

Abstract
The anti-common gamma chain (γc) mAb CP.B8 is shown to inhibit interleukin 4 (IL-4)-dependent proliferation of phytohemagglutinin (PHA) activated T cells noncompetitively with respect to cytokine by blocking the IL-4-induced heterodimerization of IL-4Rα and γc receptor chains. Affinities for the binding of IL-4 to Cos-7 cells transfected with huIL-4Rα, and to PHA blasts expressing both IL-4Rα and γc, were used to estimate the affinity of the key interaction between γc and the binary IL-4Rα⋅IL-4 complex on the cell surface. This affinity was defined in terms of the dimensionless ratio [IL-4Rα⋅IL-4⋅γc]/[IL-4Rα⋅IL-4], which we designate KR. The results show that on PHA blasts this interaction is relatively weak; KR ≈ 9, implying that ≈10% of the limiting IL-4Rα chain remains free of γc even at saturating concentrations of IL-4. This quantitative treatment establishes KR as a key measure of the coupling between ligand binding and receptor activation, providing a basis for functional distinctions between different receptors that are activated by ligand-induced receptor dimerization.
To understand, at the molecular level, how receptors allow cells to sense and respond to their external environment is a central goal of receptor research. This question can be approached by identifying the mechanism by which binding of a ligand to the extracellular portions of a receptor brings about an activated state of the receptor inside the cell membrane. However, a full understanding additionally requires quantitative information about the relationship between receptor occupancy, receptor activation, and downstream response. Insights at this level are becoming achievable for certain members of the large and diverse family of cytokine and growth factor receptors comprising two or more noncovalently associated subunits (1). A new mechanistic paradigm was introduced when it was proposed that certain of these receptors function by a mechanism of ligand-induced receptor dimerization (2), exemplified by the homodimeric receptor for human growth hormone (hGH-R) (3, 4), a class I cytokine receptor. This mechanism of receptor activation, illustrated in Fig. 1, is believed to apply to a significant number of oligomeric receptors (1, 5) and, importantly, is simple enough to be amenable to detailed experimental and theoretical analysis. The simplicity of this mechanism promises insight into how the affinity of receptor for ligand, and of the receptor chains for each other upon the binding of ligand, is coupled to the sensitivity and dynamic range of the cellular response. Improving our understanding of these quantitative features of the activation mechanism is not only of theoretical interest; the affinity between receptor chains in the presence of bound ligand has important implications for the development of inhibitors that work by blocking the interaction between receptor chains. The binding affinity between receptor chains on the surface of cells cannot easily be determined by using soluble forms of the receptor chains, because of the difficulty of accounting for the entropic consequences of the reduction in dimensionality that occurs when a binding event is constrained to the two dimensions of a cell membrane. Consequently, no quantitative measure of the interaction affinity between receptor chains on the cell surface has yet been achieved.
Figure 1.

Schematic representation of the ligand-induced receptor dimerization mechanism as it applies to class I cytokine receptors (3, 4). In the case of the IL-4 receptor, R1 is the IL-4Rα chain and R2 is γc. For homodimeric receptors such as hGH-R, R1 and R2 are identical.
We describe here an approach that uses readily obtainable experimental data to derive an estimate of the interaction affinity between receptor chains brought about by the binding of ligand to the cell, using as an example the heterodimeric receptor for interleukin 4 (IL-4) (6) comprising the IL-4 receptor α chain (IL-4Rα) and the common gamma chain (γc) (7, 8). This receptor belongs to a subfamily of class I cytokine receptors all of which use γc, and that also includes receptor subunits for IL-2, IL-7, IL-9, and IL-15. Both IL-4Rα and γc are structurally and functionally related to hGH-R, and detailed structural models of a ternary complex between IL-4, IL-4Rα, and γc have been constructed on the basis of this homology (9). Mutational analysis of IL-4 supports a mechanism of ligand-induced receptor dimerization for its receptor (10). Here, we confirm this mechanism for the IL-4 receptor on activated T cells by using a method based on monoclonal antibodies, and we use data for the binding of IL-4 to the receptor and its component subunits to estimate the affinity with which IL-4Rα interacts with γc on the cell membrane in the presence of bound IL-4. We show that this interaction is relatively weak, and that this property can be exploited by a noncompetitive inhibitor that blocks IL-4-dependent T cell proliferation without competing directly against IL-4 binding.
MATERIALS AND METHODS
Materials.
