

Abstract
This report describes a new experimental model to evaluate the effect of a recurrent systemic inflammatory challenge, after cerebral hypoxia-ischemia in immature mice, on the progression of brain injury. Treatment with a low dose of lipopolysaccharide (E. coli O55:B5, 0.2 mg/kg for 3 days, then 0.1 mg/kg for 2 days) daily for 5 days after unilateral cerebral hypoxia-ischemia (right carotid ligation followed by 35 min in 10% O2) in 10-day-old mice resulted in increased right forebrain tissue damage (35.6% reduction in right hemisphere volume compared to 20.6% reduction in saline-injected controls), in bilateral reductions in corpus callosum area (by 12%) and myelin basic protein immunostaining (by 19%), and in suppression of injury-related right subventricular zone cellular proliferation. The post-hypoxic-ischemic lipopolysaccharide regimen that amplified brain injury was not associated with increased mortality, nor with changes in body temperature, weight gain or blood glucose concentrations. The results of the present study demonstrate that systemic inflammation influences the evolution of tissue injury after neonatal cerebral hypoxia-ischemia and may also impair potential recovery mechanisms.
There is now substantial epidemiologic and experimental evidence that pre-existing intrauterine inflammation can potentiate prenatal hypoxic-ischemic (HI) brain injury (Yoon et al., 1997, Nelson et al., 1998, Wu et al., 2003, Eklind et al., 2001, Lehnardt et al., 2003, Yang et al., 2004). However, the postnatal environment of critically ill newborns is also fraught with risks of pro-inflammatory challenges. Such systemic inflammatory stimuli include sepsis, pneumonia, sustained acute lung injury, necrotizing enterocolitis and its complications, and major abdominal or thoracic surgery. Given the critical role of inflammatory mediators in the pathogenesis of experimental neonatal hypoxic-ischemic brain injury (Barks and Silverstein, 2002), and the immaturity of the blood-brain barrier (Risau and Wolburg, 1990, Stolp et al., 2005), we hypothesized that systemic inflammation might increase the severity of brain injury after an acute cerebral hypoxic-ischemic insult. The impact of post-hypoxic-ischemic systemic inflammation on the evolution of neonatal brain injury has not previously been evaluated in an experimental model.
This report describes a new neonatal mouse model in which to study the impact of postnatal systemic inflammation on the evolution of hypoxic-ischemic brain injury. Hypoxic-ischemic brain injury was elicited using a well-established model of carotid ligation followed by hypoxia exposure in 10-day-old (P10) mice (Liu et al., 1999), which is a modification of the Rice-Vannucci model originally described in immature rats (Rice et al., 1981). We utilized a regimen of five sequential daily injections of low doses of lipopolysaccharide (LPS) to elicit systemic inflammation. In contrast with previous studies, in which single doses of LPS were administered prior to a CNS insult to model the effects of pre- or perinatal systemic inflammation on HI- or seizure-induced injury to the immature brain (Eklind et al., 2001, Lehnardt et al., 2003, Sankar et al., 2007), in our model, the first LPS injection was administered at two hours after hypoxic-ischemic lesioning.
Outcomes were compared in lesioned animals that received 5 sequential daily injections of LPS or of equal volumes of saline. We examined the effects of LPS administration on body temperature, blood glucose, weight gain, survival, and brain injury. Based on recent evidence that inflammation disrupts endogenous stem cell reparative responses after stroke in adult rats (Hoehn et al., 2005), we also evaluated forebrain subventricular zone (SVZ) proliferation (Plane et al., 2004).
Methods
Lipopolysaccharide Administration Protocol
For intra-peritoneal (i.p.) injections in P10 mice, we used a 25 gauge needle attached to a 250 μl Hamilton syringe, and injected either 100 or 50 μl/10 gm body weight. In these experiments we used LPS from E. coli (O55:B5, Sigma, St. Louis, MO), dissolved (0.02 mg/ml) in 0.9% w/v NaCl in sterile water. A broad range of LPS doses, administered by intra-peritoneal, intravenous, or intra-cerebral injection, have been used in rodent models to elicit inflammation (Ahmed et al., 2000, Eklind et al., 2001, Lehnardt et al., 2003, Soucy et al., 2005, Becker et al., 2005, Herber et al., 2006). In order to establish a post-hypoxia-ischemia LPS administration protocol, we initially evaluated high doses of LPS in unlesioned P10 mice. Systemic injections of LPS (0.5–1 mg/kg) elicited temperature instability and poor weight gain; if LPS injection was followed by 30–40 min exposure to 10% oxygen, there was up to 50% mortality. Subsequently, we found that in mice that received five sequential daily LPS injections (200 microgram/kg/d for 3 consecutive days beginning on P10, and then 100 microgram/kg/day for 2 additional days), there were no overt adverse effects.
Using this dosing regimen, we measured body temperature [analog esophageal temperature, measured to the nearest half-degree (YSI Telethermometer 43TA with 554 probe, Yellow Springs Instruments, Yellow Springs, OH, USA)] 3 times daily (immediately before, and again 2 and 4 hours after each LPS injection), for 5 days, in LPS-treated animals and concurrently in non-injected controls (n=5–6/group). In additional animals, serial blood glucose values (Ascensia Elite blood glucose meter, Bayer HealthCare, Tarrytown, NY) were obtained by ear puncture on P10 immediately prior to and again 90 minutes after LPS injection, and on P11 through P14, daily at 90 minutes after LPS injections.
Hypoxia-Ischemia
Three groups of experiments were performed. Each group of experiments included LPS-treated and saline-injected control animals (CD-1 mice, Charles River, Portage MI, N = 37). Within each experiment, animals were allocated evenly into the two treatments, with similar sex distribution in each group.
All animals underwent right carotid ligation on P10, under isoflurane anesthesia (3% for duration of procedure, 2–3 min.). Animals recovered in an incubator at 36.5 ºC air temperature for 90 min, and then were placed in glass jars, partly immersed in a 36.5 ºC water bath, through which warmed 10% oxygen (balance nitrogen) flowed at a constant rate, for 35 min. We selected a duration of hypoxia exposure that we expected would produce damage of mild-moderate severity (20–35% ipsilateral hemisphere volume loss), to permit detection of either potentiation or attenuation of injury by LPS.
After the end of hypoxia exposure, mice recovered in an incubator at 36.5 °C air temperature for 30 min, and were then returned to the dams. Mice were weighed prior to surgery on P10, daily from P11-14 and again on P17.
Following hypoxia-ischemia, mice, stratified by gender, were randomly allocated to either the LPS injection group (HI + LPS) or the saline (NS) control group (HI + NS). Animals received the first LPS (N = 19) or saline (N = 18) injection at 2 h after the end of hypoxia exposure; animals received four additional daily i.p. injections of LPS or saline from P11 to P14.
In the third experiment, mice received injections of the S-phase cell proliferation marker bromodeoxyuridine (BrdU, Roche Applied Science, Indianapolis, IN, USA) 100 mg/kg i.p. (in sterile PBS) once daily for 3 consecutive days (P15-17) (n = 7/group).
