

Abstract
We have developed new, simple and efficient procedures for the synthesis of two promising histone deacetylase inhibitors ((HDIs), CI-994, (N-(2aminophenyl)-4-acetylaminobenamide), and MS-275 (N-(2-aminophenyl)4-[N-(pyridine-3-ylmethoxycarbonyl)aminomethyl]benzamide) from commercially available acetamidobenzoic acid and 3-(hydroxymethyl)pyridine, respectively. The procedures provide CI-994 and MS-275 in 80 and 72% overall yields, respectively. We found that the combination of four HDIs (CI-994, MS-275, SAHA and TSA) with retinoids all-trans-retinoic acid (ATRA) or 13-cis-retinoic acid (13-CRA) or our atypical retinoic acid metabolism blocking agents (RAMBAs) 1 (VN/14-1) or 2 (VN/66-1) produced synergistic anti-neoplastic activity on human LNCaP prostate cancer cells. The combination of 2 and SAHA induced G1 and G2/M cell cycle arrest and decrease in the S phase in LNCaP cells. 2 + SAHA treatment effectively down-regulated cyclin D1 and cdk4 and up-regulated pro-differentiation cytokeratin 8/18 and pro-apoptotic Bad and Bax. Following subcutaneous administration, 2, SAHA or 2 + SAHA were well tolerated and caused significant suppression/regression of tumor growth compared with control. These results demonstrate that compound 2 and its combination with SAHA are potentially useful agents that warrant further preclinical development for treatment of prostate cancer.
Keywords: Retinoids, RAMBAs, HDIs, prostate cancer, anti-cancer agents
Introduction
Prostate cancer (PCA) is the most common malignancy and age-related cause of cancer death worldwide. Apart from lung cancer, PCA is the most common form of cancer in men and the second leading cause of death in American men. In the United States in 2007, an estimated 218,890 new case of prostate cancer will be diagnosed and about 27,050 men will die of this disease.1 The growth of most prostate tumors depends on androgens during the initial stages of tumor development, and thus, anti-hormonal therapy by surgical or medical suppression of androgen action remains a major treatment option of the disease.2 Although this treatment may be initially successful, most tumors eventually recur due to the expansion of an androgen-refractory population of PCA cells.3 Metastatic disease that develops even after potentially curative surgery remains a major clinical challenge. Therapeutic treatments for patients with metastatic PCA are limited because current chemotherapeutic and radiotherapeutic regimens are largely ineffective.4 Hence, there is urgent need to develop new therapeutic agents with defined targets to prevent and treat this disease.
PCA tumors that arise after anti-hormonal therapy generally are less differentiated and it is believed that agents that can induce the cells to differentiate would represent a new therapeutic strategy.5 Hence, the goal of differentiation therapy is to induce malignant cells to pass the block to maturation by allowing them to progress to more differentiated cell types with less proliferative ability. Breslow and colleagues6 have led the way in the discovery of agents that inhibit the enzyme histone deacetylase (HDAC), thereby altering chromatin structure and changing gene expression patterns. Histone deacetylase inhibitors (HDIs) are potent differentiating agents towards a variety of neoplasms, including leukemia, and breast and prostate cancers. Combinations of HDIs with other known therapies including retinoic acids (RAs) have been investigated. RAs exert their effects via a nuclear receptor complex that interacts with promoters of RA-responsive genes.7 An HDAC subunit is an integral part of this co-repressor complex, which is involved in transcriptional silencing in the absence of ligand.8 This association provides a rationale for combining HDIs and RAs/retinoids therapeutically. One of the early HDIs discovered by Breslow and colleagues is N-hydroxy-N1-phenylactanediamide, also called suberoylanilide hydroxamic acid (SAHA).9,10 This compound (trade name: Vorinostat®) was recently (2006) approved by the U.S. Food and Drug Administration (FDA) for the treatment of advanced cutaneous T-cell-lymphoma.11
Recently, we reported on a family of compounds that inhibit the P450 enzyme(s) responsible for the metabolism of all-trans retinoic acid (ATRA).12 These compounds also referred to as retinoic acid metabolism blocking agents (RAMBAs) are able to enhance the antiproliferative effects of ATRA in breast and prostate cancer cells in vitro.13 In addition, the RAMBAs were shown to induce differentiation and apoptosis in these cancer cell lines. However, we also observed that the breast cancer cell lines were exquisitely more sensitive to the RAMBAs.14, 15 We also reported recently that combination of SAHA with either retinoids or RAMBAs resulted in additive/synergistic PCA (LNCaP and PC-3 cell lines) growth inhibition in vitro.16
In continuation of our research in this area, we have discovered improved syntheses of two promising HDIs,17, 18,19 CI-994, (N-(2aminophenyl)-4-acetylaminobenamide), and MS-275 (N-(2-aminophenyl)4-[N-(pyridine-3-ylmethoxycarbonyl)aminomethyl]benzamide). Furthermore, we assessed the effects of our novel RAMBAs and retinoids (see Chart 1) in combination with some HDIs in human prostate cancer model systems in vitro and in vivo. The molecular effects of compound 2 + SAHA in prostate cancer cells include inhibition of proliferation, regulation of cell cycle, and induction of differentiation and apoptosis.
Chart 1.