The murine anti-human γc mAb CP.B8, its isotype control (MOPC 21), and their respective Fab fragments were prepared as described elsewhere (11). The blocking anti-IL-4Rα mAb, MAB2309, was obtained from R & D Systems, and its isotype control, UPC10, was from Cappel. Vectors encoding the human IL-4Rα and γc receptor chains were prepared, and other reagents obtained, as described elsewhere (11).
Cell Proliferation Measurements.
Human peripheral blood mononuclear cells (PBMC) were isolated from healthy donors by Ficoll-Paque density gradient centrifugation (Pharmacia Biotech), and were enriched for T cells by negative selection as described in ref. 11. The T cell-enriched PBMC were cultured in a humidified incubator for 3 days at 37 °C, 5% CO2 at 106 cells/ml in RPMI medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 100 units/ml of penicillin, 100 μg/ml of streptomycin, and 1 μg/ml of phytohemagglutinin (PHA; Difco) to polyclonally activate T cells. The resulting PHA blasts were washed three times with fresh medium and recultured overnight at 106 cells/ml in medium without PHA. The next day the rested cells were transferred to 96-well flat-bottom plates, and cultured at 5 × 104 cells/well with mAb (CP.B8, anti-IL-4Rα, or control Ig) at the specified concentrations. After 45 min at 37 °C, recombinant human IL-4 (R & D Systems) was added to final concentrations of 0.015-100 ng/ml. Cells were cultured for 40 hr, and proliferation was measured by the incorporation of 3H-thymidine (Amersham), which was included during the last 16 hr of culture. Data were plotted as mean counts/min from triplicate wells. For each dose-response curve, the concentration of IL-4 required to give a half-maximal response (EC50) was determined by fitting the data to a standard four-parameter equation by nonlinear regression (DeltaGraph 3.5; DeltaPoint, Scotts Valley, CA).
Binding Studies.
Cos-7 cells were transiently transfected by electroporation with a pCDM8 vector containing the cDNA sequence for full-length human IL-4Rα (2 μg) or full-length human γc (20 μg) (11), or cotransfected with IL-4Rα (2 μg) plus γc (20 μg). Additional cells were mock-transfected with vector alone for use as a control. The transfected cells were cultured for 2 days at 37 °C, 5% CO2 in DMEM supplemented with 10% fetal bovine serum (FBS) and 2 mM l-glutamine. PBMC were prepared and activated as described above, except that enrichment for T cells was omitted and PHA was used at 2 μg/ml. Binding measurements were performed on day 3 after activation. Cells (5 × 105 transfected Cos-7 cells or 1.5 × 106 PBMC) were incubated in 200 μl PBS containing 1% FBS and 0-1 nM 125I-labeled IL-4 (New England Nuclear) at ambient temperature. After 1 hr, shown in control experiments to be sufficient for binding to reach equilibrium, cells were washed twice with 2 ml of PBS containing 1% FBS, and radioactivity bound to the cells was quantified by using a Wallac 1470 gamma counter. The data were corrected for nonspecific binding, which was determined in each experiment from control measurements containing 100-fold excess of unlabeled IL-4 and was found to be a linear function of 125I-IL-4 concentration. Specific counts bound were plotted as the average of duplicate measurements and were fitted by nonlinear regression to a simple hyperbolic equation representing a single site binding model.
RESULTS AND DISCUSSION
To probe the interaction between the IL-4Rα and γc receptor chains in the absence and presence of bound IL-4, we investigated the mechanisms by which the anti-γc mAb CP.B8 and a mAb directed against the IL-4Rα chain block the function of the receptor on activated T cells. CP.B8 is specific for the extracellular portion of human γc and inhibits the IL-4-dependent proliferation of PHA-activated T cells with an IC50 of ≈75 μg/ml when tested at a single subsaturating concentration of IL-4 (11). The binding site for CP.B8 has been mapped (11) to a conformational epitope close to the junction of the two fibronectin type III-like domains that comprise the extracellular portion of the γc molecule. Based on published