

Abstract
A series of 3’-(substituted phenyl)deschloroepibatidine analogs (5a–j) were synthesized. The α4β2* and α7 nicotinic acetylcholine receptor (nAChR) binding properties and functional activity in the tail-flick, hot-plate, locomotor, and body temperature tests in mice of 5a–j were compared to those of the nAChR agonist, nicotine (1), epibatidine (4), and deschloroepibatidine (13) the partial agonist, varenicline (3) and the antagonist 2’-fluoro-3’-(substituted phenyl)deschloroepibatidine analogs (7a–j). Unlike epibatidine and deschloroepibatidine, which are potent agonists in the tail-flick test, 5a–k show no or very low antinociceptive activity in the tail-flick or hot-plate test. However, they are potent antagonists in nicotine-induced antinociception in the tail-flick test, but weaker than the corresponding 2’-fluoro-3’-(substituted phenyl)deschloroepibatidines.
Keywords: nicotinic antagonist, varenicline, nAChR bindng, epibatidine analogs
1. Introduction
The use of tobacco products is believed to be in large part due to addiction to nicotine (1), which is one of the most abused reinforcing agents. It is estimated that there are four million smoking-related deaths annually from diseases such lung cancer, chronic obstructive pulmonary disease (COPD) and cardiovascular disease.1 Consequently, there is great interest in the development of pharmacotherapies for aiding people to stop smoking.2 Present FDA approved drugs for treating smoking cessation include nicotine (1) replacement medication in the form of gum, patch, lozenge, sublingual tablet, nasal spray, and vapor inhaler, the antidepressant bupropion (2) and the α4β2* nAChR partial agonist varenicline (3).2-4
During the last few years, we have conducted structure activity studies (SAR) using the potent nAChR agonist epibatidine (4) as a lead structure to identify pharmacophores for the nAChR. The studies have identified nAChR agonists more potent than epibatidine as well as analogs that showed pure antagonist or partial agonist activity.5-12 These studies showed that introduction of a substituted phenyl group at the 3’-position on the pyridine ring of epibatidine exerted a profound influence on both receptor binding (recognition) and receptor activation. Interestingly, substitution of different groups of the 2’-position distinguished between agonist and antagonist properties. In this study, we report the comparison of the nAChR binding, and pharmacological properties of the 3’-(substituted phenyl)deschloroepibatidine analogs (5a–j) to those of the nAChR agonist nicotine (1), epibatidine (4), and deschloroepibatidine (6), the partial agonist varenicline (3), and the corresponding 2’-fluoro-3’-(substituted phenyl)deschloroepibatidine analogs (7a–j). The analogs 5a–j showed α4β2* nAChR binding affinity as well as nAChR agonist and antagonist activity in mouse antinociceptive, hypothermia, and spontaneous activity test more like the partial agonist varenicline (3) than the nAChR agonist nicotine (1), epibatidine (4), and deschoroepibatdine (6a). Like the 2’-fluoro-3’-(substituted phenyl)deschloroepibatidine analogs 7a–j, most of the 3-(substituted phenyl)deschloroepibatidine analogs 5a-j were antagonist of nicotine-induced antinociception in the tail-flick test.
2. Chemistry
The synthesis of 5a–h is shown in Scheme 1. Palladium acetate catalyzed coupling of tert-butoxycarbonyl-3’-bromodeschloroepibatidine (8)10 with the appropriately substituted phenylboronic acid in dimethoxyethane (DME) in the presence of tri-(o-tolyl)phosphine and sodium carbonate gave the tert-butoxycarbonyl-protected 3’-(substituted phenyl)deschloroepibatdine analogs (9a-g). Reduction of the 4-nitrophenyl intermediate 9g with iron powder in hydrochloric acid gave the 4-aminophenyl compound 10. Treatment of 9a–g and 10 with trifluoroacetic acid in methylene chloride removed the protecting tert-butoxycarbonyl group and afforded the desired 3-(substituted phenyl)deschloroepibatidine analogs 5a-h.
Scheme 1.
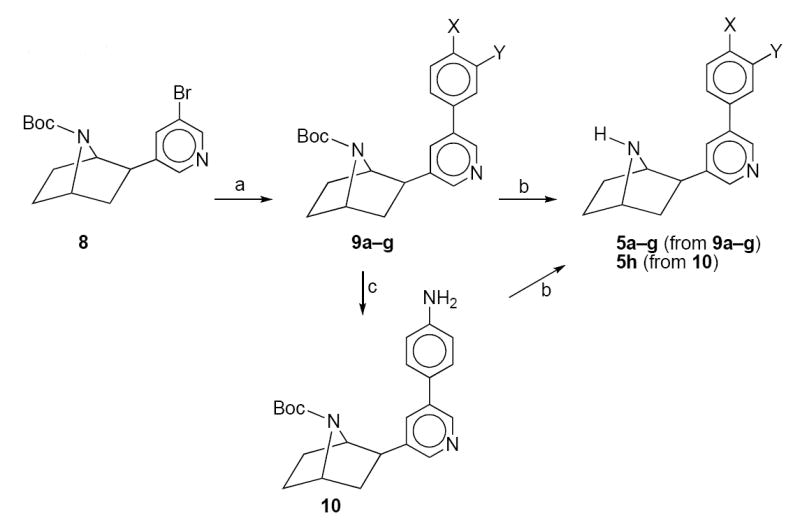
Reagents: (a) Pd(OAc)2, P(o-tolyl)3, Na2CO3, (X,Y)C6H4B(OH)2 DME; (b)CF3CO2H; (c)Fe, HCl(H2O).
The 3’-(3-aminophenyl)- and 3’-(3-methoxyphenyl)deschloroepibatidine analogs 5i and 5j, respectively, were synthesized as outline in Scheme 2. Palladium acetate catalyzed addition of 3-nitrophenylboronic acid or 3-methoxyphenylboronic acid to 7-tert-butoxycarbonyl-2-exo-2-(2-amino-3-bromo-8-pyridinyl)-7-azabicyclo[2.2.1]heptane (11)10 provided the tert-butoxycarbonyl-protected 3’-(3-nitrophenyl)- and 3’-(3-methoxyphenyl)deschloroepibatidine 12a and 12b, respectively. Diazotization of 12a and 12b using sodium nitrite in hydrochloric acid yielded 3’-(3-methoxyphenyl)epibatidine 13a and 13b, respectively. Catalytic hydrogenation of 13a and 13b using 10% palladium on carbon catalyst in methanol yielded the desired 5i and 5j, respectively.
Scheme 2.