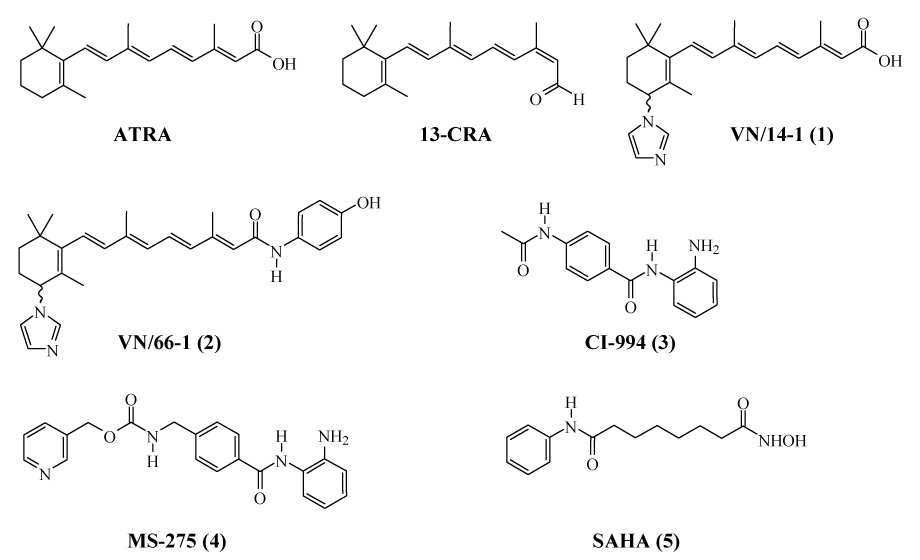
Structures of retionods, RAMBAs and HDACIs.
Results and Discussion
Chemistry
To the best of our knowledge only one method has been reported for synthesis of CI-994. This synthesis of CI-994 by Weiershausen et al20 is outlined in Scheme 1. The 3-step procedure involves the reaction of oxalyl chloride with 4-acetamidobenzoic acid to give the corresponding acid chloride that was coupled with 2-nitroaniline in situ to afford N-(21-nitrophenyl)-4-acetylaminobenzamide in 20.0% yield. This was then hydrogenated in THF using 10% palladium on activated charcoal to produce CI-994 (3) in 69.0% yield. The overall yield was only 13.8%. Apart from the low yield, this method is tedious and requires special hydrogenation conditions.
Scheme 1.
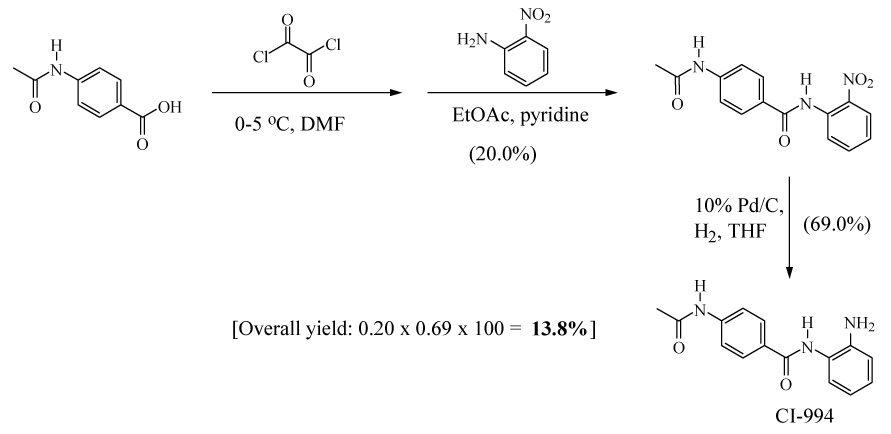
Previous procedure for synthesis of CI-994 (3)
We have developed a simple and efficient one-step procedure for the synthesis of CI-994 (Scheme 2). The readily available acetamidobenzoic acid (6) was converted into its imidazolide derivative by reaction with N, N1-carbonyldiimidazole (CDI) in THF at room temperature. This was further reacted with 1,2-phenylenediamine in the presence of TFA to obtain CI-994 (3) in 85.0% yield. The product was recrystallized from THF/methanol to give pure CI-994 (2) in 80.0% yield.
Scheme 2.

New synthesis of CI-994 (3)
Another promising HDACI N-(2-aminophenyl)4-[N-(pyridine-3-yl-methoxy-carbonyl) aminomethyl] benzamide (MS-275, 4) in currently in several phase I/II clinical trials for various solid tumors and hematological malignancies.21 MS-275 was previously synthesized by Suzuki et al. via a three-step procedure in 50.96% overall yield (Scheme 3).22 In addition to the modest overall yield, this procedure has other disadvantages such as a tedious method for the preparation of an acid chloride using oxalyl chloride and also it requires the use of column chromatography for purification of MS-275.
Scheme 3.