Histopathology
On P17, mice were deeply anesthetized with chloral hydrate (500 mg/kg) and perfusion-fixed (4% paraformaldehyde). Serial 30 μm coronal sections were sectioned on a sliding microtome and stored in cryoprotectant solution (sucrose 30% w/v and ethylene glycol 30% v/v in 0.05 M phosphate buffer pH 7.2) at −20ºC.
To estimate the severity of tissue damage in each brain, a series of regularly spaced sections, from the anterior to the posterior genu of the corpus callosum was mounted on slides, and stained with cresyl violet. Bilateral cerebral hemisphere volumes were approximated by summing bilateral hemisphere areas of intact (i.e. Nissl-stained) tissue (outlined with NIH Image) and multiplying by the distance between sections in at least 6 sections/brain.
Immunohistochemistry and Stereology
To evaluate HI white matter injury, in each brain, a series of regularly spaced coronal sections was stained for myelin basic protein (MBP) using a modification to previously published methods (Liu et al., 2002). Briefly, sections were mounted on slides, dried, rinsed in Tris-buffered saline (TBS, pH 7.6), blocked in F(ab′)2 fragment donkey anti-mouse IgG (1:400, Jackson ImmunoResearch, West Grove, PA, USA), followed by 20% normal horse serum (Vector, Burlingame, CA, USA) and then incubated overnight at 4ºC with primary antibody (1:800, anti-MBP mouse monoclonal SMI-94, Sternberger, Lutherville, MD, USA). After incubation with secondary antibody (1:200, biotinylated horse anti-mouse, Vector), signal was amplified with a Vector ABC kit and then developed with stable diaminobenzidine (Research Genetics, Huntsville, AL, USA). In negative control sections mouse IgG was substituted for the primary antibody.
The extent and intensity of MBP immunostaining in bilateral corpus callosum was assessed semi-quantitatively using NIH Image, using a modification of previously described methods (Liu et al., 2002). First, bilateral hemispheres were manually outlined in coronal sections from the level of the anterior to the posterior genu of the corpus callosum, in unaltered grayscale TIFF images. The images were then segmented and binarized, and total black pixels per hemisphere were counted. Next, to avoid the possible confounding effect of artifactual immunostaining in the rim of cystic cavities, a similar process was carried out in medial corpus callosum, which was not included in the area of overt tissue damage. Bilateral corpus callosum was manually outlined in sections at the level of striatum and SVZ (see Figure 3), in unaltered grayscale TIFF images. Corpus callosum area was measured bilaterally; the images were then segmented and binarized, and total black pixels per side of the corpus callosum were counted.
Figure 3. Effect of post-hypoxic-ischemic LPS regimen on myelin basic protein.
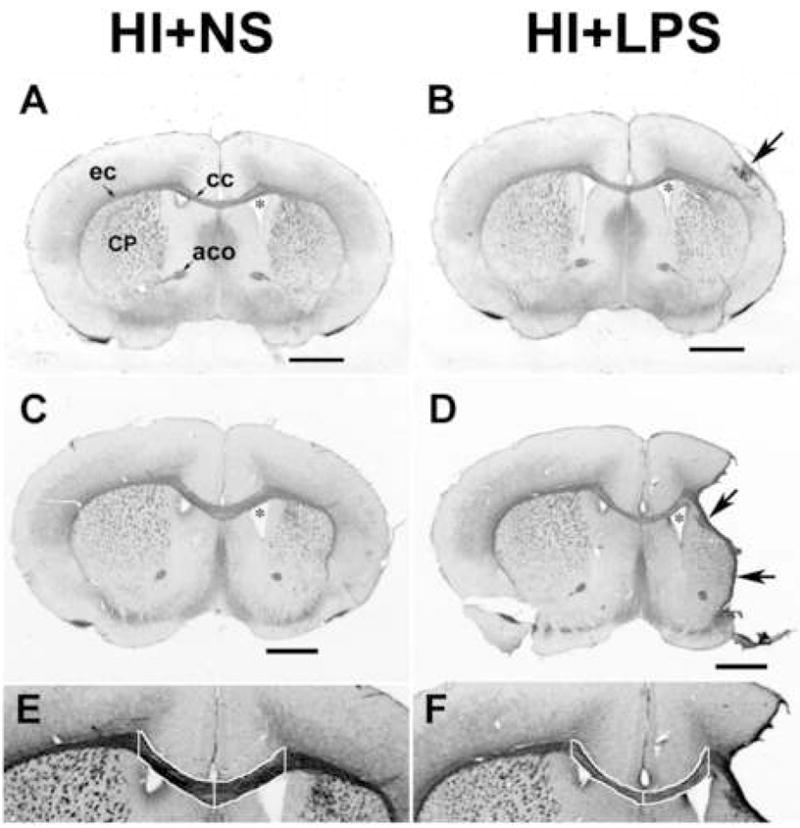
The same spectrum of pathology as in Figure 2 is illustrated in this montage of coronal sections immunostained for myelin basic protein (MBP, see Methods); each section was taken adjacent to the corresponding section in Figure 2. In addition to the ipsilateral atrophy, cystic infarction and compensatory right lateral ventriculomegaly (*), there is localized increased staining, suggestive of dysmelination, in more severely affected areas of cortex (arrow, B). In D, typical of animals with cystic infarction, there is a rim of non-specific staining lining the cavity (arrows). Panels E and F, which are corpus callosum enlargments from panels C and D respectively, illustrate the method for outlining (white lines) the corpus callosum for area measurement and density segmentation with pixel-counting. Note the reduced intensity of MBP staining in the outlined corpus callosum with post-HI LPS administration, in F, compared to E, which was stained concurrently. (Scale bar = 1 mm. Abbreviations: cc – corpus callosum; ec – external capsule; aco –anterior commisure; CP – striatum, or caudate –putamen)
Animals in the third experiment received BrdU in order to assess SVZ cell proliferation. In this neonatal HI brain injury model, there is ipsilateral SVZ proliferation at 1–2 weeks after lesioning, and, in animals with mild-moderate injury that does not directly damage the SVZ, there is a direct relationship between the severity of tissue damage and the magnitude of SVZ proliferation (Plane et al., 2004).
BrdU-labeled cells were identified immunohistochemically in a series of 4–7 regularly spaced sections through the dorsolateral SVZ and striatum, using a modification of previously described methods (Plane et al., 2004). Briefly, sections were mounted on slides, dried, rinsed in Tris-buffered saline, incubated in 2N HCl at 37ºC for 30 min, neutralized in 0.1M boric acid for 10 min., rinsed, blocked with 20% normal horse serum (Vector), and incubated overnight at 4ºC with primary antibody (1:1,000 mouse monoclonal anti mouse BrdU, Roche Applied Science, Indianapolis, IN, USA). After incubation with secondary antibody (1:200, biotinylated horse anti-mouse, Vector), signal was amplified with a Vector ABC kit and then developed with stable diaminobenzidine (Research Genetics, Huntsville, AL, USA). In negative control sections mouse IgG was substituted for the primary antibody. Labeled cells in bilateral SVZ (e.g. see outlined areas, Figure 4 A) were counted stereologically at 100X using the Optical Disector method, and bilateral SVZ volumes were estimated from the same series of sections at 2.5X using the Cavalieri point-counting method (Stereologer, Stereology Resource Center, Chester, MD, USA).