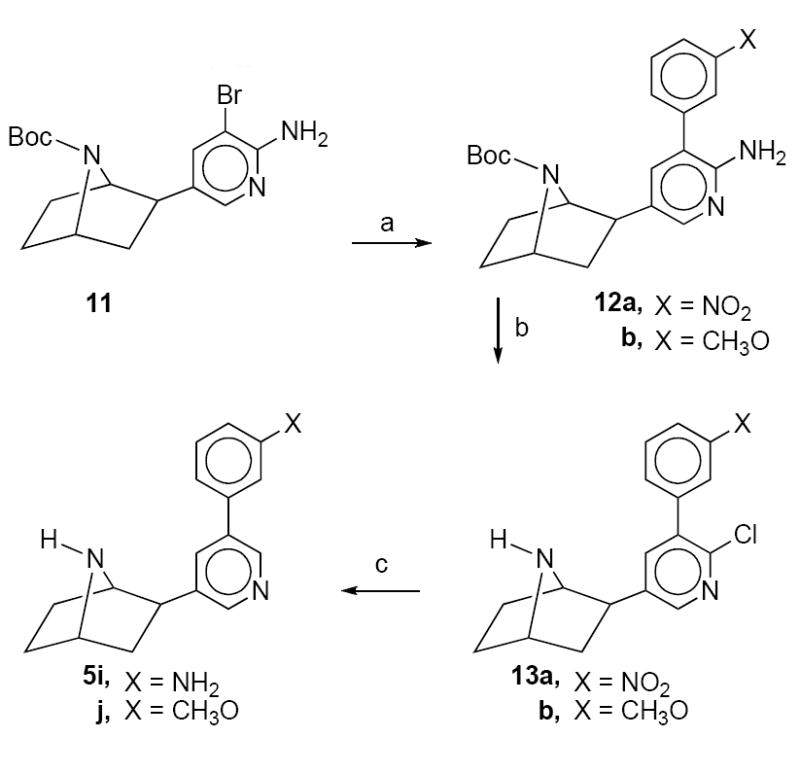
Reagents: (a) Pd(OAc)2, P(o-tolyl)3, Na2CO3, DME, H2O, 3-NO2C6H4B(OH)2 or CH3OC6H4B(OH)2; (b) NaNO2, HCl;(c)10% Pd/C, CH3OH.
3. Biology
The Ki values for the inhibition of [3H]epibatidine binding at the 〈4®2* nAChR in male rat cerebral cortex for compound 5a–j are compared to values for nicotine (1), epibatidine (4), varenicline (3) and 2’-fluoro-3’-(substituted phenyl)deschloroepibatidine analogs (7a–j) (Table 1). The binding assays were conducted and the Ki values calculated as previously described.8 Compounds (10 μM) were also evaluated for inhibition of binding to 〈7 nAChR using [125I) iodoMLA as previously reported.8
Table 1.
Comparison of epibatidine and varenicline radioligand binding and antinociception data of 3’-(substituted phenyl)deschloroepibatidine analogs to standard nAChR agonist, partial agonist, and antagonist
| Compd a | X | Y | α4β2* [3H]Epibatidine (Ki, nM)b | α7 [125I]iodo MLA (Ki, nM)b | ED 50 mg/kg Tail-Flickc | ED 50 mg/kg Hot-Platec | ED 50 mg/kg Hypothermiac | ED 50 mg/kg Spontaneous Activity c | AD 50 (μg/kg)c |
||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tail-Flick | Hot-Plate | Body Temperature | |||||||||
| Epibatidine (4) | 0.026 ± 0.002 | 198 ± 4 | 0.006 | 0.004 | 0.004 | 0.001 | |||||
| 6ad | 0.020 ± 0.001 | 0.002 | |||||||||
| 6bd | 0.027 ± 0.001 | ||||||||||
| Nicotine (1) | 1.5 ± 0.3 | 670 ± 33 | 1.3 | 0.65 | 1.0 | 0.5 | |||||
| Varenicline (3) | 0.12 ± 0.02 | 32.5 ± 1.3 | 11% @ 10 | 10% @ 10 | 2.8 | 2.1 | 0.2 | 470 | 0% @ 10,000 | ||
| 5a | H | H | 0.50 ± 0.03 | 7% @ 50 nM | 6% @ 10 | 30% @ 10 | 1.5 (1.2–3.3) | 0.5 (0.13–2) | 300 (100–3000) | 10% @ 1000 | 0% @ 1000 |
| 5b | Cl | H | 0.21 ± 0.03 | >2000 | 1% @ 10 | 11% @ 10 | 2% @ 10 | 10.5 (8.2–15.5) | 0% @ 10 | 0% @ 10 | 5% @ 1000 |
| 5c | H | Cl | 0.17 ± 0.021 | >2000 | 1% @ 20 | 10% @ 20 | 0% @ 20 | 3.8 (0.36–41) | 0.01 (0.001–0.1) | 0% @ 1000 | 0% @ 1000 |
| 5d | F | H | 0.15 ± 0.003 | >2000 | 3% @ 10 | 13% @ 10 | -1.6% @ 10 | 3.0 (2.2–3.9) | 13 (2–70) | 10% @ 100 | 0% @ 1000 |
| 5e | H | F | 0.20 ± 0.04 | >2000 | 2% @ 10 | 8% @ 10 | 0% @ 10 | 5 (3.2–7.9) | 3.1 (0.07–1) | 3000 (2500–3600) | 0% @ 10 |
| 5f | NO2 | H | 0.34 ± 0.005 | >2000 | 1% @ 10 | 26% @ 10 | 2.9 (2.1–3.5) | 0.66 (0.1–3.9) | 47(0.7–33) | 10% @ 100 | 0% @ 100 |
| 5g | H | NO2 | 0.17 ± 0.009 | >2000 | 5% @ 20 | 21% @ 20 | 37.6 (30–45) | 4.7 (0.25–8.5) | 2.5 (0.4–18) | 11,000 (900– 13,300) | 0% @ 2000 |
| 5h | NH2 | H | 0.34 ± 0.05 | >2000 | 6% @ 10 | 10% @ 10 | 0% @ 10 | 15% @ 10 | 1 (0.1–4) | 45% @ 2000 | 0% @ 20 |
| 5i | H | NH2 | 1.16 ± 0.14 | >2000 | 6.3 (3.8–10.3) | 5.4 (3.2–9.1) | 3.8 (2.8–4.8) | 1.5 (0.5–4.5) | NT | NT | |
| 5j | H | CH3O | 0.43 ± 0.05 | >2000 | 1% @ 10 | 20% @ 10 | 0% @ 10 | 2.9 (0.8–10) | 0% @ 10 | 0% @ 10 | 0% @ 10 |
| 7ae | H | H | 0.24 | >2000 | 3% @ 15 | 4% @ 15 | -0.5 °C @ 10 | 4.7 | 500 | 1200 | |
| 7be | Cl | H | 0.044 | 2% @ 10 | 7% @ 10 | 0% @ 10 | 5% @ 10 | 0.3 | 260 | 0% @ 1000 | |
| 7ce | F | H | 0.073 | 3% @ 10 | 14% @ 10 | 0% @ 10 | 15% @ 10 | 12 | 450 | 0% @ 1000 | |
| 7de | H | F | 0.029 | 2% @ 10 | 15% @ 10 | 10% @ 10 | 2 | 0.5 | 230 | 0% @ 100 | |
| 7ee | H | F | 0.087 | 3.5 | 3.3 | 1.8 | 0.36 | 5 | 2% @ 1000 | 0% @ 1000 | |
| 7fe | NO2 | H | 0.009 | 5% @ 10 | 10% @ 10 | 3.5 | 0.22 | 3 | 120 | 0% @ 1000 | |
| 7ge | H | NO2 | 0.053 | 3% @ 10 | 20% @ 10 | 0% @ 10 | 6.5 | 0.5 | 130 | 5% @ 500 | |
| 7he | NH2 | H | 0.095 | 2% @ 10 | 10% @ 10 | 0% @ 10 | 6 | 5 | 1800 | 0% @ 5000 | |
| 7ie | H | NH2 | 0.16 | 1% @ 10 | 4% @ 10 | 0% @ 10 | 8.5 | 9 | 820 | 5% @ 5000 | |
| 7je | H | CH3O | 0.12 | 0% @ 10 | 8% @ 10 | 0% @ 10 | 6 | 8 | 2000 | 0% @ 10 | |
All epibatidine analogs with the exception of 5b and 5d were tested as hydrochloride salts and all were racemates.