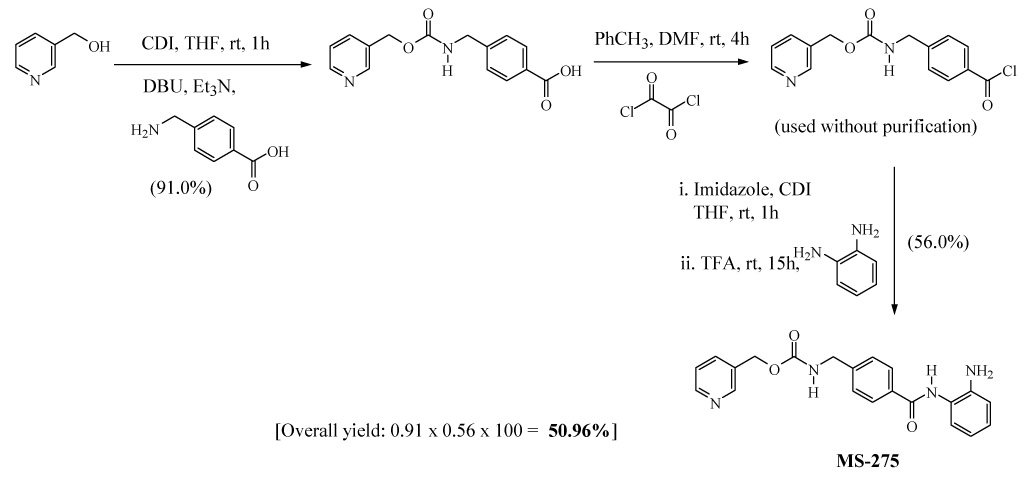
Previous procedure for synthesis of MS-275 (4)
Here again, we have developed a new two-step procedure for preparation of MS-275 as outlined in Scheme 4. Condensation of 3-(hydroxymethyl)pyridine (7) and 4-(aminomethyl)benzoic in the presence of CDI gave 4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzoic Acid (8) in 91.0% yield. In the previous method of Suzuki et al., the carboxylic acid derivative 8 was first converted into acyl chloride hydrochloride by treatment of oxalyl chloride in toluene and then reacted with imidazole to form the acylimidazole intermediate.22 However, we synthesized the imidazolide of intermediate 8 by treatment with CDI at 55–60 °C in THF. The imidazolide was then further reacted in situ with 1,2-phenylenediamine in the presence of TFA at room temperature to afford MS-275 (4). Furthermore, we developed a simpler process for large scale purification of crude MS-275 instead of using conventional column chromatography. Thus, after the completion of reaction, the solvent was evaporated and to the concentrate we added the mixture of hexane and water (2:5, v/v) and stirred for one hour. The resulting precipitate were filtered, washed with hexane and dried. The crude product was further stirred twice in dichloromethane to remove excess of 1,2-phenylenediamine, filtered and washed with hexane to give pure MS-275 in 80.0% yield (>% as determined by HPLC). The overall yield for our simple and efficient production of MS-275 (4) was 72.8%. Finally, the methods described here for the synthesis of CI-994 and MS-275 are green chemistry methods because of the greatly increased yields and fewer number of reaction steps.
Scheme 4.

New synthesis of MS-275 (4)
Biological Studies
Effects of retinoids or RAMBAs alone or in combination with HDIs on LNCaP cell proliferation
We first studied the effects of retinoids, RAMBAs and HDIs as single agents on LNCaP cell viability using the MTT assay and the IC50 values were determined from dose-response curves as shown for MS-275 (Figure 1). The growth inhibitory experiments with the other compounds gave plots that were essentially the same as in Figure 1. The HDIs, MS-275, SAHA and CI-994 and RAMBA 2 were efficacious with IC50 values of 0.36, 1.0, 7.4 and 5.5 µM, respectively. In contrast, the retinoids, ATRA and 13-CRA or RAMBA 1 were less efficacious, since each of the compounds did not significantly inhibit cell growth even at concentrations as high as 10 µM. However, the combinations of retinoids or RAMBAs with the HDIs caused dramatic, synergistic inhibitory effects as shown in Figures 2a–e. Of significance is our observation that the combination of exceptionally low doses of ATRA (0.1 nM) or 1 (0.1 nM) or 2 (0.1 nM) with MS-275 (0.1 nM) resulted in a growth inhibition of 65.0, 67.0, and 70.0%, respectively (Figure 2c–e). As we reported previously,16 treatment with 2 (5.0 µM) + SAHA (1.0 µM) resulted in > 95.0% growth inhibition of LNCaP cells (data not shown). We also found that trichostatin A (TSA), a first generation and an experimental HDI, in combination with 2 caused a synergistic inhibitory effect in LNCaP cells (data not shown). It is pertinent to state here that although we had previously found that the combination of 2 + SAHA resulted in additive growth inhibition, the effects observed in the present study with the various combinations are clearly synergistic (ref. Figures 1a–e). This assertion is based on our findings that the inhibition of cell growth by each of the combinations were greater than the sums of each of the two compounds separately; using the Valeriote and Lin analysis.23 These results clearly show that HDIs can effectively enhance the growth inhibitory activities of both retinoids and RAMBAs. It is well known that although the RAR/RXR receptors interact with HDACs, the ligands that interact with either of these proteins, i.e., the retinoids/RAMBAs and HDIs have other mechanisms of action, that are likely to cause synergistic cell growth inhibitory effects. It should also be stated that retinoid-specific signaling alone does not explain the differences in sensitivity of different cell lines to the different retinoids.24 In addition, although the HDIs are noted to resensitize certain genes that are silenced in cancer cells, thereby enhancing the functional activity of RARs, they also show other anticancer activities, such as cell cycle arrest and apoptosis.25
Figure 1.