Figure 4. Bilateral SVZ BrdU immunostaining at level of striatum.
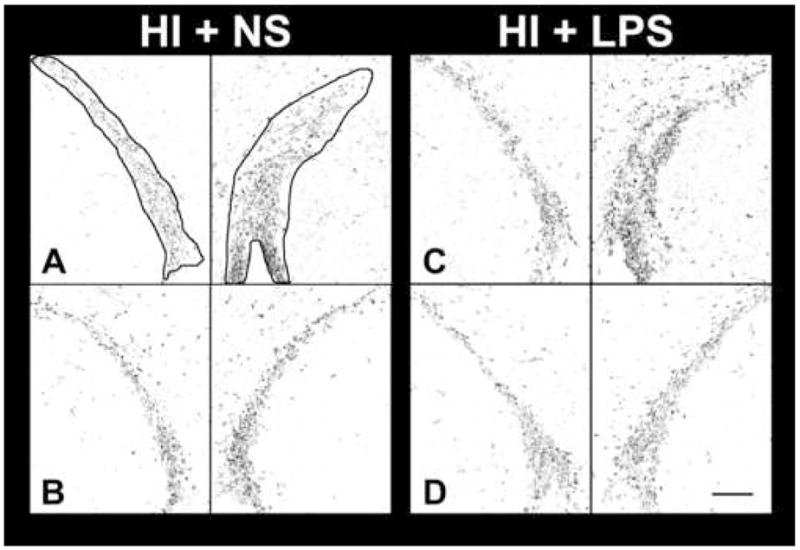
Two representative sections through bilateral SVZ are presented from each group (A,B: HI + NS; C,D: HI + LPS), from experimental group 3. Mice underwent unilateral cerebral hypoxia-ischemia (see Methods) on P10, followed by 5 daily injections of 0.9% NaCl or LPS beginning 2 hours after the end of HI (see Methods). All received daily BrdU injections of 100 mg/kg from P15-P17, and brains were perfusion fixed on P17. The orientation of each photomicrograph was adjusted such that the SVZ extended diagonally across each panel. Bilateral SVZ, as used for stereology, is outlined in A. There is no obvious difference in SVZ size or cell density between the two groups; this observation underscores the importance of systematic cell counting using stereologic methods. Bilateral SVZ BrdU-labeled cell counts (mean ± SD) were obtained using the Optical Disector method, from both the HI + normal saline controls (HI + NS: left 3457 ± 801; right 5514 ± 1940) and from mice receiving LPS injections post-HI (HI + LPS: left 4675 ± 1309; right 6146 ± 2560). The right SVZ BrdU-immunoreactive cell count was increased by 60%, relative to left, in the HI + NS group (p < 0.05, paired t-test), but the left-right difference in SVZ BrdU+ cell count was not significant in the HI + LPS treated mice. When comparing right SVZ cell counts between the HI + NS and the HI + LPS groups, there was no increase in right SVZ cell count in the HI + LPS animals, despite the fact that they had significantly worse brain injury (see Results and Table 3). (Scale bar = 100 μm)
Data Analysis
Data were analyzed using the microcomputer-based statistics programs Statview (SAS Institute, Cary, NC, USA), Systat (Systat Software Inc., San Jose, CA, USA), SAS (SAS Institute Cary, NC, USA), and InStat (GraphPad Software, San Diego, CA, USA).
Between-treatment differences in blood glucoses and body temperatures, and in serial weights of lesioned animals were evaluated by repeated-measures analysis of variance (ANOVA). Between-treatment differences in survival of lesioned animals were evaluated by Fisher exact test. Percent difference in cerebral hemisphere volumes was calculated as an index of severity of ipsilateral tissue loss (i.e. % damage) using the formula: %damage = 100*(L-R)/L. Between-treatment (i.e. HI + LPS vs. HI + NS) differences in hemisphere volumes and %damage were first evaluated by unpaired t-test. To determine whether there were experiment-related or gender effects on either HI damage severity or on the effect of post-HI LPS administration, differences were also evaluated by ANOVA factoring treatment (i.e. HI+LPS vs. HI+NS) and one or both of sex and experimental group.
The intensity of MBP immunostaining was estimated by density segmentation; measurements were obtained from the entire cerebral hemispheres and from medial corpus callosum bilaterally. LPS- and control animals in each experiment were assayed concurrently. Left-right differences in MBP immunostaining were first evaluated in each group by paired t-tests. Since MBP immunohistochemistry was carried out in batches, differences in immunostaining intensity were anticipated. Therefore, to compare hemisphere and callosal MBP pixel counts, treatment, side and experimental groups were factored as variables, using 3-way ANOVA.
Between-side differences in BrdU-labeled cell numbers within each group (i.e. HI + NS and HI + LPS) were evaluated by paired t-tests. Between-treatment differences in SVZ BrdU-labeled cell numbers were evaluated by Mann-Whitney test.
Results
Post-hypoxic-ischemic mortality did not differ between the HI + normal saline controls and the HI + LPS groups (2/18 vs. 2/19). Animals died between P14 and P17 in the controls and between P10 and P17 in the HI + LPS group (see Table 1).
Table 1.
Serial Body Weights and Survival During and After Post-Hypoxic-Ischemic LPS Regimen.
| P10 (pre-HI) | P11 | P12 | P13 | P14 | P17 | ||
|---|---|---|---|---|---|---|---|
| Weight (g) | HI + NS | 5.2 ± 0.5 | 5.4 ± 0.6 | 5.8 ± 0.7 | 5.9 ± 0.9 | 6.4 ±1.2 | 8.0 ± 0.5 |
| (mean ± SD) | HI + LPS | 5.3 ± 0.6 | 5.3 ± 0.7 | 5.7 ± 0.8 | 6.0 ± 1.1 | 6.5 ±1.3 | 8.0 ± 0.6 |
| N | HI + NS | 18 | 18 | 18 | 18 | 18 | 16‡ |
| HI + LPS | 19 | 18 | 18 | 18 | 18 | 15†‡ |
2 animals in the HI + LPS group that were exhibiting growth failure were euthanized on P14, according to IACUC guidelines.
3 animals, 2 in the HI + NS and 1 in the HI + LPS groups, died between P14 and P17.
Overall weight gain did not differ between the groups (see Table 1); in both groups, weight gain plateaued in the first day after surgery, and then increased. However, two animals in the LPS-exposed group began to lose weight later and they were euthanized on P14, according to our IACUC guidelines; both had complete right (ipsilateral to carotid ligation) hemisphere infarctions that extended to involve the medial portions of the left hemisphere. Brains from these two animals were excluded from subsequent analyses.
In preliminary experiments to assess the systemic effects of LPS, serial esophageal temperatures did not differ from concurrently measured temperatures in untreated P10-14 littermates (Figure 1). Similarly, there was no difference between LPS-and saline-injected controls in daily blood glucose measurements (obtained at 90 min after the injections, Table 2).