structural models of the IL-4 receptor complex (9), binding of CP.B8 to this position on γc would be expected to block the interaction of γc with IL-4 and possibly also with the IL-4Rα chain. A Fab fragment of CP.B8 also blocks IL-4-dependent proliferation in this assay (11). Fig. 2 shows dose-response curves for the ability of IL-4 to stimulate the proliferation of PHA-activated human T cells, measured at several fixed concentrations of anti-IL-4Rα mAb or CP.B8. Under these conditions, secretion of growth-supporting cytokines is minimal, and addition of IL-4 induces proliferation that is essentially IL-4 dependent (11). Fig. 2A shows that the blocking mAb directed against the IL-4Rα chain displays a competitive pattern of inhibition; inhibition is dose dependent, and the IL-4 dose-response curves are shifted to the right in proportion to the concentration of mAb (Fig. 2A, Inset). This result shows that the inhibitory effect of any given concentration of anti-IL-4Rα mAb can be overcome, and the full proliferative response of the cells achieved, by increasing the IL-4 concentration to sufficiently high levels. In contrast, Fig. 2B shows that CP.B8 is a noncompetitive inhibitor of IL-4-dependent T cell proliferation; the EC50 for IL-4 remains constant at ≈2 ng/ml over the entire range of mAb concentrations (Fig. 2B, Inset), whereas the level of proliferation achieved at saturating concentrations of cytokine decreases with increasing CP.B8. Thus, a given concentration of CP.B8 inhibits by the same factor—relative to the uninhibited response seen at the same IL-4 concentration—over the entire IL-4 dose range, and inhibition cannot be overcome by increasing the IL-4 dose. Fig. 2D describes an experiment similar to that in Fig. 2B, demonstrating that a sufficiently high concentration of CP.B8 can block virtually all of the IL-4-dependent proliferative response of these cells (11). This result shows that most or all of the response is mediated by γc under these conditions and rules out the involvement of IL-13 receptor components, consistent with reports that such receptors are not expressed on activated T cells (12). The inability of high concentrations of IL-4 to overcome inhibition by CP.B8 indicates that the mAb is not in direct competition with IL-4 for binding to the receptor. This finding is as expected for an antagonist that blocks the second step in receptor activation (Fig. 1) by preventing the association between γc and the binary IL-4Rα⋅IL-4 complex on the cell surface. This result additionally implies that the interaction affinity between the receptor chains at saturating IL-4 is low enough that IL-4Rα⋅IL-4 cannot effectively compete against the mAb for binding to γc, otherwise receptor occupancy by IL-4 and CP.B8 would be mutually exclusive and inhibition by CP.B8 would appear competitive. Fig. 2D also shows that a CP.B8 Fab fragment gives a pattern of inhibition that is qualitatively similar to the noncompetitive inhibition seen with CP.B8 mAb. This result shows that the noncompetitive inhibition observed with CP.B8 requires only that it bind to γc and block its participation in a productive complex; it does not require the ability to cross-link γc molecules.
Figure 2.

Overlaid dose-response curves for the IL-4-dependent proliferation of PHA-activated T cells measured at various fixed concentrations of (A) blocking anti-IL-4Rα mAb at 0 (○) 0.41 (□), 1.23 (▵), 3.7 (◊), 11.1 (▿), 33.3 (⊠), or 100 μg/ml (⊙); (B) anti-γc mAb CP.B8 at 0, (○), 1.23 (□), 3.7 (▵), 11.1 (◊), 33.3 (▿), or 100 μg/ml (⊠); (C) isotype controls MOPC 21 (mouse IgG1, for CP.B8) (□) and UPC10 (mouse IgG2a, for anti-IL-4Rα) (▵) at 100 μg/ml, or no mAb (○); or (D) anti-γc mAb CP.B8 at 0, (○), 100 (□), or 300 μg/ml (▵), MOPC 21 at 300 μg/ml (▿), or a CP.B8 Fab fragment at 100 μg/ml (◊). Data are represented as mean counts/min (cpm) determined from triplicate wells. The solid lines are best fits of the data to a standard four-parameter equation. (Inset A) EC50 for IL-4 increases linearly with anti-IL-4Rα mAb concentration. (Inset B) EC50 for IL-4 is independent of CP.B8 concentration.