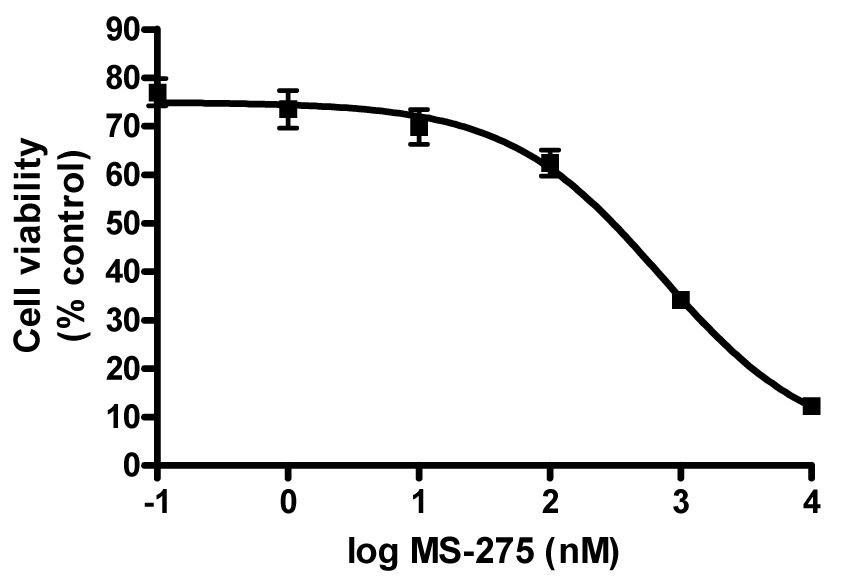
Antiproliferative effect of MS-275. LNCaP cell proliferation was measured after 6 days of treatment using a MTT assay as described in the Experimental Section. Data is mean (SEM < ± 10%) of at least three independent experiments. The experiments with the other compounds gave dose-response plots that were essentially the same as shown above.
Figure 2.


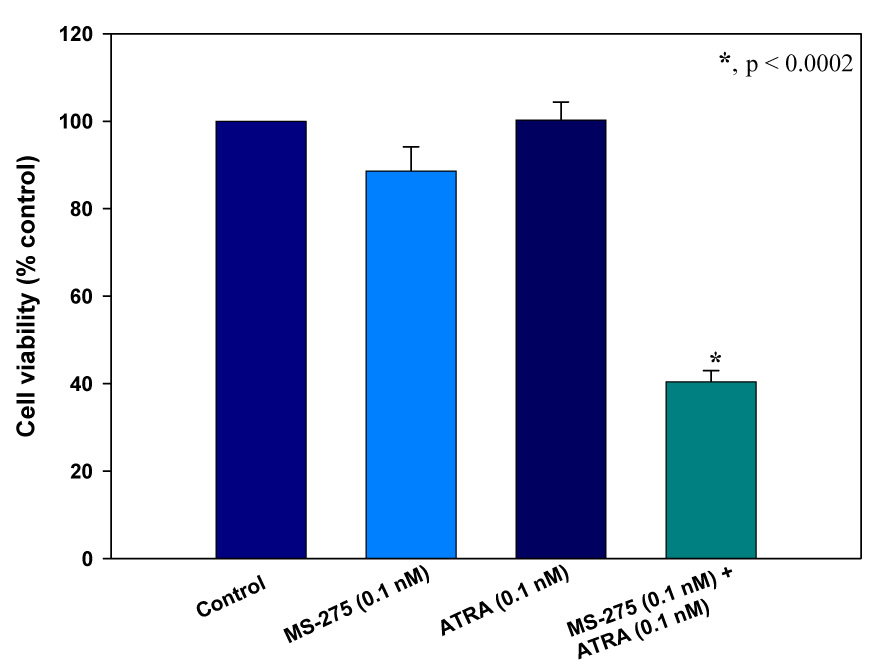


The effect of various HDACIs in combination with retinoids or RAMBAs on LNCaP cell viability as assessed by the MTT assay. (a) The effect of ATRA, CI-994, and the combination of the two at fixed concentrations. (b) The effect of 13-cis RA and CI-994. (c) The effect of ATRA and MS-275. (d) The effect of VN/14-1 and MS-275, (d) The effect of VN/66-1 and MS-275, (e) The effect of VN/66-1 and SAHA, *, p < 0.0002 (t-test) for all graphs. Data is mean (± SE) of at least three independent experiments.
A recent study by Faller and colleagues demonstrated enhanced (synergistic) suppression of androgen sensitive LNCaP and CWR22-rv1 when retinoids were combined with several HDIs.26 However, Pili and colleagues reported that phenylbutyrate in combination with 13-CRA has an additive inhibitory effect in LNCaP cells in vitro.27 These differences may be due to the phenotypes/genotypes (number of passages in culture) of the cell lines utilized.