Figure 1. Temperature measurement after serial LPS injections in immature mice.

Starting on postnatal day 10 mice received daily injections of either E. coli (O55:B5) lipopolysaccharide (LPS, solid squares, n=6) 0.2 mg/kg in 0.9% NaCl i.p. daily for 3 days, then 0.1 mg/kg for 2 days, or 0.9% NaCl (NS, open circles, n=5) 0.1 ml i.p. daily for 3 days, then 0.05 ml for 2 days. Esophageal temperature was measured to the nearest 0.5 °C with an analog telethermometer (see Methods) immediately prior to injection on day 1 (d1) of the injection course (e.g. pre d1 on x-axis), and 2 and 4 hours post-injection (e.g. 2h d1 and 4h d1, respectively), daily throughout the 5-day injection course. Error bars are ± 1 standard deviation about the mean values.
Table 2.
LPS administration protocol does not alter blood glucose concentrations in immature mice
| Mean blood glucose (mg/dl, ± SD) ‡ | ||||||
|---|---|---|---|---|---|---|
| P10 - Pre | P10 - Post | P11 | P12 | P13 | P14 | |
| NS* (n=5) | 78 ± 31 | 71 ± 16 | 68 ± 30 | 73 ±24 | 80 ± 21 | 84 ± 23 |
| LPS† (n=6) | 58 ± 14 | 81 ± 26 | 73 ± 13 | 74 ± 25 | 70 ± 15 | 93 ± 31 |
NS: 0.9% NaCl 0.1 ml i.p. daily for 3 days, then 0.05 ml for 2 days, starting on postnatal day 10 (P10).
LPS: E. coli lipopolysaccharide (LPS, O55:B5) 0.2 mg/kg in 0.9% NaCl i.p. daily for 3 days, then 0.1 mg/kg for 2 days, starting on P10.
On P10, blood glucose was measured (see Methods) immediately prior to LPS injection (P10 pre), and daily at 90 min. post-injection from P10-P14.
There was substantial inter- and intra-group variation in the severity of damage amongst the HI + normal saline (HI + NS) controls (Table 3), as is commonly observed in this model. Typical neuropathological findings in these controls included ipsilateral (right) cortical thinning, striatal atrophy and pallor, (Figure 2, panels A and C), hippocampal atrophy, and in 4 of 16 cases, cortical infarction with either cyst formation or marked cortical thinning with an underlying cavity. There was a mean 20.6 % reduction in the right hemisphere volume of controls (i.e. “% Damage”), and the extent of damage was similar in the 3 HI + NS control groups that underwent surgery on different dates. Administration of a 5-day course of LPS after HI resulted in increased severity of hypoxic-ischemic damage. There was a mean 35.6% reduction in right hemisphere volume in the HI + LPS group (p<0.01, unpaired t-test, comparing % Damage in HI + LPS to HI +NS, see Table 3). In 11/15 brains there was ipsilateral cortical infarction with cystic evolution (see Figure 2, panel D) or marked cortical thinning, and in some cases also striatal cystic infarction. Right hemisphere volumes were smaller in the HI + LPS group compared to the HI + NS group (p<0.01 unpaired t-test, see Table 3). The incidence of cortical infarction with cavitation was greater in the LPS-treated group (HI + NS 4/16 vs. HI + LPS 11/15, p<0.05 Fisher’s exact test). There was no influence of gender on HI damage severity, or on the potentiation of HI damage by LPS (2-way ANOVA factoring treatment and gender). There was a difference in left hemisphere volumes among the three groups of experiments (p < 0.005 ANOVA, for both the HI + NS controls and the HI + LPS group, Table 3) but the effects of post-HI LPS on damage severity and on right hemisphere volumes were consistent across experiments. Mean left hemisphere volumes did not differ between the HI + LPS and the HI + NS groups.
Table 3.
Post-Hypoxic-Ischemic LPS Administration Amplifies Unilateral Hypoxic-Ischemic Damage
| Experiment | Number | Left Hemisphere Volume (mm3, mean ± SD) * | Right Hemisphere Volume (mm3, mean ± SD) * | % Damage in right hemisphere volume, vs. left (mean ± SD) * | ||||
|---|---|---|---|---|---|---|---|---|
| HI + NS† | HI + LPS‡ | HI + NS || | HI + LPS || | HI + NS | HI + LPS | HI + NS | HI + LPS | |
| Group 1 | 6 | 5 | 33.3 ± 1.5 | 33.3 ± 1.9 | 28.5 ± 1.9 | 21.8 ± 7.4§ | 14.2 ± 5.7 | 35.4 ± 18.6§ |
| Group 2 | 4 | 3 | 43.0 ± 1.1 | 39.9 ± 6.0 | 30.7 ± 4.8 | 21.6 ± 6.7§ | 28.4 ± 12.5 | 46.7 ± 10.7§ |
| Group 3 | 6 | 7 | 30.3 ± 3.2 | 28.6 ± 3.1 | 23.9 ± 5.4 | 19.6 ± 5.1§ | 21.9 ± 10.4 | 31.0 ± 19.8§ |
| Total | 16 | 15 | 34.6 ± 5.6 | 32.5 ± 5.5 | 27.3 ± 4.9 | 20.7 ± 5.9¶ | 20.6 ± 10.6 | 35.6 ± 17.9¶ |
Hemisphere volume was determined by measuring cross-sectional hemisphere areas of intact cresyl violet stained tissue from regularly spaced coronal sections, from the level of the anterior genu to the posterior genu of the corpus callosum, and then multiplying the sum of the areas by the distance between sections. Percent decrease in right hemisphere volume (relative to left) was calculated using the formula %decrease = 100*(left-right)/left. Data presented are means ± standard deviation (SD).
HI + NS: Right carotid ligation followed by 35 min. in 10% O2 on P10. 0.9% NaCl 0.1 ml i.p. daily for 3 days, then 0.05 ml for 2 days, starting 2 h after end of 10% O2.
HI + LPS: Right carotid ligation followed by 35 min. in 10% O2 on P10. LPS 0.2 mg/kg in 0.9% NaCl i.p. daily for 3 days, then 0.1 mg/kg for 2 days, starting 2 h after end of 10% O2.
p<0.05, ANOVA, factoring treatment and experiment, comparing HI + LPS to HI + NS among all experiments
p<0.01, unpaired t-test, comparing HI + LPS to HI + NS.
p <0.005, ANOVA, mean left hemisphere volumes differed among the 3 groups of experiments (Fisher PLSD post-hoc test).
Figure 2. Serial LPS dosing increases damage due to prior hypoxic-ischemic insult.