Data represent means ± SE from at least three independent experimentals.
Results are provided as ED50 or AD50 values (± confidence limits) or as a percent effect at the individual dose.
Taken from reference 9.
Data take from reference 11.
The above compounds were evaluated in two acute pain models, the tail-flick and the hot-plate tests, and the results are listed in the Table 1.13 In the tail-flick method of D’Amour and Smith,14 the tail is exposed to a heat lamp and the amount of time taken for the animal to move (flick) its tail away from the heat is recorded. A control response (2–4 s) was determined for each mouse before treatment, and a test latency was determined after drug administration. The method used for the hot-plate test is a modification of those described by Eddy and Leimbach15 and Atwell and Jacobson.16 Mice were placed into a 10-cm wide glass cylinder on a hot plate (Thermojust Apparatus) maintained at 55.0 °C. Two control latencies at least 10 min apart were determined for each mouse. The normal latency (reaction time) was 8–12 s. The reaction time was scored when the animal jumped or licked its paws. The mice were tested 5 min after sc injections of nicotinic ligands for the dose-response determination. Antinociceptive response was calculated as percentage of maximum possible effect (% MPE, where % MPE = [(test–control)/(maximum latency–control) × 100]).
To measure the effect of analogs on spontaneous activity, mice were placed into individual Omnitech photocell activity cages (28 cm × 16.5 cm) 5 min after sc administration of either 0.9% saline or the epibatidine analog. Interruptions of the photocell beams (two banks of eight cells each) were then recorded for the next 10 min. Data were expressed as number of photocell interruptions. Rectal temperature was measured by a thermistor probe (inserted 24 mm) and digital thermometer (Yellow Springs Instrument Co., Yellow Springs, OH). Readings were taken just before and at different times after the sc injection of either saline or epibatidine analogs. The difference in rectal temperature before and after treatment was calculated for each mouse. The ambient temperature of the laboratory varied from 21 to 24 °C from day to day.
For the antagonist experiments, mice were pretreated sc with either saline or epibatidine analogs 10 min before nicotine. Nicotine was administered at a dose of 2.5 mg/kg, sc (an ED84 dose), and mice were tested 5 min later. ED50 and AD50 values with 95% confidence limits were determined.
4. Results and Discussion
The desired target compounds 5a–j could be synthesized by a procedure similar to that used to prepare the previously reported 2’-fluoro-3’-(substituted phenyl)deschloroepibatidine analogs (7a–k).11 The key step in both syntheses is palladium acetate catalyzed Suzuki17,18 cross coupling of a 3-bromopyridine starting material 8 and 11 with the appropriately substituted phenylboronic acids.
Binding affinities for the 3’-(3- and 4-substituted phenyl)deschloroepibatidine analogs 5a–j at α4β2* and α7 nAChRs along with data for nicotine (1), epibatidine (4), deschloroepibatidine (6a), 2’-fluorodeschloroepibatidine (6b), varenicline (3), and the corresponding 2’-fluoro-3’-(substituted phenyl)deschloroepibatidine analogs 7a–j are listed in Table 1. 3’-Phenyldeschloroepibatidine has a Ki = 0.5 nM for the α4β2* nAChR. Substitution of the 3’-phenyl group with a 3’- or 4’-position electron withdrawing or releasing substituent had only small effects on binding affinity. The Kis varied between 0.15 and 0.43 nM for 5b–i. The 3’-(4-fluorophenyl) and 3’-(3-chlorophenyl) analogs 5d and 5c with Kis = 0.15 and 0.17 nM,respectively, possessed the highest affinity and are almost identical to the Ki for varenicline (ki = 0.12 nM), but are much lower than the Kis of 0.026 and 0.020 nM for epibatidine (4) and deschloroepibatidine (6). All compounds have >2000 nM affinity for the α7 nAChR compared to a Ki = 32.5 nM for varenicline. In every case, the Ki value of the deschloroepibatidine analogs 5a–j were larger (weaker affinity) than the Ki value for the corresponding 2’-fluoro-(substituted phenyl)-deschloroepibatidine analogs 7a–j. Since deschloroepibatidine (6a) and 2’-fluorodeschloroepibatidine (6b) have almost identical Ki values (Table 1) for α4β2* AChR binding, the difference between the Ki values of 5a–j and 7a–j are apparently due to different types of effects of the 3-(substituted phenyl) groups in each series.
The 3’-(substituted phenyl)deschloroepibatidine analogs 5a–j were also evaluated for their in vivo nAChR properties in mice and compared to similar properties of varenicline and the 2-fluoro-(substituted phenyl)-deschloroepibatidine analogs 7a–j (Table 1). With the exception of the 3’-(3-aminophenyl) analog 5i, which has an ED50 = 6.3 mg/kg, no compound possessed antinociceptive activity in the tail-flick or hot-plate test. This contrasts sharply with descloroepibatidine (6), which has an ED50 = 0.002 mg/kg in this test, but is very similar to the results obtained with the 2-fluoro-(substituted phenyl)-deschloroepibatidine analogs 7a–j. Compounds 5a, 5f, and 5i showed weak activity in the hypothermia test (ED50 = 0.5 to 3.8 mg/kg) similar to that of varenicline (ED50 = 2.8 mg/kg). All compounds with the exception of 5b and 5h showed activity in the spontaneous activity test similar to varenicline; ED50 = 0.5 to 5 mg/kg compared to 2.1 mg/kg for varenicline. Even though there was not a one-to-one correlation between the analogs 5a–j and 7a–j, most of the 2-fluoro-(substituted phenyl)-deschloroepibatidine analogs (7a–j) showed similar potency in this test.
All analogs with the exception of the 3’-(3-methoxyphenyl) analog 5j were potent antagonists in the tail-flick test. The 3-substituted phenyl analogs 5c, 5e, and 5g were more potent antagonists than the 4-substituted analogs 5c, 5d, and 5f. The 3’-(3-chlorophenyl) analog 5c with and AD50 = 0.01 μg/kg was the most potent analog. The 3’-(3-fluorophenyl) and 3’-(3-nitrophenyl) analogs 5e and 5g with AD50s = 3000 and 11,000 μg/kg, respectively, were weak antagonists in the hot-plate test; the other compounds were inactive in this test. No compound antagonized nicotine-induced hypothermia.