The noncompetitive mode of inhibition displayed by CP.B8 in Fig. 2B, supporting a sequential model of IL-4 receptor assembly, was further demonstrated by examining the effect of CP.B8 on IL-4 binding. Fig. 3A shows data for the binding of IL-4 to Cos-7 cells transfected with IL-4Rα, or cotransfected with both the IL-4Rα chain and γc. There is very little specific binding of IL-4 to mock-transfected cells or to cells expressing γc in the absence of IL-4Rα, as has been shown previously (8). Three such independent experiments established that cells transfected with IL-4Rα alone bind IL-4 with an affinity of KD = 600 ± 150 pM, and that cells cotransfected with both γc and IL-4Rα bind IL-4 with a ≈3-fold higher affinity of KD(App) = 200 ± 100 pM. These results agree with those of Russell et al. (8), who similarly found IL-4 to bind with ≈3-fold higher affinity to Cos-7 cells cotransfected with IL-4Rα and γc compared to cells transfected with IL-4Rα alone. Contribution to this binding by IL-13 receptor components can be precluded because Cos-7 cells express only very low levels of IL-13 binding sites (13). The binding of IL-4 to PHA-activated PBMC is shown by the open symbols in Fig. 3B. Multiple determinations consistently showed that the binding data strictly fit a simple hyperbolic equation (solid line in Fig. 3B), and that IL-4 binds to the cells with an affinity of KD(App) = 60 ± 10 pM (n = 6), comparable to published values for IL-4 binding to a variety of cell types (6, 14). The filled symbols in Fig. 3B show that there is very little binding of IL-4 to unactivated PBMC, in agreement with published data that show a significant up-regulation of IL-4Rα upon activation with PHA (14). Fig. 4A shows that, as expected, the anti-IL-4Rα antibody blocks binding of IL-4 to activated PBMC competitively with respect to IL-4. In contrast, experiments such as that shown in Fig. 4B showed that CP.B8 does not block binding at high levels of IL-4, even at a mAb concentration of 100 μg/ml, which inhibits IL-4-dependent proliferation by ≥50% (see Fig. 2 B and D). CP.B8 does, however, appear to bring about a small decrease in the affinity of the cells for binding IL-4, consistent with a partial loss of the modest enhancement of affinity for IL-4 that γc confers upon IL-4Rα. Taken together, the data in Figs. 2 and 4 provide strong evidence that the effect of CP.B8 is to block the recruitment of γc into a ternary complex with IL-4 and the alpha chain. In so doing, these results provide confirmation that the IL-4 receptor comprising IL-4Rα and γc functions by a mechanism of ligand-induced receptor dimerization (10).
Figure 3.

Binding of 125I-labeled IL-4 (A) to Cos-7 cells transiently transfected with a pCDM8 vector containing the cDNA sequence for full-length huIL-4Rα (2 μg, ○), full-length human γc (20 μg, ◊), cotransfected with IL-4Rα (2 μg) plus γc (20 μg, □), or to mock-transfected cells (▵); or (B) to PHA-activated (○) or resting (•) PBMC. The data are plotted as the average of duplicate measurements after correction for nonspecific binding. Data are fitted to a simple hyperbolic equation representing a single site binding model. (Inset A) Data for IL-4 binding to cells transfected with IL-4Rα alone (○), or cotransfected with IL-4Rα plus γc (□), represented as Scatchard plots of bound/free 125I-IL-4 × 10−4 (B/F) versus bound 125I-IL-4 (B, in cpm).
Figure 4.

Binding of 125I-labeled IL-4 to PHA-activated PBMC in the presence of various fixed concentrations of (A) blocking anti-IL-4Rα mAb at 0 (○) 0.01 (□), 0.1 (▵), 1.0 (◊), 10 (▿), 100 (⊠), or 1,000 ng/ml (⊙); (B) anti-γc mAb CP.B8 at 0, (○), 0.01 (□), 0.1 (▵), 1.0 (◊), 10 (▿), or 100 μg/ml (⊠); or (C) isotype controls MOPC 21 (for CP.B8, □) and UPC10 (for anti-IL-4Rα, ▵) at 100 μg/ml, or no mAb (○). Data are fitted to a simple hyperbolic binding curve.
Estimating the Interaction Affinity Between γc and IL-4⋅IL-4Rα on the Cell Surface.
The sequential assembly of a heterodimeric receptor comprising receptor chains R1 and R2, according to the mechanism of Fig. 1, can be thought of in terms of a thermodynamic scheme in which the binding of ligand (L) to R1 and the dimerization of R1 and R2 are considered as distinct processes that make up two orthogonal sides of a thermodynamic box (Scheme S1) (15). In the case of the IL-4 receptor, R1 corresponds to IL-4Rα, R2 to γc, and L to IL-4. The key feature of Scheme S1 is its recognition that the overall equilibrium constant (designated K5) for the binding of L to cells that express both R1 and R2 to produce R1⋅L⋅R2 complexes—i.e. for going from the bottom left corner to the top right corner of Scheme S1—constitutes one side of a closed thermodynamic cycle of which the binding of L to R1 and the subsequent association of R1⋅L with R2 make up the other two sides. Thus, K5 is a direct function of K1 and K2 as shown in Eq. 1, in which {R1}free, {R2}free, and {R1⋅L⋅R2} represent the concentrations on the cell surface, in units of molecules/μm2, of free IL-4Rα, free γc, and IL-4Rα⋅IL-4⋅γc, respectively.