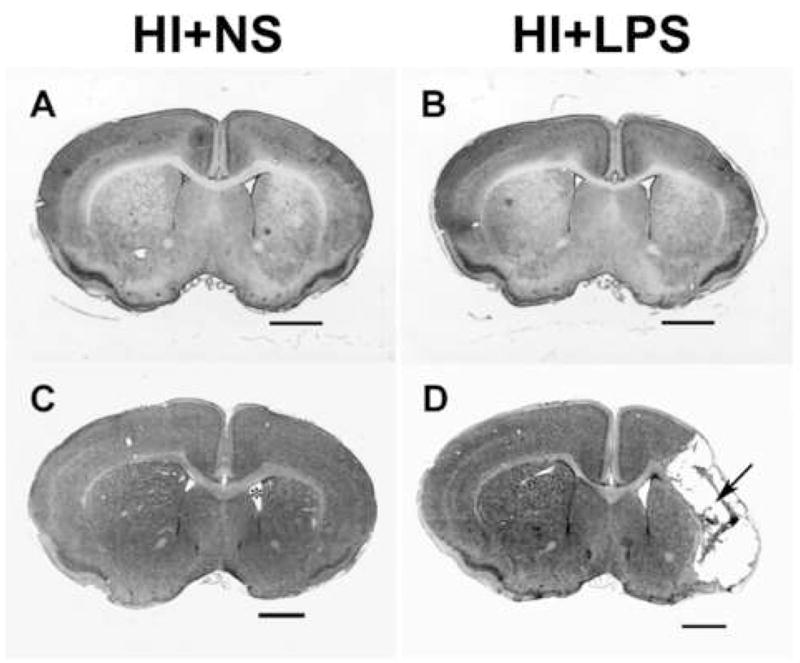
The spectrum of hypoxic-ischemic neuropathology at the level of striatum is illustrated in cresyl violet stained coronal sections of P17 mice that underwent HI on P10 and then received either normal saline injections for 5 days post-HI (A, C) or daily injections of LPS (see Methods) for 5 days post-HI (B, D). At the milder end of the injury spectrum in both groups (A, B) there is subtle right hemisphere atrophy with striatal shrinkage and pallor, and cortical thinning. At the more severe end of the spectrum (C,D), there is readily evident right hemisphere atrophy, striatal atrophy, and compensatory lateral ventriculomegaly (* in C), and, more commonly in the HI + LPS group (see Results), cystic cortical infarction (arrow in D). (Scale bar = 1 mm)
In P17 mice, MBP immunostaining is most intense in the corpus callosum, the external capsule, the anterior commisure, and in the myelinated bundles in the striatum, and myelination is beginning to extend radially into the cortex (see Figure 3). In animals with mild HI injury (see Figure 3A and C) there was reduced myelin staining in the atrophied right striatum and disruption of the radial myelination pattern in the cortex. In animals with moderate (see Figure 3 B) damage there were, in addition, foci of intense myelin staining in more thinned areas of cortex. In animals with cystic infarction, which was more prevalent in the HI + LPS group, there was often a narrow rim of intense non-specific staining on the lesion edge (see Figure 3 D). This artifactual staining, which was most pronounced in animals with more severe damage, confounded quantitative analysis of hemisphere optical densities (see below). As has been previously reported in this HI model in immature rats and mice (Liu et al., 2002, Hedtjarn et al., 2005), right hemisphere MBP immunostaining was reduced in both the HI + NS group (black pixels/hemisphere, mean ± SD, Left 7631 ± 2494, Right 6404 ± 2014, p<0.001, paired t-test; mean 14% decrease) and in the HI + LPS group (Left 7335 ± 2683, Right 5399 ± 2216, p<0.005, paired t-test; mean 23% decrease). The non-specific staining in the many severely affected animals in the HI + LPS group posed a challenge in quantitative comparison of MBP immunostaining staining between the HI + NS and HI + LPS groups. To avoid this potentially confounding effect we focused on quantitative analysis of MBP immunostaining in the corpus callosum (at the level of the striatum); this region, which is outside the typical lesion territory, is well-myelinated at this age and is an area of post-HI oligodendrogliogenesis (Zaidi et al., 2004). In the medial corpus callosum (see Figure 3) there were no right-left differences in MBP staining intensity in either the HI + NS or HI + LPS groups. However, at P17, in the HI + LPS groups, there were bilateral reductions in corpus callosum area (−12%) and in MBP immunostaining (−19%) (see Figure 3), as measured by density segmentation of MBP-immunostained sections (p<0.05, 3-way ANOVA factoring treatment, side and group, see Table 4). Although pixel counts in controls differed among experiments, reflecting variation in intensities of background staining among the three assay batches, LPS consistently suppressed callosal MBP immunostaining (HI + LPS vs. HI + NS, p<0.05, Fisher PLSD post-hoc test, see Table 4).
Table 4.
Post-Hypoxic-Ischemic LPS Exposure Reduces Corpus Callosum Myelin Basic Protein Immunostaining
| Experiment | Number | Black Pixels per Corpus Callosum (mean ± SD)* | ||||||
|---|---|---|---|---|---|---|---|---|
| Left | Right | Total | ||||||
| HI + NS† | HI + LPS‡ | HI + NS | HI + LPS§ | HI + NS | HI + LPS§ | HI + NS | HI + LPS¶ | |
| Group 1 | 6 | 5 | 215 ± 35 | 186 ± 22 | 199 ± 54 | 164 ± 17 | 414 ± 88 | 350 ± 34 |
| Group 2 | 4 | 3 | 399 ± 52 | 376 ± 66 | 415 ± 56 | 331 ± 99 | 814 ± 95 | 707 ± 128 |
| Group 3 | 6 | 7 | 158 ± 29 | 130 ± 25 | 167 ± 37 | 146 ± 29 | 325 ± 64 | 276 ± 48 |
| Corpus Callosum Area (mm2, mean ± SD)* | ||||||||
| Left | Right | Total | ||||||
| HI + NS† | HI + LPS‡ | HI + NS | HI + LPS§ | HI + NS | HI + LPS§ | HI + NS | HI + LPS | |
| Group 1 | 6 | 5 | 0.21 ± 0.04 | 0.19 ± 0.01 | 0.20 ± 0.05 | 0.17 ± 0.02 | 0.40 ± 0.08 | 0.36 ± 0.03 |
| Group 2 | 4 | 3 | 0.19 ± 0.03 | 0.19 ± 0.02 | 0.20 ± 0.02 | 0.18 ± 0.03 | 0.39 ± 0.05 | 0.37 ± 0.03 |
| Group 3 | 6 | 7 | 0.14 ± 0.02 | 0.12 ± 0.02 | 0.13 ± 0.02 | 0.12 ± 0.02 | 0.27 ± 0.04 | 0.24 ± 0.04 |
Bilateral medial corpus callosum was manually outlined (see Fig. 3) in unaltered grey-scale TIFF images of MBP stained sections; area and mean optical density were measured, followed by binary segmentation and pixel counting, using NIH Image. Samples from the 3 experimental groups were assayed in independent batches.
HI + NS: Right carotid ligation followed by 35 min. in 10% O2 on P10. 0.9% NaCl 0.1 ml i.p. daily for 3 days, then 0.05 ml for 2 days, starting 2 h after end of 10% O2.