Effects of 2 and SAHA on cell cycle and pro-differentiation and pro-apoptotic proteins
Treatments of 2 (1 µM) + SAHA (10 nM) on LNCaP cells were used to further investigate the mechanisms of action. We elected to use SAHA in these studies because the agent is currently in clinical use.11 The effects of 2, SAHA or the combination of both agents on cell cycle profile of LNCaP cells were examined by flow cytometry as we have previously described.12, 15, 28 Compared to control, 2 and SAHA alone induced G1 cell cycle arrest, while simultaneous treatment of both agents induced both G1 and G2/M cell cycle arrest with concomitant and significant decreases in percentage of cells in S phase (Table 1). The effects of these agents on protein expression on G1 phase cell cycle regulatory proteins, differentiation and apoptosis was also examined in LNCaP cells by Western blot analysis following treatment for 24 h. As can be seen in Figure 3, treatment with 2 + SAHA caused significant up-modulation of cytokeratin 8/18 (4.5 fold), Bad (2.4 fold), Bax (5.4 fold) and down-modulation of cyclin D1 (>10 fold) and cdk4 (10 fold). Clearly, this treatment resulted in induction of differentiation, apoptosis and cell cycle arrest. A lack of cellular differentiation, uncontrolled cell cycle progression and evasion of apoptosis are hallmarks of many human cancers.29, 30 Therefore, therapeutic agents such as 2 + SAHA that can simultaneously impede cell cycle progression and promote differentiation and apoptosis in cancer cells are highly desirable. The cytostatic and cytotoxic properties of 2 + SAHA in prostate cancer indicate that the biological mechanisms of action of these two agents are diverse, thus rendering them more attractive for preclinical and clinical development for prostate cancer therapy.
Table 1.
Cell cycle analysis of effects of VN/66-1, SAHA or their combination on LNCaP cells.
| Table 1: Cell cycle distribution in LNCaP cellsa | |||
|---|---|---|---|
| Treatment | G1 (%) | S (%) | G2/M (%) |
| Control | 72.61 | 27.39 | 0 |
| VN/66-1 (5 mM) | 89.14 | 10.86 | 0 |
| SAHA (1 mM) | 81.90 | 18.10 | 0 |
| VN/66-1 + SAHA | 87.34 | 4.94 | 7.72 |
Percent of cells in each phase of the cell cycle after an 18 hour treatment of LNCaP cells with either VN/66-1 (5.0 µM), SAHA (1.0 µM) or VN/66-1 + SAHA (5.0 + 1.0 µM), respectively. Percentage distribution of cells in each of the cell cycle phase are the mean obtained from experiments performed in triplicate of at least two independent experiments, SEM < ± 10%.
Figure 3.

Western blot analysis for various proteins in LNCaP whole cell lysates after a 24 hour treatment with VN/66-1 (1.0 µM), SAHA (10 nM) or their combination. Densitometry was carried out relative to loading control (β-actin). Blots are representative of at least three experiments.
Inhibition of human LNCaP prostate cancer xenografts by 2 and SAHA in SCID mice
To confirm our in vitro findings, we evaluated the in vivo anticancer efficacy of 2 and SAHA in a human LNCaP tumor xenograft model. LNCaP xenografts were grown in SCID mice and treated (subcutaneous administration) with 2 (10 → 20 mg/kg/day) or SAHA (20 → 10 mg/kg/day) or the combination of the two agents. As shown in Figure 4, growth of tumors was significantly inhibited with either 2 or SAHA as compared to control (81.6 and 97.1% inhibition, respectively), and tumors in the group treated with 2 + SAHA actually regressed by 22% over the duration of the experiment. In addition, these treatments did not cause any overt toxicity as evaluated by weight gain. A recent study by Pili and colleagues27 reported that treatment of LNCaP tumor xenografts with phenylbutyrate (PB) or 13-CRA resulted in modest tumor growth inhibition that was generally not statistically significant as compared with control, but that the combination of PB and 13-CRA resulted in a significant additive inhibitory effect (up to 90% growth inhibition as compared with control). Given the excellent efficacies of 2 or SAHA and their combination reported in this study, it seems probable that 2 or its combination with SAHA may be more effective anticancer agents. In addition, the anti-tumor efficacies of 2 and SAHA appear to be superior to the reported efficacy of SAHA on related androgen-dependent CWR22 prostate tumors.31 This is the first report of combination of a RAMBA/retinoid with an HDACI that cause LNCaP tumor regression.
Figure 4.

The effect of VN/66-1 (10 mg/kg), SAHA (20 mg/kg), and the combination (10 + 20 mg/kg, respectively) in a LNCaP xenograft model in male SCID mice. Doses were switched on day 12 to VN/66-1 (20 mg/kg), SAHA (10 mg/kg) and the combination (20 + 10 mg/kg, respectively) as indicated by the red arrow. Mice (n=7) were injected subcutaneously QD, tumors were measured twice a week. Statistical significance was determined by the t-test. *, p < 0.005 and **, p < 0.001; ***, p < 0.0005 vs control. Not shown on graph: p < 0.05 for VN/66-1 vs. SAHA; p < 0.01 for VN/66-1 vs. VN/66-1 + SAHA; p < 0.05 for SAHA vs. VN/66-1 + SAHA.