For comparison, varenicline19 blocked nicotine-induced antinociception in the tail-flick and hot-plate tests with ED50s of 0.2 and 470 μg/kg, respectively. Interestingly, compound 5c was almost 20 times more potent in blocking nicotine’s effects on the tail-flick test than varenicline, suggesting that 5c is a more potent antagonist or partial agonist. However, our in vivo models cannot discriminate between these two properties. Interestingly, compared to varenicline, the differential in vivo blockade potency in the different tests, suggest a better in vivo and possibly in vitro selectivity at nAChRs. While there was a lack of a good correlation, the 2-fluoro-(substituted phenyl)-deschloroepibatidine analogs 7a–j had potencies as antagonist in the tail-flick similar to those of 5a–j. In contrast, 7a–j tended to be more potent antagonist in the hot-plate test than 5a–j.
In summary, the addition of a 3’-phenyl or 3’-(substituted phenyl) group to deschloroepibatidine (6) provided compounds 5a–j, which showed binding affinity at the α4β2* nAChR similar to that of nicotine (1) and varenicline (3), but considerably less than that of deschloroepibatidine (6), epibatidine (4), and 2’-fluoro-3’-(substituted phenyl)deschloroepibatidine analogs 7a–j. In contrast to varenicline (3), the analogs 5a–k had no affinity for the α7 nAChR. Unlike deschloroepibatidine (6), which is a potent agonist in the tail-flick test, 5a–h and 5i show no antinociceptive activity in the tail-flick or hot-plate test. Similar to varenicline and 2’-fluoro-3’-(substituted phenyl)deschloroepibatidine analogs 7a–j are modest activators of locomotor activity and are potent antagonist of nicotine-induced antinociception in the tail-flick test. However, unlike varenicline (3) and the 2-fluoro-(substituted phenyl)-deschloroepibatidine analogs 7a–j, which had good potency as antagonist in the hot-plate test, the 3-phenyldeschloroepibatidine analogs 5a–j had weak or no activity in this test. Additionally, the 3-substituted phenyl analogs exhibited greater antagonistic activity than the corresponding 4-substituted phenyl analogs.
5. Experimental
Melting points were determined on a Mel-temp (Laboratory Devices, Inc.) capillary tube apparatus. NMR spectra were recorded on a Bruker Avance 300 or AMX 500 Spectrometer using tetramethylsilane as internal standard. Thin layer chromatography was carried out on Whatman silica gel 60 plates. Visualization was accomplished under UV or in an iodine chamber. Microanalysis was carried out by Atlantic Microlab, Inc. Flash chromatography was carried out using silica gel 60 (230–400 mesh) using various solvents combined with a solvent mixture of 80% chloroform, 18% methanol, and 2% concentration ammonium hydroxide (CMA80).
The [ 3H]epibatidine was purchased from Perkin Elmer Inc., (Boston, MA). The [125]iodo-MLA was synthesized as previously reported.
7-tert-Butoxycarbonyl-2-exo-[3-phenyl-5-pyridinyl)]-7-azabicyclo[2.2.1]heptane (9a)
To a resealab1e reaction tube under nitrogen was added 810 (300 mg, 0.85 mmol), Pd(OAc)2 (16 mg, 0.071 mmol), P(o-tolyl)3 (43 mg, 0.2 mmol), sodium carbonate (190 mg, 1.8 mmo1), phenylboronic acid (171 mg, 1.4 mmol), degassed water (1 mL), and DME (10 mL). The reaction mixture was heated at 80 °C overnight, cooled and added to a saturated sodium bicarbonate solution. The mixture was extracted with CH2C12, 4 × 50 mL. The extracts were dried (Na2SO4)and concentrated. The resulting residue was purified by column chromatography on silica gel, eluting with hexane-ethyl acetate (3:1) to yield 0.28 g, (95%) of 9a as an oil. 1H NMR (CDCl3) δ (ppm): 1.42 (s, 9H), 1.53-1.66 (m, 2H), 1.87-1.93 (br s, 3H), 2.00-2.07 (m, 1H, CH2), 2.93-2.98(m, 1H, CH), 4.28 (br s, 1H), 4.41 (br s, 1H), 7.36-7.48 (m, 3H), 7.57-7.59 (d, 2H), 7.87 (s, 1H), 8.48 (s, 1H), 8.70 (s, 1H).
3-Phenyldeschloroepibatidine (5a) Dihydrochloride
To a stirred solution of 9a (280 mg, 0.797 mmol) in methylene chloride (5 mL) at 0 °C was added trifluroacetic acid (5 mL). After stirring at 25 °C for 2 h, the reaction mixture was poured into 150 mL of a solution of concentrated NH4OH-H2O (1:1). The mixture was extracted with CH2Cl2 (4 × 50 mL), dried (Na2SO4) and concentrated. The residue was purified by silica gel column chromatography,eluting with CMA80-ethyl acetate (1:3) to yield 180 mg, (90%) of 5a .1H NMR (CDCl3) δ 1.51-1.76 (m, 5H), 1.92-2.00 (m, 1H, CH2), 2.85-2.90 (m, 1H, CH), 3.65 (br s, 1H), 3.81 (br s, 1H),7.35-7.48 (m, 3H), 7.57-7.60 (d, 2H), 7.91 (s, 1H), 8.50 (s, 1H), 8.66 (s, 1H). 13C NMR (CDCl3) δ (ppm) (I7-C): 30.464, 31.747, 40.692, 45.767, 56.873, 63.169, 127.625(2C), 128.297, 129.345(2C), 133.457, 136.703, 138.539, 142.274, 146.329, 148.302.
The free base was dissolved in CH3OH and excess 2M ethereal HCl was added. The solvents were removed and the resulting solid was recrystallized from a CH3OH and Et2O mixture. The dihydrochloride salt had mp 170–175 °C, anal (C17H20Cl2N2•1.5H2O) C, H, N.
7-tert-Butoxycarbonyl-2-exo-[3-(4-chlorophenyl)-5-pyridinyl)]-7-azabicyclo[2.2.1]heptane (9b)
Using a procedure similar to that described for 9a, compound 8 (400 mg, 1.13 mmol) was coupled to 292 mg (1.86 mmol) of 4-chlorophenylboronic acid to give 0.40 g (97%) of 9b as a white solid; mp 119–120 °C. 1H NMR (CDCl3,) δ 1.42 (s, 9H), 1.53-1.66 (m, 2H), 1.86-1.93 (m, 3H), 2.00-2.08 (m, 1H), 2.93-2.98 (m, 1H), 4.27 (br, s, 1H), 4.41 (br, s, 1H), 7.43 (d, J = 10.5 Hz, 2H), 7.51 (d, J = 10.5, 2H), 7.83 (t, J = 2.1 Hz, 1H), 8.48 (s, J = 2.1 Hz, 1H), 8.65 (s, J = 2.1 Hz, 1H); 13C NMR (CDCl3) δ 155.3, 148.5, 146.4, 136.8, 135.6, 134.6, 132.8, 129.5, 128.8, 80.1, 62.2, 56.3, 45.9, 40.8, 30.2, 29.2, 28.7 ESI-MS (m/z): 385.5 (M+1);Anal (C22H25ClN2O2) C, H, N.