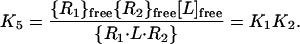 |
1 |
A rigorous analysis of experimental binding data in terms of the thermodynamic relationships implied by Scheme S1 is complicated by the fact that the experimentally observed affinity for the binding of ligand to cells expressing both receptor chains, KD(App), is only indirectly related to K5. The problem is one of dimensionality: the interaction between receptor chains on the cell surface is constrained to the two dimensions of the membrane, and thus K2 has units of molecules/μm2. Similarly, the formation of ternary R1·L·R2 complexes upon the binding of ligand to cells expressing both R1 and R2 as nonpreassociated (i.e., independently diffusing) components is a termolecular process when taken overall, and K5 has units of M.molecules/μm2, distinguishing it from KD(App), which has units of M.
Scheme 1.

To circumvent this problem, expression levels of IL-4Rα and γc on PHA blasts were determined by flow cytometry, using a standard curve generated with calibrated beads that bind defined numbers of antibody molecules (data not shown). The results showed that IL-4Rα is expressed at 1,000–1,600 copies/cell, while γc is expressed at the ≈5-fold higher level of 5,000-8,500 copies/cell. This finding, and the fact that the binding of IL-4 to PHA blasts is hyperbolic (Fig. 3B), allows us to define KD(App) in terms of bound forms of ligand (R1⋅L and R1⋅L⋅R2), total ligand ([L]T = [L]free), and limiting free R1 (Eq. 2). Substituting Eq. 1 and the definition of K1 into this expression gives KD(App) as a function of K1, K5, and {R2}free (Eq. 2).
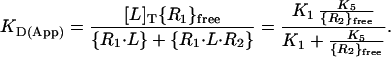 |
2 |
Eq. 2 shows that KD(App) is a function of {R2}free, indicating that the apparent affinity for IL-4 binding to cells expressing both IL-4Rα and γc may vary from one cell type to another depending on the level of γc expression. The ≈3-fold difference in KD(App) found for PHA blasts compared to cotransfected Cos-7 cells (Fig. 3) is an example of this effect. Furthermore, hyperbolic binding would not be expected unless R2 is present in sufficient excess over R1 such that free {R2} is not greatly decreased upon IL-4 binding to the cells (15). This condition is met by the experimentally determined ≈5-fold excess of γc over IL-4Rα, which establishes that the pool of free γc is sufficient, in relation to the IL-4Rα chain, that it is depleted by no more than about ≈20% upon binding of IL-4 to the cells. This result allows us to make the approximation that {R2}free is roughly constant over the entire IL-4 binding curve.
The dissociation constant K2 for the interaction between IL-4Rα⋅IL-4 and γc on the cell surface can be described in terms of {R2}free and a dimensionless apparent association constant KR, as shown in Eq. 3.
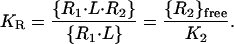 |
3 |
Because {R2}free ≈ constant in Fig. 3B, KR has a unique value on these cells that represents the equilibrium distribution of total IL-4Rα⋅IL-4 between the binary state, i.e. IL-4Rα⋅IL-4 itself, and the ternary state in which it has bound γc to form the ternary complex IL-4Rα⋅IL-4⋅γc. Thus, KR represents the affinity of γc for binding to IL-4Rα⋅IL-4. Because KR depends on the local or effective concentration of γc (Eq. 3), it is not an intrinsic property of the receptor, as K2 is, but depends on the level of expression of γc, which is specific to the particular cell type—and sometimes to the specific cell population—under study. The magnitude of KR can be estimated from experimental values for K1 and KD(App) by combining Eqs. 1–3 to give the relationship shown in Eq. 4.
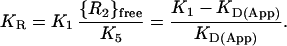 |
4 |
The affinity for IL-4 binding to Cos-7 cells transfected with IL-4Rα alone (Fig. 3A) establishes that K1 has a value of ≈600 pM (n = 3; range = 460–760 pM). Using KD(App) ≈ 60 pM for the binding of IL-4 to activated PBMC (from Fig. 3B), the relationship in Eq. 4 gives a value of KR ≈ 9 for these cells. An earlier treatment of aggregating receptor systems, focusing on the analysis of curvature in Scatchard plots (16), contains an equation that can be adapted to allow calculation of KR by an alternative approach. At the limit R2 ≫ R1 (under which condition Scatchard plots appear linear), application of this equation adapted from ref. 16 to our experimental data results in a value of KR ≈ 9, identical to the value obtained above by using our method.