HI + LPS: Right carotid ligation followed by 35 min. in 10% O2 on P10. LPS 0.2 mg/kg in 0.9% NaCl i.p. daily for 3 days, then 0.1 mg/kg for 2 days, starting 2 h after end of 10% O2.
p<0.05, 3-way ANOVA, factoring treatment, side and experiment, comparing HI + LPS to HI + NS among all experiments.
p<0.05, 2-way ANOVA factoring treatment and experiment, comparing HI + LPS to HI + NS among all experiments.
In animals that had received BrdU, BrdU-immunoreactive cells were concentrated in the forebrain subventricular zone (SVZ) bilaterally, adjacent to the striatum. The SVZ was not directly injured in any of the animals that were analyzed. Although subtle increases in right-sided SVZ cells numbers could be discerned, there were no apparent differences between the distributions of BrdU-labeled cells in the SVZ’s of animals from both groups (see Figure 4). However, systematic stereologic cell counting revealed surprising trends. In the HI+saline controls, there was a mean 60% increase in BrdU-immunoreactive cells in the right, compared with the left SVZ (left 3457 ± 801; right 5514 ± 1940, p < 0.05 paired t-test). In contrast, in the HI+LPS group, although right hemisphere tissue damage was more severe, there was no significant increase in BrdU-labeled cells in the right SVZ (left 4675 ± 1309; right 6146 ± 2560; p: NS, paired t-test). Based on our prior study (Plane et al., 2004), an increase in the mean right SVZ cell count in the HI + LPS group would be predicted, because of greater right hemisphere injury (than in controls). Yet, despite more extensive tissue damage, stimulation of SVZ cell proliferation was attenuated in the HI+LPS group.
Discussion
In the past decade, epidemiological data heightened awareness of pathophysiological links between inflammatory mediators and neonatal neuropathology (Yoon et al., 1997, Nelson et al., 1998, Wu et al., 2003). Concurrently, strong experimental evidence emerged demonstrating both that a pro-inflammatory milieu may increase the susceptibility of neonatal brain to hypoxic-ischemic injury (Eklind et al., 2001, Lehnardt et al., 2003), and that brain-derived inflammatory mediators can amplify tissue damage (Martin et al., 1994, Liu et al., 1999).
This report describes features of a new experimental model in which the impact of indolent inflammation (elicited by repeated systemic administration of LPS) on recovery after neonatal hypoxic-ischemic brain injury can be studied. This model replicates clinically relevant pathophysiological mechanisms that may evolve in neonates with acute hypoxic-ischemic brain injury who subsequently incur inflammatory challenges, as a result of infection or other organ damage. The results demonstrate that systemic inflammation influences the evolution of tissue injury, and may also modify intrinsic potentially reparative responses.
Systemic LPS administration is a widely accepted strategy to model the impact of inflammation on pathological processes. In neonates, the immaturity of the blood-brain barrier may increase accessibility of inflammatory mediators to brain parenchyma. The dose, developmental stage, route of administration, timing (relative to onset of CNS injury), genetic background of the animal, and environmental factors can all influence the impact of LPS-induced inflammation on brain pathology. The complexities inherent in understanding the effects of inflammation on brain pathology are epitomized by the observations that LPS administration pre-ischemia can elicit either a protective (preconditioning) response (Ikeda et al., 2006) or amplification of injury (Eklind et al., 2001). In neonatal rats, systemic injection of LPS four hours prior to hypoxic-ischemic lesioning amplifies tissue damage (Eklind et al., 2001); some deleterious effects of LPS are attenuated with glucose supplementation, but increased striatal damage persists (Eklind et al., 2004). In addition to hypoglycemia, other pathophysiological confounds in interpretation of the effects of LPS in vivo include LPS-induced fever, hypotension, anorexia, and malaise; these adverse effects are minimized by use of low LPS doses. The doses selected for our study stemmed from preliminary experiments in which we sought to identify the highest dose that did not result in poor feeding, growth impairment, or excessive mortality. This LPS administration protocol is not associated with fever, overt systemic illness, poor growth, or increased mortality in CD-1 mice at this age. It should be noted that other mouse strains might respond differently (Sheldon et al., 1998).
Our results are congruent with and complement results in several other models that have examined the impact of pro-inflammatory stimuli on neonatal brain injury. Systemic administration of a single dose of LPS, several hours prior to an hypoxic-ischemic insult, increases the ultimate severity of brain damage in immature rats (Eklind et al., 2001) and mice (Lehnardt et al., 2003); activated microglia were strongly implicated as effectors. In newborn rats, five injections of LPS (0.2 mg/kg) in the first post-natal week resulted in selective increases in blood-brain barrier permeability and loss of white matter (Stolp et al., 2005). Direct intracerebral LPS (3 injections, 1 mg/kg/dose) in neonatal rats resulted in extensive white matter injury and ventricular enlargement; treatment with minocycline suppressed microglial activation and attenuated LPS-induced brain injury (Fan et al., 2005). It is likely that activated microglia also contribute to the LPS-related amplification of hypoxic-ischemic brain damage that was observed in this study. However, it was not feasible to test the efficacy of minocycline directly to address this question, because this drug exacerbates hypoxic-ischemic injury in neonatal mice (Tsuji et al., 2004).
Immature OL are particularly vulnerable to hypoxic-ischemic and inflammatory injury. Although OL precursors are vulnerable to cytokine-mediated injury (Andrews et al., 1998, Molina-Holgado et al., 2001), CNS inflammation can also stimulate SVZ oligodendrogliogenesis (Wu et al., 2000, Arnett et al., 2001). In neonatal rats, we found that HI injury induced robust oligodendrogliogenesis; new OL survive for up to 4 weeks after injury in the injured striatum and in the corpus callosum (Zaidi et al., 2004). Analysis of the impact of chronic inflammation on hypoxic-ischemic white matter injury was challenging. Semi-quantitative methods to assess white matter integrity, based on intensity of MBP immunostaining (Liu et al., 2002), have been applied successfully to assess white matter injury in neonatal mice (Hedtjarn et al., 2005). However, in many LPS-exposed animals, marked tissue damage was associated with increased non-specific staining in the rim of cystic cavities, and this confounded cerebral hemisphere optical density measurements. It was feasible to evaluate MBP immunostaining in the corpus callosum, and we found that LPS administration resulted in bilateral reductions in callosal myelin integrity. We speculate that this, at least in part, reflects cytokine-mediated oligodendroglial injury.
The SVZ contains neural stem cells and more mature progenitors of neurons, astrocytes, and oligodendrocytes. Diverse forms of brain injury stimulate cell proliferation in the adult rodent SVZ (Parent, 2003). Initial studies in neonatal mice highlighted the susceptibility of SVZ cells (in particular OL precursors) to HI injury (Skoff et al., 2001); a subsequent study identified differential effects of neonatal HI on SVZ stem cells (that were resistant) and neural progenitors (that were susceptible) in a similar injury model in neonatal rats (Romanko et al., 2004).
In neonatal mice, using the same lesioning model as in this study, we found that moderate HI injury markedly stimulated SVZ cell proliferation in the first week of recovery (Plane et al., 2004). Moreover, if cases with hemispheric infarction extending into the ipsilateral SVZ were excluded, the magnitude of SVZ cell proliferation was directly related (i.e. there was a positive linear correlation) to the severity of adjacent striatal and cortical tissue loss.