3-(4-Chlorophenyl)deschloroepibatidine (5b)
To a solution of 9b (400 mg, 1.04 mmol) in CH2Cl2 (7 mL) at 0 °C was added trifluoroacetic acid (7 mL). After 2 h at 25 °C, the reaction mixture was poured into 200 mL of concentrated NH4OH-H2O (1:1), extracted with CH2Cl2 (5 x 70 mL), dried (Na2SO4) and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc-MeOH (2:1 to 2:3) to give 184 mg (62 %) of 5b as a crystalline solid; mp 118–119 °C.1H NMR (CDCl3) δ 1.50-1.76 (m, 5H), 1.92-2.00 (m, 1H), 2.84-2.89 (m, 1H), 3.65 (br, s, 1H), 3.82 (br, s, 1H), 7.04 (d, J = 8.04 Hz, 2H), 7.52 (d, J = 7.52, 2H), 7.90 (t, J = 2.1 Hz, 1H), 8.52 (d, J = 2.1 Hz, 1H), 8.63 (d, J = 2.l Hz, 1H); 13C NMR (CDCl3) δ 30.6, 31.9, 40.8, 45.7, 56.9, 63.2, 128.9, 129.5, 133.3, 134.5, 135.6, 137.1, 142.5, 146.1, 148.7; ESI-MS (m/z):285.5 (M+1); Anal (C17H17ClN2) C, H, N.
7-tert-Butoxycarbonyl-2-exo-[3-(3-chlorophenyl)-5-pyridinyl)]-7-azabicyclo(2.2.1]heptane (9c)
Using a procedure similar to that described for 9a, compound 8 (600 mg, 1.70 mmol) was coupled to 440 mg (2.80 mmol) of 3-chlorophenylboronic acid to give 0.67 g (>100%) of 9c as a noncrystalline solid.1H NMR (CDCl3) δ 1.34 (s, 9H), 1.39-1.57 (m, 2H), 1.78-1.82 (m, 3H), 1.91-1.98 (m, 1H), 2.84-2.89 (m, 1H), 4.20 (br, s, 1H), 4.32 (br, s, 1H), 7.227.46 (m, 3H), 7.46 (s, br, 1H), 7.76 (s, br, 1H), 8.40 (s, br, 1H), 8.56 (s, br, 1H; 13C NMR (CDCl3) δ 28.6, 29.1, 30.1, 40.7, 45.7, 56.2, 62.2, 80.0, 125.6, 127.5, 128.3, 130.6, 133.0, 135.2, 135.3, 140.0, 141.6, 146.2, 148.5, 155.2.
3’-(3-Chlorophenyl)deschloroepibatidine (5c) Dihydrochloride
To a stirred solution of 9c (600 mg, 1.56 mmol) in CH2Cl2 (10 mL) at 0 °C was added trifluoroacetic acid (10 mL). After 2 h at 25 °C, the reaction mixture was poured into 300 mL of concentrated NH4OH-H2O (1:1) extracted with CH2Cl2 (5 × 80 mL), dried (Na2SO4) and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc-MeOH (2:1 to 4:3) to yield 430 mg, (98 %) of 5c. 1H NMR (CDCl3) δ 1.47-1.75 (m, 5H), 1.89-1.96 (m, 1H), 2.02-2.05 (m, 1H), 2.81-2.86 (m, 1H), 3.62 (br, s, 1H), 3.80 (br, s, 1H), 7.29-7.45 (m, 3H), 7.54 (s, br, 1H), 7.91 (s, br, 1H), 8.52 (5, br, 1H), 8.61 (5, br, 1H); 13C NMR (CDCl3) δ 30.5, 31.8, 40.5, 45.6, 56.8, 63.1, 125.8, 127.6, 128.3, 130.6, 133.4, 135.2, 135.3, 140.3, 142.4, 146.1, 148.9.
The dihydrochloride salt had mp 227–230 °C; Anal (C17H19Cl3N2•1.5H2O) C, H, N.
7-tert-Butoxycarbonyl-2-exo-[3-(4-fluorophenyl)-5-pyridinyl)]-7-azabicyclo[2.2.1]heptane (9d)
Using a procedure similar to that described for 9a, compound 8 (1.0 g, 0.0028 mol) was coupled to 650 mg (4.67 mmol) of 4-fluorophenylboronic acid to give 1.04 g (97%) of 9d as a noncrystalline solid. 1H NMR (CDCl3) δ 1.43 (5, 9H), 1.58-1.64 (m, 2H), 1.87-1.93 (m, 3H), 2.00-2.08 (m, 1H), 2.96-2.98 (m, 1H), 4.29 (br, s, 1H), 4.42 (br, s, 1H), 7.14 (m, 2H), 7.55 (m, 2H), 7.84 (m, 1H), 8.05 (br, s, 1H), 8.65 (br, s, 1H).
3’-(4-Fluorophenyl)deschloroepibatidine (5d)
To a stirred solution of 9d (1.38 g, 0.0038 mmol) in CH2Cl2 (23 mL) at 0 °C was added trifluoroacetic acid (23 mL). After 2 h, the reaction mixture was poured into 500 mL of concentrated NH4OH-H2O (1:1) extracted with CH2Cl2 (5 × 100 mL), dried (Na2SO4), and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc-MeOH (2:1 to 2:3) to yield 0.81 g, (81 %) of 5d as a crystalline solid; mp 115–116 °C. 1H NMR (CDCl3) δ 1.46-1.76 (m, 5H), 1.92-1.99 (m, 1H), 2.84-2.89 (m, 1H), 3.65 (br, s, 1H), 3.82 (br, s, 1H), 7.16 (t, J = 9.6 Hz, 2H), 7.55 (m, 2H), 7.89 (m, 1H), 8.50 (br, s, 1H), 8.62 (br, s, 1H); 13C NMR (CDCl3) δ 30.6, 31.9, 40.8, 45.7, 56.9, 63.2, 116.3 (d, JCF = 21.6 Hz), 129.3 (d, JCF = 8.1 Hz) 133.2, 134.7, 135.8, 142.4, 146.2, 148.4, 163.0 (d, JCF = 236.3) ; ESI-MS (m/z); 269.1 (M+l); Anal (C17H17FN2) C, H, N.
7-tert-Butoxycarbonyl-2-exo-[3-(3-fluorophenyl)-5-pyridinyl)]-7-azabicyclo[2.2.1]heptane (9e)
Using a procedure similar to that described for 9a, compound 8 (600 mg, 1.70 mmol) was coupled to 392 mg (2.80 mmol) of 3-fluorophenylboronic acid to give 0.63 g (96%) of 9e as a noncrystalline solid. 1H NMR (CDCl3) δ 1.44 (s, 9H), 1.49-1.67 (m, 2H), 1.87-1.94 (m, 3H), 2.01-2.08 (m, 1H), 2.94-2.99 (m, 1H), 4.29 (br, s, 1H), 4.42 (br, s, 1H), 7.07 (m, 1H), 7.26-7.46 (m, 3H), 7.87 (m, 1H), 8.50 (br, s, 1H), 8.68 (br, s, 1H); 13C NMR (CDCl3) δ 28.3, 28.8, 29.8, 40.4, 45.4, 55.9, 61.9, 79.7, 114.4 (m), 122.8, 130.6, 132.7, 135.1, 140.2, 141.3, 145.9, 148.2, 154.9, 161.6, 164.8.