The modest value we estimate for KR implies that, on PHA blasts, the affinity of γc for binding to IL-4Rα⋅IL-4 is such that ≈90% [i.e. KR/(KR + 1)] of the IL-4Rα chains exist as complexes with γc at saturating IL-4. On these cells, therefore, the binding interaction between IL-4Rα and γc in the presence of bound IL-4 is evidently relatively weak; even at full occupancy of all IL-4Rα chains by IL-4, a small but significant fraction of IL-4Rα chains remains dissociated from γc even though γc is present in excess. Although some responses of cells to IL-4 may require activation of only a small fraction of the available receptor, Fig. 5 shows that close to 50% receptor occupancy is required to achieve 50% of the maximal proliferation of activated T cells in response to IL-4, and that the response is affected by occupancy of even the last few percent of free receptor by IL-4. Thus, in mediating a dose-dependent modulation of heterodimeric receptor complexes from low residual levels to levels that involve ≈90% of the limiting receptor chain, the binding affinity between γc and IL-4Rα⋅IL-4 is just sufficient to access essentially the full dynamic range that is available for signaling through the IL-4 receptor on these cells. The value of KR ≈ 9 for the interaction between γc and IL-4Rα⋅IL-4 suggests that these receptor components have evolved a binding affinity for each other that is strong enough—but no stronger than is necessary—to allow essentially all of the IL-4Rα on the cell to form signaling complexes with γc at saturating IL-4.
Figure 5.

IL-4-dependent proliferation of PHA-activated T cells (as % maximal 3H-thymidine incorporation) over a range of IL-4 concentrations, plotted as a function of the fractional occupancy of the receptor at each concentration of IL-4. Proliferation data were taken from the zero mAb data set in Fig. 2B. Receptor occupancy at each IL-4 concentration was calculated by using KD(App) = 60 pM for the binding of 125I-IL-4 to PHA-activated PBMC (see Fig. 3B and text). The solid line is an arbitrary interpolation of the data points.
The finding that PHA blasts express 5,000–8,500 γc molecules per cell allows us to estimate that {γc} ≈1–20 molecules/μm2 (assuming spherical cells of diameter ≈12 μm and a membrane roughness that increases the surface area by a factor of 1–10). Eq. 3 thus gives a value for K2 of roughly 0.1–2.0 molecules/μm2. Clearly, K2 can be defined with much less precision than KR. Moreover, KR gives much more direct insight into the strength of the interaction between the receptor chains on the surface of the cell population at hand. Estimates obtained from radioligand binding data and from FACS analysis (data not shown) indicate that cotransfection of Cos-7 cells with IL-4Rα and γc (Fig. 3A) results in the expression of several-fold higher levels of γc than is seen on PHA blasts. However, the much greater size of Cos-7 cells means that the surface density of γc is probably significantly lower than on PHA blasts. This finding is consistent with the higher value of KD(App) measured for cotransfected Cos-7 cells (Fig. 3A, Eq. 2), and with the lower value of KR ≈ 2 calculated from Eq. 4.
To determine whether the characteristics described above might be unique to the IL-4 receptor, the effect of CP.B8 on IL-2-dependent proliferation of PHA-activation T cells was tested. Activated T cells express IL-2 receptor α, β, and γc (6, 7); however, IL-2Rα and IL-2Rβ have been shown to exist as a preassociated complex (17). Thus, activation of the IL-2 receptor can be thought of as a heterodimerization event in which the preassociated IL-2Rα/β complex is the component responsible for the initial binding of IL-2, and activation of the receptor occurs upon the subsequent recruitment of γc into an IL-2Rα/β⋅IL-2⋅γc complex (17). A value of KR ≈ 10 was calculated for the IL-2 receptor by using Eq. 4 together with KD(App) values for the binding of IL-2 to activated T cells (KD(App) ≈ 10 pM) compared to cells expressing IL-2Rα and IL-2Rβ but not γc (KD ≈ 100 pM) (7). This modest value for KR suggested that the binding of γc to IL-2Rα/β⋅IL-2, like the interaction of the corresponding components of the IL-4 receptor, is relatively weak. This expectation was supported by experiments similar to those shown in Fig. 2D that showed that CP.B8 and its Fab fragment inhibit IL-2-dependent proliferation of activated T cells noncompetitively with respect to IL-2 (data not shown).
Mechanistic Differences Between Heterodimeric and Homodimeric Receptors.