Several prior studies prompted us to evaluate the effects of indolent inflammation on SVZ cell proliferation. In two distinct adult rat models, inflammation suppressed hippocampal dentate gyrus neurogenesis (Monje et al., 2003, Ekdahl et al., 2003). A subsequent study in an adult rat stroke model provided evidence that the endogenous inflammatory response to acute brain injury suppressed SVZ cell proliferation and neurogenesis (Hoehn et al., 2005). Our results in lesioned controls demonstrated a 60% increase in BrdU-labeled cells in the right SVZ, as expected from prior studies. Yet, paradoxically, in the more severely injured LPS-treated animals, ipsilateral SVZ cell proliferation was relatively blunted. This trend suggests that inflammation suppressed hypoxia-ischemia-induced SVZ proliferation.
Much remains to be learned about the roles of inflammatory mediators in brain injury and repair. In light of the findings of the present study, it is tempting to speculate that anti-inflammatory drugs could ultimately represent new neuroprotection modalities. A recent retrospective study of MRI-detectable white matter injury in premature infants found that those who had received indomethacin had less damage (Miller et al., 2006). However, it will be critically important to delineate the risks inherent in anti-inflammatory drug therapy. For example, although earlier evidence had suggested a detrimental role of monocyte chemoattractant protein-1 (MCP-1) in neonatal brain injury (Galasso et al., 2000), a recent study identified the chemokine MCP-1 as a potential positive regulator of cortical neurogenesis after neonatal HI injury (Yang et al., 2007). Inflammatory signaling may exert beneficial effects that enhance some endogenous reparative responses after brain injury.
Our findings offer important insights into the complex effects of systemic illness in critically ill neonates. Systemic inflammation in response to tissue injury may increase vulnerability to CNS injury, may amplify antecedent acute brain injury and may also disrupt endogenous recovery mechanisms.
Acknowledgments
Supported by US PHS award NS 35059.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- AHMED SH, HE YY, NASSIEF A, XU J, XU XM, HSU CY, FARACI FM. Effects of lipopolysaccharide priming on acute ischemic brain injury. Stroke. 2000;31:193–9. doi: 10.1161/01.str.31.1.193. [DOI] [PubMed] [Google Scholar]
- ANDREWS T, ZHANG P, BHAT NR. TNFalpha potentiates IFNgamma-induced cell death in oligodendrocyte progenitors. J Neurosci Res. 1998;54:574–83. doi: 10.1002/(SICI)1097-4547(19981201)54:5<574::AID-JNR2>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- ARNETT HA, MASON J, MARINO M, SUZUKI K, MATSUSHIMA GK, TING JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001;4:1116–22. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- BARKS JDE, SILVERSTEIN FS. Inflammation and neonatal brain injury. In: DONN SM, SINHA SK, CHISWICK ML, editors. Birth Asphyxia and the Brain: Basic Science and Clinical Implications. Armonk, NY: Futura Publishing Inc; 2002. [Google Scholar]
- BECKER KJ, KINDRICK DL, LESTER MP, SHEA C, YE ZC. Sensitization to brain antigens after stroke is augmented by lipopolysaccharide. J Cereb Blood Flow Metab. 2005;25:1634–44. doi: 10.1038/sj.jcbfm.9600160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EKDAHL CT, CLAASEN JH, BONDE S, KOKAIA Z, LINDVALL O. Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A. 2003;100:13632–7. doi: 10.1073/pnas.2234031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EKLIND S, ARVIDSSON P, HAGBERG H, MALLARD C. The role of glucose in brain injury following the combination of lipopolysaccharide or lipoteichoic acid and hypoxia-ischemia in neonatal rats. Dev Neurosci. 2004;26:61–7. doi: 10.1159/000080713. [DOI] [PubMed] [Google Scholar]
- EKLIND S, MALLARD C, LEVERIN AL, GILLAND E, BLOMGREN K, MATTSBY-BALTZER I, HAGBERG H. Bacterial endotoxin sensitizes the immature brain to hypoxic-ischaemic injury. Eur J Neurosci. 2001;13:1101–6. doi: 10.1046/j.0953-816x.2001.01474.x. [DOI] [PubMed] [Google Scholar]
- FAN LW, PANG Y, LIN S, TIEN LT, MA T, RHODES PG, CAI Z. Minocycline reduces lipopolysaccharide-induced neurological dysfunction and brain injury in the neonatal rat. J Neurosci Res. 2005;82:71–82. doi: 10.1002/jnr.20623. [DOI] [PubMed] [Google Scholar]
- GALASSO JM, LIU Y, SZAFLARSKI J, WARREN JS, SILVERSTEIN FS. Monocyte chemoattractant protein-1 is a mediator of acute excitotoxic injury in neonatal rat brain. Neuroscience. 2000;101:737–44. doi: 10.1016/s0306-4522(00)00399-7. [DOI] [PubMed] [Google Scholar]
- HEDTJARN M, MALLARD C, ARVIDSSON P, HAGBERG H. White matter injury in the immature brain: role of interleukin-18. Neurosci Lett. 2005;373:16–20. doi: 10.1016/j.neulet.2004.09.062. [DOI] [PubMed] [Google Scholar]
- HERBER DL, MALONEY JL, ROTH LM, FREEMAN MJ, MORGAN D, GORDON MN. Diverse microglial responses after intrahippocampal administration of lipopolysaccharide. Glia. 2006;53:382–91. doi: 10.1002/glia.20272. [DOI] [PubMed] [Google Scholar]
- HOEHN BD, PALMER TD, STEINBERG GK. Neurogenesis in rats after focal cerebral ischemia is enhanced by indomethacin. Stroke. 2005;36:2718–24. doi: 10.1161/01.STR.0000190020.30282.cc. [DOI] [PubMed] [Google Scholar]
- IKEDA T, YANG L, IKENOUE T, MALLARD C, HAGBERG H. Endotoxin-induced hypoxic-ischemic tolerance is mediated by up-regulation of corticosterone in neonatal rat. Pediatr Res. 2006;59:56–60. doi: 10.1203/01.pdr.0000191140.87314.ce. [DOI] [PubMed] [Google Scholar]