Conclusions
We have developed new methods that enabled us to synthesize two promising HDIs, CI-994 and MS-275 in excellent and improved overall yields. In addition, we have shown that retinoids and RAMBAs interact with HDIs to cause synergistic inhibition of growth of LNCaP prostate cancer cells. These studies are the first to specifically explore the biological mechanisms of action of a RAMBA in combination with HDACI. The combination of 2 and SAHA inhibit the growth of LNCaP cancer cells by inducing G1 and G2/M cell cycle arrest and induction of differentiation and apoptosis. Compound 2, SAHA and the combination exhibited potent anti-tumor efficacy in vivo. On the basis of these impressive results, further preclinical studies are warranted to develop RAMBAs and HDIs such as 2 and SAHA for prostate cancer treatment.
Experimental Section
Chemistry
General procedures and techniques were identical to those previously reported.12, 16 Infrared spectra were recorded on a Perkin-Elmer 1600 FTIR spectrometer using Nujol paste or KBr pellets. High-resolution mass spectra (HRMS) were determined on a Bruker 12T APEX-Qe FTICR-MS with an Apollo II ion source (College of Sciences Major Instrumentation Cluster, Old Dominion University, Norfolk, VA). 1H NMR Spectra were performed in CDCl3 and DMSO-d6 at 500 MHz with Me4Si as an internal standard using a Varian Inova 500 MHz spectrometer. Melting points (mp) were determined with a Fischer Johns melting point apparatus and are uncorrected.
N-(2-Aminophenyl)-4-acetylaminobenzamide (3, CI-994)
To the suspension of acetamido benzoic acid (6, 7.5 gm, 0.041 mol) in 75 ml THF, was added CDI (7.5 gm, 0.046 mol) portion wise at room temperature. The reaction mixture was stirred for one hr to form acylimidazole followed by addition of 1,2-phenylenediamine (36.2 gm, 0.335 mol) and TFA (4.19 gm, 0.036 mol, 2.8 ml) and stirring for 16 hours. The reaction mixture was then filtered to afford crude CI-994 which was re-crystallized from THF/methanol to give 9.0 gm of CI-994 (3) as white crystals (yield, 80%). HPLC analysis showed purity >99%. mp 207–208 °C; IR (Nujol): cm−1; 1H-NMR: δ 2.08 (s, 3H, CH3), 4.86 (s, 2H, NH2), 6.59 (s, 1H, Ar-H), 6.78 (d, 1H, J = 7.5 Hz, Ar-H), 6.96 (s, 1H, Ar-H), 7.16 (d, 1H, J = 7.5 Hz, Ar-H), 7.69 (d, 2H, J = 7.5, Ar), 7.94 (d, 2H, J = 8.0, Ar-Hs), 9.55 (s, 1H, NH), 10.188 (s, 1H, NH). HRMS calcd 269.1164 (C15H15N3O2), found 269.1161. These spectral and analytical data are as previously reported.20
4-[N-(Pyridin-3-ylmethoxycarbonyl)aminomethyl]benzoic Acid (8)
To a suspension of 1,1′- carbonyldiimidazole (CDI, 25.6 g, 158 mmol) in THF (120 mL) was added 3-pyridinemethanol (7, 17.3 g, 158 mmol) in THF (50 mL) at 10 °C, and the mixture was stirred for 1 h at rt. The resulting solution was added to a suspension of 4-(aminomethyl)benzoic acid (22.6 g, 158 mmol), DBU (24.3 g, 158 mmol), and triethylamine (22.2 mL, 158 mmol) in THF (250 mL). After stirring for 5 h at rt, the mixture was evaporated to remove THF and then dissolved in water (300 mL). The solution was acidified with HCl (pH 5) to precipitate a white solid which was collected by filtration, washed with water (300 mL) and methanol (50 mL), respectively, and dried to give pure 8 (41.1 g, 91% yield): mp 207–208 °C; IR (KBr) 3043, 1718, 1568, 1434, 1266, 1108, 1037, 984, 756 cm−1; 1H NMR (DMSO-d6) δ 4.28 (d, 2H, J = 5.9 Hz), 5.10 (s, 2H), 7.3–7.5 (m, 3H), 7.7–8.1 (m, 4H), 8.5–8.7 (m, 2H). These spectral and analytical data are as previously reported.22
N-(2-Aminophenyl)-4-[N-(pyridin-3-ylmethoxycarbonyl) aminomethyl] benzamide (4, MS-275)
To a suspension of 8 (5.0 g, 0.017 mol) in THF (100 mL) was added CDI (3.12 g, 0.019 mol), and the mixture was stirred for 3 h at 60 °C. After formation of acylimidazole the clear solution was cooled to rt. To this was added 1,2-phenylenediamine (15.11 g, 0.14 mmol) and trifluoroacetic acid (1.2 mL, 0.015 mol) and then stirred for 16 h. The reaction mixture was evaporated to remove THF and the crude product was stirred in a mixture of hexane and water (2:5, v/v) for 1 h and filtered and dried. The residue was triturated in dichloromethane twice to afford pure MS-275 (4) as off white powder 5.25 g, 80% yield: mp 159–160 °C; IR (KBr) 3295, 1648, 1541, 1508, 1457, 1309, 1183, 742 cm−1. 1H NMR (DMSO-d6) δ 4.28 (d, 2H, J = 5.9 Hz), 4.86 (s, 2H), 5.10 (s, 2H), 6.60 (t, 1H, J = 7.3 Hz), 6.78 (d, 1H, J = 7 Hz), 6.97 (t, 1H, J = 7 Hz), 7.17 (d, 1H, J = 8 Hz), 7.3–7.5(m, 3H), 7.78 (d, 1H, J = 8 Hz), 7.93 (d, 2H, J = 8 Hz), 8.53 (d, 1H, J = 3.7 Hz), 8.59 (s, 1H), 9.61 (s, 1H); HRMS: calcd 376.1560 (C21H20N4O3), found 376.1558. These spectral and analytical data are as previously reported.22
Cell Growth Inhibition Assay (MTT Colorimetric Assay)
LNCaP cell lines were maintained in RPMI 1640 medium containing 10% fetal bovine serum, 1% penicillin and streptomycin, as the complete culture medium.2 × 104 cells were seeded in 24 well plates and incubated in a 5% CO2 incubator at 37°C for 1 day. Cultures were treated with various compounds as listed, alone and in combination on day 2 and 4.Cells were washed on day 2 and media was changed. Mitochondrial metabolism was measured as a marker for cell growth by adding 100 µl /well MTT (5 mg /ml in medium) with 2 h incubation at 37°C on Day 6.Crystals formed were dissolved in 500µl of DMSO. The absorbance was determined using a microplate reader at 560 nm. The absorbance data was converted into cell proliferation percentage. Each assay was performed in triplicate.