Homodimeric receptors (i.e. receptors in which R1 and R2 are identical) that are activated by a mechanism of ligand-induced receptor dimerization, such as hGH-R, possess the potential to give a bell-shaped dose-response curve for activation by their natural ligand (4, 18, 19). This tendency exists because very high ligand concentrations can potentially force the system into a state in which each receptor chain binds a separate ligand molecule, thus depleting the pool of free receptor available to be recruited into ternary R1⋅L⋅R1 complexes (4). This phenomenon is governed by competition between exogenous cytokine and membrane-bound R1⋅L complexes for binding to free R1, and thus depends directly on KR. The concentration difference that separates the agonist and antagonist limbs of the bell-shaped curve defines the range of ligand concentrations over which an agonist response will occur. Published data for several homodimeric cytokine receptors suggests that they achieve a broad effective dose range at least in part through having evolved a relatively high interaction affinity between receptor chains on the cell surface in the presence of bound ligand, i.e. a high KR. For example, published data on hGH-R suggest that hGH has an effective dose range spanning up to 4–6 logs (4, 18, 20), consistent with KR ≈ 102–103 (A.W., unpublished data). Similarly, the homodimeric receptors for erythropoietin and for prolactin display broad effective dose ranges of 6 logs (19) and >6 logs (21), respectively. Although in some of these cases data were obtained by using transfected cells, and in some spare receptors may be partly responsible for broadening the effective dose range (4), the results nevertheless suggest that KR for these homodimeric receptors is relatively high. The relatively low affinities observed for binding of soluble receptor ectodomains to receptor/ligand dimers in solution (3, 22) do not contradict this conclusion, but rather highlight the large entropic advantage that is conferred when the components are constrained to the cell surface. Although IL-4Rα homodimerization can be induced by artificial means (23, 24), the ability of IL-4 to induce these complexes appears unlikely (25); moreover, the heterodimeric composition of the receptor in our systems is supported by the data in Fig. 2D and by the hyperbolic form of the binding curves in Fig. 3 (16). The potential for self-inhibition at high ligand concentrations does not exist for the IL-4Rα/γc receptor, or for other heterodimeric receptors in which the ligand has an intrinsically very low affinity for binding to one of the two receptor chains. For heterodimeric receptors such as IL-4R there is therefore no obvious need for the affinity between receptor chains in the presence of bound ligand to exceed a value of KR ≈ 10 that is sufficient to bring virtually all of the receptors into functional complexes upon the binding of ligand. It is, indeed, possible that the formation of a receptor complex that is relatively weak and rapidly dissociable may confer advantages for the regulation of receptor activity, especially in cases where a receptor chain is shared in common with other receptors of related function.
An implication of our results is that receptors such as IL-4R that possess a low KR in a given cellular context may be particularly amenable to pharmaceutical intervention using noncompetitive agents that block the second step in receptor activation. Because such inhibitors are not in direct competition with the natural ligand, which often binds its receptor with very high affinity, they may effectively inhibit receptor activation at high local concentrations of the activating ligand even if themselves possessing only moderate affinity for the receptor. Moreover, the level of inhibition achieved by such an agent will be independent of the concentration of the agonist cytokine, suggesting the possibility that a relatively low affinity drug might be loaded and maintained at an effective level, regardless of local or systemic up-regulation of ligand. Both of these properties are illustrated in Fig. 2, for inhibition of IL-4-dependent T cell proliferation by CP.B8. Receptors displaying a high value of KR, and therefore possessing a high binding affinity between receptor chains once ligand has bound, are predicted to be less susceptible to inhibitors that work in this way.
Acknowledgments
We thank Cenk Sumen for technical assistance, Dr. Joseph Rosa for encouragement and support, and one of the anonymous reviewers for insightful comments and suggestions that led to substantial improvements in the manuscript.