3’-(3-Fluorophenyl)deschloroepibatidine (5e) Dihydrochloride
To a solution of 9e (600 mg, 1.63 mmol) in CH2Cl2 (27 mL) at 0 °C was added trifluoroacetic acid (27 mL). After 2 h, the reaction mixture was poured into 300 mL of NH4OH-H2O (1:1), extracted with CH2Cl2 (5 × 80 mL), dried (Na2SO)4, and concentrated. The residue was purified by silica gel chromatography eluting with (EtOAc-MeOH) (2:1 to 1:1) to give 0.39 g (90%) of 5e. 1H NMR (CDCl3) δ 1.48-1.61 (m, 5H), 1.68-1.76 (m, 1H, CH2), 1.90-1.97 (m, 1H, CH), 2.82-2.87 (m, 1H), 3.63 (br, s,1H), 3.80 (br, s, 1H), 7.02-7.08 (m, 1H), 7.26-7.30 (m, 1H), 7.34-7.44 (m, 2H), 7.93 (m, 1H), 8.53 (br, s, 1H), 8.63 (br, s, 1H); 13C NMR (CDCl3) δ 30.2, 31.4, 40.4, 42.3, 56.5, 62.8, 113.9 (d, JCF = 22.4 Hz), 114.7 (d, JCF = 21.1 Hz), 122.9 (d, JCF = 2.9 Hz), 131.0 (d, JCF = 8.2 Hz), 133.0, 135.1, 140.0 (d, JCF = 9.1 Hz), 138.3, 148.4, 148.1, 163.2 (d, JCF = 246.2 Hz).
The dihydrochloride salt had mp 165–167 °C; Anal (C17H19Cl2N2•1.5H2O) C, H, N.
7-tert-Butoxycarbonyl-2-exo-[3-(4-nitrophenyl)-5-pyridinyl)]-7-azabicyclo[2.2.1]heptane (9f)
Using a procedure similar to that described for 9a, compound 8 (400 mg, 2.40 mmol) was coupled to 312 mg (1.87 mmol) of 4-nitrophenylboronic acid to give 0.40 g (90%) of 9f as a solid. 1H NMR (CDCl3) × 1.43 (s, 9H), 1.58-1.75 (m, 2H), 1.89-1.94 (m, 3H), 2.04-2.11 (m, 1H), 2.97-3.01 (m, 1H), 4.29 (br, s, 1H), 4.42 (br, s, 1H), 7.75 (d, J = 8.7 Hz, 2H), 7.93 (br, s, 1H), 8.33 (d, J = 8.4 Hz, 2H), 8.56 (br, s, 1H), 8.72 (br, s, 1H); 13C NMR (CDCl3) δ 28.6, 29.2, 30.0, 40.7, 45.7, 56.3, 62.1, 80.0, 124.5, 128.2, 133.1, 134.3, 141.8, 144.7, 146.4, 147.8, 149.6, 155.2.
3’-(4-Nitrophenyl)deschloroepibatidine (5f) Dihydrochloride
To a stirred solution of 9f (390 mg, 0.99 mmol) in CH2Cl2 (6 mL) at 0 °C was added trifluoroacetic acid (6 mL). After 2 h,the reaction mixture was poured into 150 mL of concentrated NH4OH-H2O (1:1) extracted with CH2Cl2 (5 × 70 mL), dried (Na2SO4), and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc-MeOH (2:1 to 2:3) to yield 0.25 g (77%) of 5f. 1H NMR (CDCl3) δ 1.52-1.86 (m, 5H), 1.95-2.02 (m, 1H), 2.88-2.93 (m, 1H), 3.61 (br, s, 1H), 3.84 (br, s, 1H), 7.78 (d, J = 8.7 Hz, 2H), 8.07 (br, s, 1H), 8.30 (d, J = 8.7, 2H), 8.61 (s, 1H), 8.70 (s, 1H); 13C NMR (CDCl3) δ 28.6, 29.9, 38.8, 43.5, 54.8, 61.2, 122.5, 126.3, 131.7, 132.3, 140.9, 143.0, 144.1, 145.7, 147.8.
The dihydrochloride salt had mp 215–216 °C; Anal (C17H19Cl2N3O2) C, H, N.
7-tert-Butoxycarbonyl-2-exo-[3-(3-nitrophenyl)-5-pyridinyl)]-7-azabicyclo[2.2.1]heptane (9g)
Using a procedure similar to that described for 9a, compound 8 (600 mg, 1.70 mmol) was coupled to 382 mg (3.60 mmol) of 3-nitrophenylboronic acid to give 0.53 g (79%) of 9g as a oil. 1H NMR (CDCl3) δ 1.44 (s, 9H), 1.49-1.71 (m, 2H), 1.90-1.95 (m, 3H), 2.04-2.13 (m, 1H), 3.00-3.05 (m, 1H), 4.32 (br, s, 1H), 4.44 (br, s, 1H), 7.67 (t, J = 7.8 Hz, 1H), 7.94 (br, s, 2H), 8.23-8.26 (m, 1H), 8.44 (s, br, 1H), 8.58 (s, br, 1H), 8.73 (s, br 1H); 13C NMR (CDCl3,) δ 28.3, 28.8, 29.8, 40.4, 45.5, 56.1, 61.9, 79.8, 121.9, 122.7, 130.1, 132.8, 133.1, 134.1, 139.8, 141.5, 146.0, 148.8, 149.0, 155.0.
3’-(3-Nitrophenyl)deschloroepibatidine (5g) Dihydrochloride
To a stirred solution of 9g (0.47 mg, 1.19 mmol) in CH2Cl2 (8 mL) at 0 °C was added trifluoroacetic acid (8 mL). After 2 h,the reaction mixture was poured into 300 mL of concentrated NH4OH-H2O (1:1), extracted with CH2Cl2 (5 × 50 mL), dried (Na2SO4), and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc-MeOH (2:1 to 1:1) to yield 0.25g (71%) of 5g as an oil. 1H NMR (CDCl3) δ 1.91-2.64 (m, 6H), 3.29-3.34 (m, 2H), 3.77-3.82 (m, 1H), 4.42-3.32 (m, 1H), 4.72-4.89 (m, 1H), 7.85-7.90 (m, 1H), 8.31-8.34 (m, 1H), 8.42-8.45 (m, 1H), 8.78-8.79 (m, 1H), 9.00 (br, 2, 2H), 9.23 (br, s, 1H); 13C NMR (CDCl3) δ 25.6, 27.6, 35.8, 42.8, 59.2, 62.4, 122.6, 124.5, 130.8, 133.9, 135.7, 137.0, 139.1, 140.3, 142.1, 143.4, 149.3.