Western Immunoblotting
LNCaP cells were treated for 24 hours and harvested thereafter. The cells were washed with ice-cold DPBS, scraped, processed, and the supernatant separated and stored at −80C. Western blotting was carried out as described previously.32 Antibodies against cytokeratins 8/18 and cyclin D1 were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA ) and Cdk4, Bad, and Bax were purchased from Cell Signaling Technology, Inc. (Danvers, MA).
Cell Cycle Analysis
Cells were plated in T-75 flasks containing complete RPMI 1640 medium for 24 hours. Cells were serum starved after washing with phosphate buffered saline and incubating with RPMI 1640 (minus phenol red) with 0.2% FBS for 48 hours. Under these conditions cells were arrested in the G0/G1 phase as determined by flow cytometry. Cells were then stimulated by the addition of complete RMPI 1640 medium containing 10% FBS. Cells were treated with VN/66-1 or SAHA for various times. Cells were washed with PBS, trypsinized, resuspended in 10 ml PBS and counted. They were then centrifuged (10 min, 2500 rpm at 4°C), resuspended in PBS fixed in 70% ice-cold ethanol and stored in −20°C until staining. Cells were stained for at least one hour in the dark with a solution containing 20 µg/ml propidium iodide (Sigma), 0.02 µg/ml RNAse and 1 % Triton-X 100 (Sigma). The DNA content in the treated and mock-treated groups was measured by flow cytometry analysis using a FACSort flow cytometer (Becton Dickinson, San Jose, CA); 15,000 events were analyzed for each sample. ModFit LT version 3.1 (Verity Software House Ind., ME) was used to analyze cell cycle distribution. The mean of two independent experiments is reported.
Animal Studies
All animal studies were performed according to the guidelines approved by the Institution of Animal Care and Use Committee (IACUC) of the University of Maryland School of Medicine. Male SCID mice 4–6 weeks of age were obtained from the National Cancer Institute (Fredrick, MD). The animals were housed in a pathogen-free environment under controlled conditions of light and humidity and received food and water ad libidum. Subconfluent cells were scraped into Dulbecco’s phosphate-buffered saline, collected by centrifugation, and resuspended in Matrigel (10 mg/mL) at 5.0 × 107 cells/ml. Each animal received subcutaneous inoculations in one site per flank with 100 µl of cell suspension. Animals were randomly grouped with 7 mice per group. Tumors were measured twice weekly with calipers, and tumor volume was calculated by the formula (4/3 π r12 × r2), where r1 is the smaller radius and r2 is the larger radius. Treatments began when the tumors reached a measurable size (approximately 100 mm3). VN/66-1 (2) and SAHA were prepared in sterile conditions as suspensions in 0.3% hydroxypropyl cellulose (HPC).
Statistical Analysis
All experiments were carried out in at least triplicates and are expressed as mean ± SE where applicable. Treatments were compared to controls using the Students t test with either GraphPad Prism or Sigma Plot. Various treatment groups were compared using the analysis of variance (ANOVA). P values less than 0.05 were considered to be statistically significant.