- LEHNARDT S, MASSILLON L, FOLLETT P, JENSEN FE, RATAN R, ROSENBERG PA, VOLPE JJ, VARTANIAN T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A. 2003;100:8514–9. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU XH, KWON D, SCHIELKE GP, YANG GY, SILVERSTEIN FS, BARKS JD. Mice deficient in interleukin-1 converting enzyme are resistant to neonatal hypoxic-ischemic brain damage. J Cereb Blood Flow Metab. 1999;19:1099–108. doi: 10.1097/00004647-199910000-00006. [DOI] [PubMed] [Google Scholar]
- LIU YQ, SILVERSTEIN FS, SKOFF R, BARKS JDE. Hypoxic-ischemic oligodendroglial injury in neonatal rat brain. Pediatr Res. 2002;51:25–33. doi: 10.1203/00006450-200201000-00007. [DOI] [PubMed] [Google Scholar]
- MARTIN D, CHINOOKOSWONG N, MILLER G. The interleukin-1 receptor antagonist (rhIL-1ra) protects against cerebral infarction in a rat model of hypoxia-ischemia. Exp Neurol. 1994;130:362–367. doi: 10.1006/exnr.1994.1215. [DOI] [PubMed] [Google Scholar]
- MILLER SP, MAYER EE, CLYMAN RI, GLIDDEN DV, HAMRICK SE, BARKOVICH AJ. Prolonged indomethacin exposure is associated with decreased white matter injury detected with magnetic resonance imaging in premature newborns at 24 to 28 weeks’ gestation at birth. Pediatrics. 2006;117:1626–31. doi: 10.1542/peds.2005-1767. [DOI] [PubMed] [Google Scholar]
- MOLINA-HOLGADO E, VELA JM, AREVALO-MARTIN A, GUAZA C. LPS/IFN-gamma cytotoxicity in oligodendroglial cells: role of nitric oxide and protection by the anti-inflammatory cytokine IL-10. Eur J Neurosci. 2001;13:493–502. doi: 10.1046/j.0953-816x.2000.01412.x. [DOI] [PubMed] [Google Scholar]
- MONJE ML, TODA H, PALMER TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–5. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- NELSON KB, DAMBROSIA JM, GRETHER JK, PHILLIPS TM. Neonatal cytokines and coagulation factors in children with cerebral palsy. Ann Neurol. 1998;44:665–675. doi: 10.1002/ana.410440413. [DOI] [PubMed] [Google Scholar]
- PARENT JM. Injury-induced neurogenesis in the adult mammalian brain. Neuroscientist. 2003;9:261–72. doi: 10.1177/1073858403252680. [DOI] [PubMed] [Google Scholar]
- PLANE JM, LIU R, WANG TW, SILVERSTEIN FS, PARENT JM. Neonatal hypoxic-ischemic injury increases forebrain subventricular zone neurogenesis in the mouse. Neurobiol Dis. 2004;16:585–95. doi: 10.1016/j.nbd.2004.04.003. [DOI] [PubMed] [Google Scholar]
- RICE JE, VANNUCCI RC, BRIERLEY JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol. 1981;9:131–141. doi: 10.1002/ana.410090206. [DOI] [PubMed] [Google Scholar]
- RISAU W, WOLBURG H. Development of the blood-brain barrier. Trends Neurosci. 1990;13:174–8. doi: 10.1016/0166-2236(90)90043-a. [DOI] [PubMed] [Google Scholar]
- ROMANKO MJ, ROTHSTEIN RP, LEVISON SW. Neural stem cells in the subventricular zone are resilient to hypoxia/ischemia whereas progenitors are vulnerable. J Cereb Blood Flow Metab. 2004;24:814–25. doi: 10.1097/01.WCB.0000123906.17746.00. [DOI] [PubMed] [Google Scholar]
- SANKAR R, AUVIN S, MAZARATI A, SHIN D. Inflammation contributes to seizure-induced hippocampal injury in the neonatal rat brain. Acta Neurol Scand. 2007;115:16–20. doi: 10.1111/j.1600-0404.2007.00804.x. [DOI] [PubMed] [Google Scholar]
- SHELDON RA, SEDIK C, FERRIERO DM. Strain-related brain injury in neonatal mice subjected to hypoxia-ischemia. Brain Res. 1998;810:114–122. doi: 10.1016/s0006-8993(98)00892-0. [DOI] [PubMed] [Google Scholar]
- SKOFF RP, BESSERT DA, BARKS JD, SONG D, CERGHET M, SILVERSTEIN FS. Hypoxic-ischemic injury results in acute disruption of myelin gene expression and death of oligodendroglial precursors in neonatal mice. Int J Dev Neurosci. 2001;19:197–208. doi: 10.1016/s0736-5748(00)00075-7. [DOI] [PubMed] [Google Scholar]
- SOUCY G, BOIVIN G, LABRIE F, RIVEST S. Estradiol is required for a proper immune response to bacterial and viral pathogens in the female brain. J Immunol. 2005;174:6391–8. doi: 10.4049/jimmunol.174.10.6391. [DOI] [PubMed] [Google Scholar]
- STOLP HB, DZIEGIELEWSKA KM, EK CJ, POTTER AM, SAUNDERS NR. Long-term changes in blood-brain barrier permeability and white matter following prolonged systemic inflammation in early development in the rat. Eur J Neurosci. 2005;22:2805–16. doi: 10.1111/j.1460-9568.2005.04483.x. [DOI] [PubMed] [Google Scholar]
- TSUJI M, WILSON MA, LANGE MS, JOHNSTON MV. Minocycline worsens hypoxic-ischemic brain injury in a neonatal mouse model. Exp Neurol. 2004;189:58–65. doi: 10.1016/j.expneurol.2004.01.011. [DOI] [PubMed] [Google Scholar]
- WU JP, KUO JS, LIU YL, TZENG SF. Tumor necrosis factor-alpha modulates the proliferation of neural progenitors in the subventricular/ventricular zone of adult rat brain. Neurosci Lett. 2000;292:203–6. doi: 10.1016/s0304-3940(00)01472-5. [DOI] [PubMed] [Google Scholar]
- WU YW, ESCOBAR GJ, GRETHER JK, CROEN LA, GREENE JD, NEWMAN TB. Chorioamnionitis and cerebral palsy in term and near-term infants. JAMA. 2003;290:2677–84. doi: 10.1001/jama.290.20.2677. [DOI] [PubMed] [Google Scholar]
- YANG L, SAMESHIMA H, IKEDA T, IKENOUE T. Lipopolysaccharide administration enhances hypoxic-ischemic brain damage in newborn rats. J Obstet Gynaecol Res. 2004;30:142–7. doi: 10.1111/j.1447-0756.2003.00174.x. [DOI] [PubMed] [Google Scholar]
- YANG Z, COVEY MV, BITEL CL, NI L, JONAKAIT GM, LEVISON SW. Sustained neocortical neurogenesis after neonatal hypoxic/ischemic injury. Ann Neurol. 2007;61:199–208. doi: 10.1002/ana.21068. [DOI] [PubMed] [Google Scholar]
- YOON BH, JUN JK, ROMERO R, PARK KH, GOMEZ R, CHOI JH, KIM IO. Amniotic fluid inflammatory cytokines (interleukin-6, interleukin-1β, and tumor necrosis factor-α), neonatal brain white matter lesions, and cerebral palsy. Am J Obstet Gynecol. 1997;177:19–26. doi: 10.1016/s0002-9378(97)70432-0. [DOI] [PubMed] [Google Scholar]
- ZAIDI AU, BESSERT DA, ONG JE, XU H, BARKS JD, SILVERSTEIN FS, SKOFF RP. New oligodendrocytes are generated after neonatal hypoxic-ischemic brain injury in rodents. Glia. 2004;46:380–90. doi: 10.1002/glia.20013. [DOI] [PubMed] [Google Scholar]