ABBREVIATIONS
- IL
interleukin
- IL-4Rα
IL-4 receptor α chain
- γc
common gamma chain
- hGH-R
human growth hormone receptor
- PBMC
peripheral blood mononuclear cells
- PHA
phytohemagglutinin
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1. Heldin C-H. Cell. 1995;80:213–223. doi: 10.1016/0092-8674(95)90404-2. [DOI] [PubMed] [Google Scholar]
- 2.Ulrich A, Schlessinger J. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 3.Cunningham B C, Ultsch M, De Vos A M, Mulkerrin M G, Clauser K R, Wells J A. Science. 1991;254:821–825. doi: 10.1126/science.1948064. [DOI] [PubMed] [Google Scholar]
- 4.Fuh G, Cunningham B C, Fukunaga R, Nagata S, Goeddel D V, Wells J A. Science. 1992;256:1677–1680. doi: 10.1126/science.256.5064.1677. [DOI] [PubMed] [Google Scholar]
- 5.Stahl N, Yancopoulos G D. Cell. 1993;74:587–590. doi: 10.1016/0092-8674(93)90506-l. [DOI] [PubMed] [Google Scholar]
- 6.Beckman M P, Cosman D, Fanslow W, Maliszewski C R, Lyman S D. Chem Immunol. 1992;51:107–134. [PubMed] [Google Scholar]
- 7.Sugamura K, Asao H, Kondo M, Tanaka N, Ishii N, Nakamura M, Nakamura T. Adv Immunol. 1995;59:225–277. doi: 10.1016/s0065-2776(08)60632-x. [DOI] [PubMed] [Google Scholar]
- 8.Russell S M, Keegan A D, Harada N, Nakamura Y, Noguchi M, Leland P, Friedmann M C, Miyajima A, Puri R K, Paul W E, Leonard W J. Science. 1993;262:1880–1883. doi: 10.1126/science.8266078. [DOI] [PubMed] [Google Scholar]
- 9.Bamborough P, Hedgecock C J R, Richards W G. Structure. 1994;2:839–851. doi: 10.1016/s0969-2126(94)00085-9. [DOI] [PubMed] [Google Scholar]
- 10.Kruse N, Shen B-J, Arnold S, Tony H-P, Müller T, Sebald W. EMBO J. 1993;12:5121–5129. doi: 10.1002/j.1460-2075.1993.tb06207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raskin, N., Jakubowski, A., Sizing, I. D., Olson, D. L., Kalled, S. L., Hession, C. A., Benjamin, C. D., Baker, D. P. & Burkly, L. C. (1998) J. Immunol., in press. [PubMed]
- 12.Gauchet J-F, Schlagenhauf E, Feng N-P, Moser R, Yamage M, Jeannin P, Alouani S, Elson G, Notarangelo L D, Wells T, Eugster H-P, Bonnefoy J-Y. Eur J Immunol. 1997;27:971–978. doi: 10.1002/eji.1830270425. [DOI] [PubMed] [Google Scholar]
- 13.Obiri N I, Leland P, Murata T, Debinski W, Puri R K. J Immunol. 1997;158:756–764. [PubMed] [Google Scholar]
- 14.Park L S, Friend D, Sassenfeld H M, Urdal D L. J Exp Med. 1987;166:476–488. doi: 10.1084/jem.166.2.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee T W, Sole M J, Wells J W. Biochemistry. 1985;25:7009–7020. doi: 10.1021/bi00370a038. [DOI] [PubMed] [Google Scholar]
- 16.Wofsy C, Goldstein B. Math Biosci. 1992;112:115–154. doi: 10.1016/0025-5564(92)90090-j. [DOI] [PubMed] [Google Scholar]
- 17.Myszka D G, Arulanantham P R, Sana T, Wu Z, Morton T A, Ciardelli T L. Protein Sci. 1996;5:2468–2478. doi: 10.1002/pro.5560051209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ilonda M M, Damholt A B, Cunningham B A, Wells J A, De Meyts P, Shymko R M. Endocrinology. 1994;134:2397–2403. doi: 10.1210/endo.134.6.8194466. [DOI] [PubMed] [Google Scholar]
- 19.Schneider H, Chaovapong W, Matthews D J, Karkaria C, Cass R T, Zhan H, Boyle M, Lorenzini T, Elliott S G, Giebel L B. Blood. 1997;89:473–482. [PubMed] [Google Scholar]
- 20.Ishizaka-Ikeda E, Fukunaga R, Wood W I, Goeddel D V, Nagata S. Proc Natl Acad Sci USA. 1993;90:123–127. doi: 10.1073/pnas.90.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fuh G, Colosi P, Wood W I, Wells J A. J Biol Chem. 1993;268:5376–5381. [PubMed] [Google Scholar]
- 22.Philo J S, Aoki K H, Arakawa T, Owers Narhi L, Wen J. Biochemistry. 1996;35:1681–1691. doi: 10.1021/bi9524272. [DOI] [PubMed] [Google Scholar]
- 23.Lai S Y, Molden J, Liu K D, Puck J M, White M D, Goldsmith M A. EMBO J. 1996;15:4506–4514. [PMC free article] [PubMed] [Google Scholar]
- 24.Kammer W, Lischke A, Morriggl R, Groner B, Ziemiecki A, Gurniak C B, Berg L J, Friedrich K. J Biol Chem. 1996;271:23634–23637. doi: 10.1074/jbc.271.39.23634. [DOI] [PubMed] [Google Scholar]
- 25.Hoffmann R C, Schalk-Hihi C, Castner B J, Gibson M G, Rasmussen B D, Zdanov A, Gustchina A, March C J, Wlodawer A. FEBS Lett. 1994;347:17–21. doi: 10.1016/0014-5793(94)00496-x. [DOI] [PubMed] [Google Scholar]