

Abstract
The relatively rapid development of microbial resistance after the entry of every new antimicrobial into the marketplace necessitates a constant supply of new agents to maintain effective pharmacotherapy. Despite extensive efforts to identify novel lead compounds from molecular targets, only the peptide deformylase inhibitors (PDIs) have shown any real promise, with some advancing to phase I human trials. Bacterial peptide deformylase, which catalyzes the removal of the N-formyl group from N-terminal methionine following translation, is essential for bacterial protein synthesis, growth, and survival. The majority of PDIs are pseudopeptide hydroxamic acids and two of these (IV BB-83698 and oral NVP LBM-415) entered phase I human trials. However, agents to the present have suffered from major potential liabilities. Their in vitro activity has been limited to gram-positive aerobes and some anaerobes and has been quite modest against the majority of such species (MIC90 values ranging from 1–8 mg/L). They have exerted bacteriostatic, not bacteriocidal, activity, thus reducing their potential usefulness in the management of serious infections in the immunocompromised. The relative ease with which microorganisms have been able to develop resistance and the multiple available mechanisms of resistance (mutations in fmt, defB, folD genes; AcrAB/TolC efflux pump; overexpression of peptide deformylase) are worrisome. These could portend a short timespan of efficacy after marketing. Despite these current liabilities, further pursuit of more potent and broader spectrum PDIs which are less susceptible to bacterial mechanisms of resistance is still warranted.
Keywords: peptide deformylase, peptide deformylase inhibitors, actinonin, LBM-415, BB-83698, BB-3497
Natural products have played pivotal roles in the development of antimicrobials since the early years of the twentieth century. However, the relatively rapid onset of microbial resistance with every new antimicrobial introduced into the marketplace requires a constant supply of new agents for effective therapy of infectious diseases. Table 1 illustrates natural product-derived antimicrobial compounds undergoing investigation (including natural product templates and new templates present in recently-discovered lead antimicrobials) [Butler and Buss 2006]. Despite extensive efforts to identify novel lead compounds from molecular targets, only the peptide deformylase inhibitors (PDIs) are currently in clinical trials.
Table 1.
Natural product-derived antimicrobial compounds under investigationa
| Name (synonym) | Class (lead compound) | Developer |
|---|---|---|
| NVP LBM 415 (NVP PDF 713) | Peptide deformylase inhibitor (actinonin) | Novartis |
| Rifalazil (ABI-1648, KRM-1648) | Ansamycin (rifamycin B) | ActivBiotics |
| Ceftobripole medocaril (BAL-5788) | β-lactam-cephalosporin | Basilea and J & J |
| PPI-0903 (TAK-599) | β-lactam-cephalosporin | Cerexa |
| RWJ-442831 | β-lactam-cephalosporin | J & J |
| CS-023 (R1558) | β-lactam-cephalosporin | Roche/Sankyo |
| Tebipenem pivoxil (ME1211) | β-lactam-carbapenem | Meiji Seika Kaisha |
| ME1036 (CP5069) | β-lactam-carbapenem | Meiji Seika Kaisha |
| Faropenem daloxate | β-lactam-penem | Replidyne |
| Dalbavancin | glycopeptide (A40926) | Vicuron |
| Telavancin (TD-6424) | glycopeptide (vancomycin) | Theravance |
| Cethromycin | macrolide (erythromycin) | Advanced Life Sciences |
| EP-013420 | macrolide (erythromycin) | Enanta/Shionogi |
| Pleuromutilin derivative (565154) | new (pleuromutilin) | GlaxoSmithKline |
| Ramoplanin | new (ramoplanin) | Oscient |
| NXL103 (XRP2868)-RPR132552A and RPR202698 | streptogramin | Novexel |
| PTK0796 | tetracycline | Paratek |
| Tiacumicin B (PAR-101, OPT-80) | new (tiacumicin) | Par |
14/19 are derivates of known drugs. only NVP LBM-415, pleuromutilin derivative (565154), ramoplanin, and tiacumicin B are not related to drugs previously marketed for human use.