The dihydrochloride salt had mp 248–250 °C; Anal (C17H19Cl2N3O2) C, H, N.
7-tert-Butoxycarbonyl-2-exo-[3-(4-aminophenyl)-5-pyridinyl)]-7-azabicyclo[2.2.1]heptane (10)
To a stirred solution of 9g (0.34 g, 0.86 mmol) in EtOH (3 mL), water (0.6 mL), and conc. HCl (0.1 mL), excess Fe powder was added in one portion over 1 min. After heating at 90 °C for 1 h, the reaction mixture was poured into 100 mL of NH4OH-H2O (1:1) solution, extracted with CH2Cl2 (5 × 50 mL), dried (Na2SO4), and concentrated. The residue was purified by silica gel chromatography eluting with hexanes-EtOAc (2:1 to 1:2) to yield 0.25 g (80%) of 10 as a solid. 1H NMR (CDCl3) δ 1.42 (s, 9H), 1.51-1.64 (m, 2H), 1.85-1.91 (m, 3H), 1.97-2.04 (m, 1H), 2.89-2.93 (m, 1H), 3.82 (br, s, 2H), 4.27 (br, s, 1H), 4.40 (br, s, 1H), 6.74 (d, J = 8.4 Hz, 2H), 7.38 (d, J = 8.4 Hz, 2H), 7.79 (br, s, 1H), 8.38 (br, s, 1H), 8.63 (br, s, 1H); 13C NMR (CDCl3) δ 28.7, 29.2, 30.3, 40.7, 45.9, 56.3, 62.3, 80.0, 115.8, 128.0, 128.4, 132.1, 136.7, 141.2, 145.9, 146.9, 147.2, 155.3.
3-(4-Aminophenyl)deschloroepibatidine (5h) Trihydrochloride
To a stirred solution of 10 (240 mg, 0.66 mmol) in CH2Cl2 (4 mL) at 0 °C was added trifluoroacetic acid (4 mL). After 2h, the reaction mixture was poured into 100 mL of concentrated NH4OH-H2O (1:1) solution, extracted with CH2Cl2 (5 × 50 mL), dried (Na2SO4), and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc-MeOH (2:1 to 1:1) to yield 0.17 g (95 %) of 5h. 1H NMR (CDCl3) δ 1.48-1.78 (m, 5H), 1.92-1.99 (m, 1H), 2.85-2.89 (m, 1H), 3.65 (br, s, 1H), 3.80 (br, s, 1H), 6.77 (d, J = 8.4 Hz, 2H), 7.40 (d, J = 8.4 Hz, 2H), 7.81 (s, 1H), 8.41 (s, 1H)), 8.61 (s, 1H); 13C NMR (CDCl3) δ 30.4, 31.7, 40.6, 45.9, 56.9, 63.2, 115.8, 128.3, 128.5, 132.5, 136.7, 142.1, 145.7, 147.1, 147.2.
The trihydrochloride salt had mp 238–240 °C; Anal (C17H21Cl3N3•1.5H2O) C, H, N.
7-tert-Butoxycarbonyl-2-exo-[5’-(3’-(3-nitrophenyl)-2’-aminopyridinyl)]-7-azabicyclo[2.2.1]heptane (12a)
To a resealable reaction tube under nitrogen was added 11 (600 mg, 1.6 mmol), Pd(OAc)2 (29.1 mg, 0.129 mmol), P(o-tolyl)3 (78.5 mg, 0.258 mmol), sodium carbonate (348 mg, 3.28 mmol), 3-nitrophenylboronic acid (411 mg, 2.47 mmol), degassed water (1.65 mL) and DME (8.2 mL). The reaction mixture was heated at 80°C overnight, cooled and poured into saturated sodium bicarbonate solution and extracted with ethyl acetate (3 × 20). The organic combination was dried (Na2SO4) and concentrated. The resulting residue was purified by silica gel column chromatography, eluting with hexane-ethyl acetate (1:2) to yield 0.56 g, (85%) of 12a as an oil. 1H NMR (CDCl3) δ 1.39 (s, 9H), 1.49-1.62 (m, 2H), 1.76-1.85 (br s, 3H), 1.94- 2.01 (m, 1H), 2.79-2.84 (m, 1H), 4.16 (br s, 1H), 4.36 (br s, 1H), 4.56 (br s, 1H), 7.40 (s, 1H), 7.61-7.66 (t, 1H), 7.81-7.84 (d, 1H), 7.99 (s, 1H), 8.21-8.24 (d, 1H), 8.33 (s, 1H)
3-(3-Nitrophenyl)epibatidine (13a)
To a solution of 12a (158 mg, 0.192 mmol) in concentrated hydrochloric acid (2 ml) was added sodium nitrite 473 mg (6.86 mmol) in portions over 30 min and stirring continued for 1 h at 0 °C, then to room temperature for additional 3 h. The mixture was poured into 100 mL solution of concentrated NH4OH-H2O (1:1), extracted with CHCl3 (3 × 20 mL), dried (Na2SO4), and concentrated. The residue was purified by silica gel column chromatography, eluting with CMA80-ethyl acetate (1:3) to yield 80 mg (63%) of 13a as an oil. 1H NMR (CDCl3) δ 1.54-1.71 (m, 5H), 1.92-2.02 (dd, 1H), 2.80-2.85 (dd, 1H), 3.62 (br s, 1H), 3.81 (br s, 1H), 7.64-7.67 (t, 1H), 7.80-7.87 (m, 2H), 8.27-8.37 (m, 3H).
3’-(3-Aminophenyl)deschloroepibatidine (5i) Trihydrochloride
Compound 13a, (70 mg, 0.211 mmol), 10% Pd/C (110 mg), and methanol (20 m1) were placed into a Fisher-Porter tube under nitrogen. The tube was evacuated, refilled with hydrogen gas (50 psi). The reaction was allowed to shake for 7 h, filtered through a celite pad and the solvent was removed. The resulting residue was purified by flash chromatography eluting with CMA80-ethyl acetate (1:1) to yield 45 mg (80%) of 5i. 1H NMR (CD3OD) δ 1.50-1.68 (m, 5H), 1.91-1.98 (dd, 1H), 2.89-2.94 (dd, 1H), 3.57 (br s, 1H), 3.66 (br s, 1H), 6.64-6.67 (d, 1H), 6.84-6.90 (m, 2H), 7.07-7.12 (t, 1H), 7.85 (s, 1H), 8.31 (s, 1H), 8.44 (s, 1H); 13C NMR (CD3OD) δ 30.2, 32.1, 41.3, 46.8, 58.3, 63.9, 115.2, 116.8, 118.2, 131.3, 135.2, 139.2, 139.9, 143.6, 146.3, 148.2, 150.1.
The trihydrochloride salt had mp 209 °C (dec); Anal (C17H22Cl3N3•1.25H 2O) C, H, N.