Acknowledgment
This research was supported by a grant from the U.S. National Institutes of health (NIH) and the National Cancer Institute (NCI), grant No: CA117991 to Vincent C. O. Njar. We are grateful for the generous support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. CA Cancer J Clin. 2007;57:43. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Denmeade SR, Isaacs JT. Nat Rev Cancer. 2002;2:389. doi: 10.1038/nrc801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barry MJ, Fowler FJ, Jr, Bin L, Oesterling JE. J Urol. 1997;158:488. doi: 10.1016/s0022-5347(01)64510-5. [DOI] [PubMed] [Google Scholar]
- 4.Feldman BJ, Feldman D. Nat Rev Cancer. 2001;1:34. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 5.Sartorelli AC. Br J Cancer. 1985;52:293. doi: 10.1038/bjc.1985.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Nat Rev Cancer. 2001;1:194. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 7.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. Cell. 1995;83:835. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagy L, Kao HY, Chakravarti D, Lin RJ, Hassig CA, Ayer DE, Schreiber SL, Evans RM. Cell. 1997;89:373. doi: 10.1016/s0092-8674(00)80218-4. [DOI] [PubMed] [Google Scholar]
- 9.Richon VM, Webb Y, Merger R, Sheppard T, Jursic B, Ngo L, Civoli F, Breslow R, Rifkind RA, Marks PA. Proc Natl Acad Sci U S A. 1996;93:5705. doi: 10.1073/pnas.93.12.5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelly WK, Richon VM, O'Connor O, Curley T, MacGregor-Curtelli B, Tong W, Klang M, Schwartz L, Richardson S, Rosa E, Drobnjak M, Cordon-Cordo C, Chiao JH, Rifkind R, Marks PA, Scher H. Clin Cancer Res. 2003;9:3578. [PubMed] [Google Scholar]
- 11.Bolden JE, Peart MJ, Johnstone RW. Nat Rev Drug Discov. 2006;5:769. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 12.Patel JB, Huynh CK, Handratta VD, Gediya LK, Brodie AM, Goloubeva OG, Clement OO, Nanne IP, Soprano DR, Njar VC. J Med Chem. 2004;47:6716. doi: 10.1021/jm0401457. [DOI] [PubMed] [Google Scholar]
- 13.Njar VC, Gediya L, Purushottamachar P, Chopra P, Vasaitis TS, Khandelwal A, Mehta J, Huynh C, Belosay A, Patel J. Bioorg Med Chem. 2006;14:4323. doi: 10.1016/j.bmc.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 14.Belosay A, Brodie AM, Njar VC. Cancer Res. 2006;66:11485. doi: 10.1158/0008-5472.CAN-06-2168. [DOI] [PubMed] [Google Scholar]
- 15.Patel JB, Mehta J, Belosay A, Sabnis G, Khandelwal A, Brodie AM, Soprano DR, Njar VC. Br J Cancer. 2007;96:1204. doi: 10.1038/sj.bjc.6603705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gediya LK, Chopra P, Purushottamachar P, Maheshwari N, Njar VC. J Med Chem. 2005;48:5047. doi: 10.1021/jm058214k. [DOI] [PubMed] [Google Scholar]
- 17.Acharyaf MR, Sparreboom A, Venitz J, Figg WD. Mol Pharmacol. 2005;68:917. doi: 10.1124/mol.105.014167. [DOI] [PubMed] [Google Scholar]
- 18.Miller TA, Witter DJ, Belvedere S. J Med Chem. 2003;46:5097. doi: 10.1021/jm0303094. [DOI] [PubMed] [Google Scholar]
- 19.Mork CN, Faller DV, Spanjaard RA. Curr Pharm Des. 2005;11:1091. doi: 10.2174/1381612053507567. [DOI] [PubMed] [Google Scholar]
- 20.Weiershausen USGV, K-O, Herrmann W. N-(2′-Aminophenyl)-benzamide derivatives: process for the preparation thereof and pharmaceutical composition containig them. 5,137,918. USA. 1992
- 21.Hess-Stumpp H, Bracker TU, Henderson D, Politz O. Int J Biochem Cell Biol. 2007;39:1388. doi: 10.1016/j.biocel.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki T, Ando T, Tsuchiya K, Fukazawa N, Saito A, Mariko Y, Yamashita T, Nakanishi O. J Med Chem. 1999;42:3001. doi: 10.1021/jm980565u. [DOI] [PubMed] [Google Scholar]
- 23.Valeriote F, Lin H. Cancer Chemother Rep. 1975;59:895. [PubMed] [Google Scholar]
- 24.Mongan NP, Gudas LJ. Differentiation. 2007;75:853. doi: 10.1111/j.1432-0436.2007.00206.x. [DOI] [PubMed] [Google Scholar]
- 25.Richon VM. Br. J. Cancer. 2006;95:S2. [Google Scholar]
- 26.Gu J, Zhao X, Spanjaard RA, Chen TC, Flanagan JN, Boosalis M, Perrine SP, Faller DV. Journal of Cancer Molecules. 2006;2:25. [Google Scholar]
- 27.Pili R, Kruszewski MP, Hager BW, Lantz J, Carducci MA. Cancer Res. 2001;61:1477. [PubMed] [Google Scholar]
- 28.Huynh CK, Brodie AM, Njar VC. Br J Cancer. 2006;94:513. doi: 10.1038/sj.bjc.6602971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanahan D, Weinberg RA. Cell. 2000;100:57. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 30.Tenen DG. Nat Rev Cancer. 2003;3:89. doi: 10.1038/nrc989. [DOI] [PubMed] [Google Scholar]
- 31.Butler LM, Agus DB, Scher HI, Higgins B, Rose A, Cordon-Cardo C, Thaler HT, Rifkind RA, Marks PA, Richon VM. Cancer Res. 2000;60:5165. [PubMed] [Google Scholar]
- 32.Purushottamachar P, Khandelwal A, Chopra P, Maheshwari N, Gediya LK, Vasaitis TS, Bruno RD, Clement OO, Njar VC. Bioorg Med Chem. 2007;15:3413. doi: 10.1016/j.bmc.2007.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]