Bacterial peptide deformylase (EC 3.5.1.31) is an enzyme responsible for catalyzing the removal of the N-formyl group from N-terminal methionine following translation. This enzyme is encoded by the def gene, which is present in all pathogenic bacteria, including Mycoplasma and Chlamydia species, and which does not share a functionally equivalent gene in mammalian cells. The def gene is an essential gene for bacterial growth and survival (this has been validated for Streptococcus pneumoniae and Escherichia coli) [Mazel et al 1994; Chan et al 2003]. The enzyme contains three highly conserved catalytic domains and belongs to the matrix metallo-protease (MMP) family of enzymes. Blockade of bacterial peptide deformylase produces inhibition of protein synthesis (similar to the mechanism of the tetracyclines, macrolides, streptogramins, lincosamides, and chloramphenicol).
The only naturally-occurring PDIs are actinonin and macrolactin N [Chen et al 2000; Yoo et al 2006]. Although actinonin is too weak an inhibitor to use clinically, it did form the framework for the synthesis, purification, and evaluation of more potent PDIs [Chen et al 2000].
Individual papers have reviewed the discoveries of BB-3497 [Clements et al 2001], VRC 3852 [Hackbarth et al 2002], VRC 3375 [Jain et al 2003; Chen et al 2004], Ro 66-0376 and Ro 66-6976 [Apfel et al 2000], and PDF-611 (LBK611) [Yoo et al 2006]. Table 2 outlines the peptide deformylase inhibitors of greatest potential. The pseudopeptidic hydroxamic acids (or N-formyl-N-hydroxylamines) constitute the largest group and two of these compounds were investigated for some time in human clinical trials (intravenous BB-83698 and oral LBM-415). The chemical structures of these latter two PDIs are illustrated in the Figure 1.
Table 2.
Peptide deformylase inhibitors
| A. Peptidic Inhibitors |
|
B. Non-Actinonin-Based, Non-Peptidic Inhibitors
|
Figure 1.
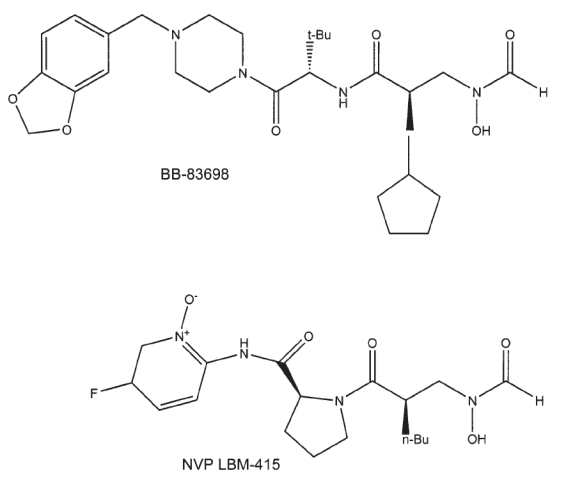
Chemical structures of the peptide deformylase inhibitors BB-83698 and NVP LBM-415.
Chemistry
Peptide deformylase inhibitors are selective for bacterial enzyme and exhibit activity against similar mammalian (including human) enzymes only at extremely high concentrations unlikely to be achievable in vivo. For example, BB-81384 inhibits the peptide deformylase (Ni complex) of Streptococcus pneumoniae, Haemophilus influenzae, E. coli, and Staphylococcus aureus with IC50 values of 9, 11, 60, and 300 nM, respectively [Gross et al 2004], where IC50 refers to the concentration inhibiting enzyme activity by 50 percent. Corresponding IC50 values for the human metalloenzymes collagenase (MMP-1), gelatinase (MMP-2) and angiotensin converting enzyme (ACE) were 10000, 60000, and 5000 nM, respectively [Gross et al 2004]. In the development of VRC 3852, 20/21 related compounds (95%) exhibited IC50 values of 100 nM or less for E. coli peptide deformylase and 18/20 (90%) exhibited IC50 values of 75 nM or less for S. pneumoniae peptide deformylase. All 21 compounds were very selective, with IC50 values for tested human metalloenzymes being 200000 nM or greater [Hackbarth et al 2002]. Two PDIs exhibited potent activity against the peptide deformylase of Mycobacterium bovis BCG: PDF-611 (IC50 of 69.5 nM) and BB-3497 (IC50 of 24.9 nM) [Teo et al 2006].
