOI] [PubMed]
- Karapetian, R. N., Evstafieva, A. G., Abaeva, I. S., Chichkova, N. V., Filonov, G. S., Rubtsov, Y. P., Sukhacheva, E. A., Melnikov, S. V., Schneider, U., Wanker, E. E. & Vartapetian, A. B. (2005). Mol. Cell. Biol. 25, 1089–1099. [DOI] [PMC free article] [PubMed]
- Karetsou, Z., Kretsovali, A., Murphy, C., Tsolas, O. & Papamarcaki, T. (2002). EMBO Rep. 3, 361–366. [DOI] [PMC free article] [PubMed]
- Kobayashi, M., Itoh, K., Suzuki, T., Osanai, H., Nishikawa, K., Katoh, Y., Takagi, Y. & Yamamoto, M. (2002). Genes Cells, 7, 807–820. [DOI] [PubMed]
- Kobayashi, A., Kang, M. I., Watai, Y., Tong, K. I., Shibata, T., Uchida, K. & Yamamoto, M. (2006). Mol. Cell. Biol. 26, 221–229. [DOI] [PMC free article] [PubMed]
- Kobayashi, M. & Yamamoto, M. (2006). Adv. Enzyme Regul. 46, 113–140. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Li, X., Zhang, D., Hannink, M. & Beamer, L. J. (2004). J. Biol. Chem. 279, 54750–54758. [DOI] [PubMed]
- Lo, S.-H., Li, X., Hanzl, M. T., Beamer, L. J. & Hannink, M. (2006). EMBO J. 25, 3605–3617. [DOI] [PMC free article] [PubMed]
- Martini, P. G., Delage-Mourroux, R., Kraichely, D. M. & Katzenellenbogen, B. S. (2000). Mol. Cell. Biol. 20, 6224–6232. [DOI] [PMC free article] [PubMed]
- Motohashi, H. & Yamamoto, M. (2004). Trends Mol. Med. 10, 549–557. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Neer, E. J. & Smith, T. F. (1996). Cell, 84, 175–178. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Padmanabhan, B., Scharlock, M., Tong, K. I., Nakamura, Y., Kang, M.-I., Kobayashi, A., Matsumoto, T., Tanaka, A., Yamamoto, M. & Yokoyama, S. (2005). Acta Cryst. F61, 153–155. [DOI] [PMC free article] [PubMed]
- Padmanabhan, B., Tong, K. I., Ohta, T., Nakamura, Y., Scharlock, M., Ohtsuji, M., Kang, M. I., Kobayashi, A., Yokoyama, S. & Yamamoto, M. (2006). Mol. Cell, 21, 689–700. [DOI] [PubMed]
- Stogios, P. J., Downs, G. S., Jauhal, J. J., Nandra, S. K. & Privé, G. G. (2005). Genome Biol. 6, R82. [DOI] [PMC free article] [PubMed]
- Tong, K. I., Katoh, Y., Kusunoki, H., Itoh, K., Tanaka, T. & Yamamoto, M. (2006). Mol. Cell. Biol. 26, 2887–2900. [DOI] [PMC free article] [PubMed]
- Tong, K. I., Kobayashi, A., Katsuoka, F. & Yamamoto, M. (2006). Biol. Chem. 387, 1311–1320. [DOI] [PubMed]
- Tong, K. I., Padmanabhan, B., Kobayashi, A., Shang, C., Hirotsu, Y., Yokoyama, S. & Yamamoto, M. S. (2007). Mol. Cell. Biol. 27, 7511–7521. [DOI] [PMC free article] [PubMed]
- Wakabayashi, N., Itoh, K., Wakabayashi, J., Motohashi, H., Noda, S., Takahashi, S., Imakado, S., Kotsuji, T., Otsuka, F., Roop, D. R., Harada, T., Engel, J. D. & Yamamoto, M. (2003). Nature Genet. 35, 238–245. [DOI] [PubMed]
- Zhang, D. D. & Hannink, M. (2003). Mol. Cell. Biol. 23, 8137–8151. [DOI] [PMC free article] [PubMed]
- Zipper, L. M. & Mulcahy, R. T. (2002). J. Biol. Chem. 277, 36544–36552. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: α peptide complex, 2z32