7-tert-Butoxycarbonyl-2-exo-[2-amino-3-(3-methoxyphenyl)-5-pyridinyl]-7-azabicyclo[2.2.1]heptane (12b)
Using a procedure similar to that described for 12a, (997 mg, 2.7 mmol) of 11 was coupled to 3-methoxyphenylboronic acid (649 mg, 4.3 mmol) to give 959 mg (90%) of 12b as a colorless oil. 1H NMR (CDCl3) δ 1.38 (s, 9H), 1.45-1.65 (m, 3H), 1.7-20 (m, 3H), 2.78 (dd, J = 4.9, 7.7 Hz, 1H), 3.82 (s, 3H), 4.16 (s, 1H), 4.34 (s, 1H), 4.66 (s, 2H), 6.85-7.07 (m, 3H), 7.28-7.40 (m, 2H), 7.92 (s, 1H); 13C NMR (CDCl3) δ 28.1 (3C), 28.6, 29.7, 40.1, 44.8, 55.1, 55.7, 62.1, 79.3, 113.1, 114.1, 120.8, 121.4, 129.9, 131.5, 136.5, 139.5, 145.5, 154.3, 154.9, 159.8.
3’-(3-Methoxyphenyl)epibatidine (13b)
Using a procedure similar to that described for 13a, (167 mg, 0.422 mmol) of 12b was converted to 83 mg (63%) of 13b as a colorless oil. 1H NMR (CDCl3) δ 1.4-1.8 (m, 4H), 1.8-2.05 (m, 2H), 2.81 (dd, J = 5.0, 8.8 Hz, 1H), 3.62 (br s,1H), 3,78 (br s, 1H), 3.84 (s, 3H), 6.8-7.1 (m, 3H), 7,31 (t, J = 7.9 Hz, 1H), 7.76 (s, 1H), 8.34 (s,1H); 13C NMR (CDCl3) δ 30.0, 31.3, 40.2, 44.5, 55.3, 56.4, 62.7, 113.5, 115.1, 121.7, 129.2, 136.1, 138.5, 139.0, 141.2, 146.9, 147.4, 159.2.
3-(3-Methoxyphenyl)deschloroepibatidine (5j) Dihydrochloride
Dechlorohydrogenation of 13b (80 mg, 0.254 mmol) using conditions similar to that described for 12b yielded 28 mg (39%) of 5j as a colorless oil. 1H NMR (CDCl3) δ 1.45-1.80 (m, 4H), 1.86 (s, 1H), 1.94 (dd, J = 8.9, 12.3 Hz, 1H), 2.87 (dd, J = 5.1, 8.7 Hz, 1H), 3.65 (br s, 1H), 3.80 (br s, 1H), 3.86 (s, 3H), 6.9-7.5 (m, 4H), 7.88 (s, 1H), 8.50 (s, 1H), 8.64 (s, 1H); 13C NMR (CDCl 3) δ 30.1, 31.3, 40.3, 45.5, 55.4, 56.5, 62.8, 113.2, 113.2, 119.7 (2C), 130.0, 133.1, 136.2, 139.7, 141.8, 146.0, 148.0
The dihydrochloride salt had mp 242 °C (dec); Anal (C18H22Cl2N2O•H2O) C, H, N.
Supplementary Material
Elemental analysis data. This material is available free of charge via the internet at http://pubs.acs.org.
Acknowledgments
This research was supported by the National Institute on Drug Abuse, Grant DA12001.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ezzati M, Lopez AD. Lancet. 2003;362:847–852. doi: 10.1016/S0140-6736(03)14338-3. [DOI] [PubMed] [Google Scholar]
- 2.Henningfield JE, Fant RV, Buchhalter AR, Stitzer ML. CA Cancer J Clin. 2005;55:281–299. doi: 10.3322/canjclin.55.5.281. [DOI] [PubMed] [Google Scholar]
- 3.Keating GM, Siddiqui MAA. CNS Drugs. 2006;20:945–960. doi: 10.2165/00023210-200620110-00007. [DOI] [PubMed] [Google Scholar]
- 4.Niaura R, Jones C, Kirkpatrick P. Nat Rev Drug Discov. 2006;5:537–538. doi: 10.1038/nrd2088. [DOI] [PubMed] [Google Scholar]
- 5.Carroll FI. Bioorg & Med Chem Ltrs. 2004;14:1889–1896. doi: 10.1016/j.bmcl.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 6.Carroll FI, Brieaddy LE, Navarro HA, Damaj MI, Martin BR. J Med Chem. 2005;48:7491–5. doi: 10.1021/jm058243v. [DOI] [PubMed] [Google Scholar]
- 7.Carroll FI, Lee JR, Navarro HA, Brieaddy LE, Abraham P, Damaj MI, Martin BR. J Med Chem. 2001;44:4039–4041. doi: 10.1021/jm015561v. [DOI] [PubMed] [Google Scholar]
- 8.Carroll FI, Lee JR, Navarro HA, Ma W, Brieaddy LE, Abraham P, Damaj MI, Martin BR. J Med Chem. 2002;45:4755–4761. doi: 10.1021/jm0202268. [DOI] [PubMed] [Google Scholar]
- 9.Carroll FI, Liang F, Navarro HA, Brieaddy LE, Abraham P, Damaj MI, Martin BR. J Med Chem. 2001;44:2229–2237. doi: 10.1021/jm0100178. [DOI] [PubMed] [Google Scholar]
- 10.Carroll FI, Ma W, Yokota Y, Lee JR, Brieaddy LE, Navarro HA, Damaj MI, Martin BR. J Med Chem. 2005;48:1221–1228. doi: 10.1021/jm040160b. [DOI] [PubMed] [Google Scholar]
- 11.Carroll FI, Ware R, Brieaddy LE, Navarro HA, Damaj MI, Martin BR. J Med Chem. 2004;47:4588–4594. doi: 10.1021/jm040078g. [DOI] [PubMed] [Google Scholar]
- 12.Abdrakhmanova GR, Damaj MI, Carroll FI, Martin BR. Mol Pharmacol. 2006;69:1945–1952. doi: 10.1124/mol.105.021782. [DOI] [PubMed] [Google Scholar]
- 13.Damaj MI, Glassco W, Aceto MD, Martin BR. J Pharmacol Exp Ther. 1999;291:390–398. [PubMed] [Google Scholar]
- 14.D’Amour FE, Smith DL. J Pharmacol Exp Ther. 1941;72:74–79. [Google Scholar]
- 15.Eddy NB, Leimbach D. J Pharmacol Exp Ther. 1953;107:385–939. [PubMed] [Google Scholar]
- 16.Atwell L, Jacobson AE. Laboratory Animal. 1978;7:42–47. [Google Scholar]
- 17.Miyaura N, Suzuki A. Chem Commun. 1979:866. [Google Scholar]
- 18.Miyaura N, Yamada K, Suzuki A. Tetrahedron Lett. 1979;20:3437–3440. [Google Scholar]
- 19.Rollema H, Chambers LK, Coe JW, Glowa J, Hurst RS, Lebel LA, Lu Y, Mansbach RS, Mather RJ, Rovetti CC, Sands SB, Schaeffer E, Schulz DW, Tingley FD, 3, Williams KE. Neuropharmacology. 2007;52:985–994. doi: 10.1016/j.neuropharm.2006.10.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Elemental analysis data. This material is available free of charge via the internet at http://pubs.acs.org.