The majority of PDIs are hydroxamic acid derivatives Figure 1), in which the PDI coordinates with the active-site metal atom [Huo et al 1999; Clements et al 2001; Guilloteau et al 2002; Hackbarth et al 2002]. These data have been obtained during crystallography studies of enzyme-substrate complexes at resolutions of 2Å or less. Two PDIs produce time-dependent inhibition of peptide deformylase: actinonin and BB-3497 (including its 15-membered macrocyclic ring analogue) [Hu et al 2003, 2004; Van Aller et al 2005]. With these agents, binding to the enzyme occurs in two steps, wherein the initial encounter complex tightens into a final encounter complex with an extremely slow rate of dissociation (half-life for dissociation ≥0.77 days [actinonin] and ≥1.9 days [BB-3497]) [Van Aller et al 2005].
The effect of NVP LBM 415 on the proteomes of S. aureus and S. pneumoniae has been studied using two-dimensional electrophoresis. During exposure to PDI, similar findings were noted with both microorganisms. Many N-terminal formylated peptides/proteins were seen, their accumulation being time-dependent and the degree of accumulation differing for different peptides/proteins. Upon removal of PDI, these peptides/proteins underwent deformylation in a time-dependent manner. However, if sub-MIC (minimum inhibitory concentration) concentrations of the PDI were maintained over time, high levels of formylated peptides/proteins were present for a longer period and this correlated with a prolonged post-antibiotic effect (PAE) in vitro [Wang et al 2006].
The PDIs may also work via stimulation of the innate immune system. The innate immune system uses the formylation of bacterial proteins as a target and professional phagocytes express receptors for bacterial-derived formylated peptides. Activation of formyl peptide receptors (FPR) mediates phagocyte (neutrophil) migration and release of free radicals and other antimicrobial substances from phagocytes. Theoretically, PDIs should enhance this response and, hence, innate immunity. This has been demonstrated with actinonin in animal models. In subtherapeutic doses, actinonin enhances the production and secretion of neutrophil-activating peptides that work via FPR [Fu et al 2003].
In vitro antibacterial activity
Peptide deformylase inhibitors are much less active against intact bacteria than predicted by their kinetics of inhibition of purified enzyme, likely due to the barrier effects of the cell wall and outer membrane and the presence of active efflux pump mechanisms [Apfel et al 2000].
These agents generally lack useful activity against Enterobacteriaceae and non-fermentative gram-negative bacilli [Jones and Rhomberg 2003]. However, BB-3497 was active against single isolates of E. coli, Enterobacter cloacae, and Klebsiella pneumoniae (MIC = 8 mg/L for each) [Clements et al 2001]. In addition, Ro 66-0376 and Ro 66-6976 were active against one isolate of Stenotrophomonas maltophilia (MIC < 0.25 mg/L and 4 mg/L, respectively) [Apfel et al 2000]. Structure-activity relationship studies with the VRC series of compounds suggest the feasibility of extending the spectrum of activity of the PDIs to include gram-negative microorganisms [Hackbarth et al 2003].
Table 3 and 4 illustrate the in vitro antibacterial activities of the British Biotech (BB) and Vicuron/Novartis (NVP) series of PDIs, respectively [Wootton et al 2001; Wise et al 2002; Bowker et al 2003; Jones and Rhomberg 2003; Roblin and Hammerschlag 2003; Credito et al 2004; Cynamon et al 2004; Ednie et al 2004; Jones et al 2004; Lofland et al 2004; Bell et al 2005; Edelstein et al 2005; Fritsche et al 2005; Jones et al 2005; Snydman et al 2005; Waites et al 2005; Teo et al 2006; Watters et al 2006].
Table 3.
Antibacterial activity of British Biotech (BB) series of peptide deformylase inhibitorsa
| MIC90 (mg/L) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Organism (N) | BB-83698 | BB-3497 | BB-83815 | BB-83857 | BB-84518 | BB-84416 | BB-85318 | BB-84888 | BB-85128 | BB-84879 | BB-84880 | BB-84885 | References |
| Streptococcus pneumoniae (40) | 0.5 | ≥16 | 0.5 | 1 | 2 | 2 | – | – | – | – | – | – | [1] |
| penicillin-sensitive (113) | 0.25–0.5 | – | – | – | – | – | 0.5 | 0.06 | 1 | 1 | 0.5 | 0.25 | [2,3] |
| penicillin-intermediate (47) | 0.5 | – | – | – | – | – | – | – | – | – | – | – | [2] |
| penicillin-resistant (75) | 0.25 | – | – | – | – | – | – | – | – | – | – | – | [2] |
| Group A streptococci (41) | 0.12 | – | – | – | – | – | 0.25 | 0.03 | 0.5 | 0.5 | 0.25 | 0.06 | [2,3] |
| Group B streptococci (21) | 0.12 | – | – | – | – | – | – | – | – | – | – | – | [2] |
| S. viridans(26) | 0.5 | – | – | – | – | – | – | – | – | – | – | – | [2] |
| MS Staphylococcus aureus (89) | 4–8 | – | – | – | – | – | 2 | 2 | 2 | 2 | 1 | 2 | [2,3] |
| MR S. aureus (95) | 4–8 | – | – | – | – | – | 2 | 2 | 2 | 2 | 1 | 1 | [2,3] |
| h GISA (33) | 4 | – | – | – | 0.5 | – | – | – | – | – | – | – | [4] |
| GISA (10) | 2 | – | – | – | 2 | – | – | – | – | – | – | – | [4] |
| Haemophilus influenzae (35) | ≥16 | 2 | ≥16 | ≥16 | 4 | ≥16 | – | – | – | – | – | – | [1] |
| beta-lactamase-neg. (85) | 8–32 | – | – | – | – | – | 2 | 8 | 2 | 2 | 4 | 8 | [2,3] |
| beta-lactamase-pos. (65) | 16–64 | – | – | – | – | – | 4 | 8 | 2 | 2 | 8 | 16 | [2,3] |
| Moraxella catarrhalis (58) | 0.06–0.12 | 0.12 | 0.12 | 0.12 | 0.12 | 0.25 | 0.06 | 0.03 | 0.12 | 0.06 | 0.03 | 0.015 | [2,3] |
| beta-lactamase-neg. (25) | 0.12 | – | – | – | – | – | – | – | – | – | – | – | [2] |
| beta-lactamase-pos. (25) | 0.12 | – | – | – | – | – | – | – | – | – | – | – | [2] |
| Mycobacterium tuberculosis (17) | >8 | 0.5–1 | – | – | >8 | – | – | – | – | – | – | – | [5,6] |
Abbreviations: MIC90 = minimum medium concentration inhibiting the growth of 90% of isolates; ms = methicillin-susceptible; MR = methicillin-resistant; h = heterogeneous; GISA = glycopeptide-intermediate S.aureus
Range of pooled MIC90 values are shown, where available. Studies were only pooled when each study met all of the following criteria: study used National Committee for Clinical Laboratory Standards (NCCLS) methodology, a minimum of 10 isolates were tested for each organism-of-interest, and the test inoculum ranged from 104–106 CFU per spot.
1 = Wise et al 2002, 2 = Lofland et al 2004, 3 = Bowker et al 2003, 4 = Wootton et al 2001, 5 = Cynamon et al 2004, 6 = Teo et al 2006.
Table 4.
Antibacterial activity of NVP series of peptide deformylase inhibitorsa
| MIC90 (mg/L) |
|||
|---|---|---|---|
| Organism (N) | NVP PDF-386 (VRC 4887) | NVP PDF-713 (LBM-415) | References |
| Staphylococcus aureus (9865) | 1 | 1 | [1–3] |
| methicillin-sensitive (5990) | – | 1–2 | [3–6] |
| methicillin-resistant (3323) | – | 1–4 | [3–6] |
| Coagulase-negative staphylococci (3105) | 1 | 1–2 | [1–3] |
| methicillin-sensitive (693) | – | 2 | [3–6] |
| methicillin-resistant (2143) | – | 1–4 | [3–6] |
| β-hemolytic streptococci (963) | – | 0.5–1 | [2–4,6] |
| Viridans streptococci (445) | – | 0.5–2 | [2–4,6] |
| Streptococcus pneumoniae (2421) | 0.5 | 2 | [1–3] |
| penicillin-sensitive (1594) | – | 1–2 | [3,4,6,7] |
| penicillin-intermediate (533) | – | 1 | [3,4,6,7] |
| penicillin-resistant (624) | – | 0.5–1 | [3,4,6,7] |
| MDRb (77) | 1 | [6] | |
| Enterococcus sp. (3758) | 2 | 4 | [1–3] |
| E. faecalis (2348) | – | 4 | [3] |
| VS E. faecalis (2262) | – | 4 | [3,6] |
| VR E. faecalis (118) | – | 4 | [3] |
| E. faecium (914) | – | 2 | [3] |
| VS E. faecium (393) | – | 2 | [3,6] |
| VR E. faecium (531) | – | 2 | [3] |
| Haemophilus influenzae (3153) | 32 | 4 | [1,3] |
| beta-lactamase-neg. (2369) | – | 4 | [3,4] |
| beta-lactamase-pos. (746) | – | 8 | [3,4] |
| Moraxella catarrhalis (341) | ≤0.25 | 0.5 | [1,3,4] |
| Chlamydia pneumoniae (21) | 0.008 | – | [8] |
| Mycoplasma pneumoniae (100) | – | 0.001 | [9] |
| Legionella sp. (20) | – | 16 | [10] |
| L. pneumophila (50) | – | 0.12 | [4] |
| Neisseria sp. | |||
| N. gonorrhoeae (157) | – | 8 | [11] |
| N. meningitidis (100) | – | 2 | [11] |
| Bacteroides sp. | |||
| B. fragilis (209) | – | 0.5 | [12] |
| B. distasonis (28) | – | 0.5 | [12] |
| B. ovatus (32) | – | 0.25 | [12] |
| B. uniformis (22) | – | 0.25 | [12] |
| B. vulgatus (22) | – | 0.5 | [12] |
| Peptostreptococcus sp. (25) | – | 1 | [12] |
| Clostridium perfringens (25) | – | 2 | [12] |
| Propionibacterium sp. (15) | – | 2 | [12] |
Abbreviations: MIC90 = minimum medium concentration inhibiting the growth of 90% of isolates; MDR = multidrug-resistant (ie, resistant to ≥3 of penicillin, erythromycin, tetracycline, chloramphenicol, levofloxacin, or trimethoprim-sulfamethoxazole); VS = vancomycin-susceptible; VR = vancomycin-resistant.
Range of pooled MIC90 values are shown, where available. Studies were only pooled when each study met all of the following criteria: study used National Committee for Clinical Laboratory Standards (NCCLS) methodology, a minimum of 10 isolates were tested for each organism-of-interest, and the test inoculum ranged from 104–106 CFU per spot.
1 = Jones and Rhomberg 2003, 2 = Jones et al 2004, 3 = Watters et al 2006, 4 = Fritsche et al 2005, 5 = Credito et al 2004, 6 = Bell et al 2005, 7 = Ednie et al 2004, 8 = Roblin & Hammerschlag 2003, 9 = Waites et al 2005, 10 = Edelstein et al 2005, 11 = Jones et al 2005, 12 = Snydman et al 2005.
Within a series of five British Biotech compounds evaluated for in vitro activity against C. pneumoniae (strains TW 183, CT 815, CT 712), the activity was quite variable: for TW 183 and CT815, MIC range of 0.5 to 2 mg/L and minimum lethal concentration (MLC) range of 0.5 to 4 mg/L and for CT 712, MIC range of 0.25 to 4 mg/L and MLC range of 0.25 to 8 mg/L [Wise et al 2002].
NVP LBM-415 has demonstrated modest-moderate activity against linezolid-resistant staphylococci (N = 6, MIC range of 0.25–2 mg/L), Streptococcus oralis (N = 1, MIC 0.5 mg/L), E. faecalis (N = 3, MIC range of 2–4 mg/L), and E. faecium (N = 10, MIC range of 0.5–4 mg/L) [Jones et al 2004a]. It has also demonstrated a similar degree of activity against quinupristin/dalfopristin-resistant E. faecium(N = 6, MIC range of 1–2 mg/L) and staphylococci (N = 19, MIC range of 0.12–2 mg/L) [Jones et al 2004a]. Two PDIs exhibit reasonable in vitro activity against mycobacteria: PDF-611 (MIC90 = 0.25 mg/L vs. M. bovis BCG) and BB-3497 (MIC90 = 0.5–1 mg/L vs. M. tuberculosis) [Cynamon et al 2004; Teo et al 2006].
An inoculum effect has been noted with NVP LBM-415, with 10- to 100-fold increases in inoculum producing 2- to 4-fold increases in MIC [Fritsche et al 2005]. However, varying the incubation environment, pH, or calcium concentration and medium supplementation have negligible effects on NVP LBM-415 activity [Fritsche et al 2005]. Alterations in medium cation content and inoculum (over a range of 102 to 106 colony-forming units [CFU] per mL) do not have a significant effect on BB-83698 activity [Lofland et al 2003]. Alteration in medium pH does not affect its activity against S. pneumoniae but does reduce it against S. aureus and H. influenzae. For example, a medium pH of 6 increases S. aureus MIC’s about four-fold while the identical pH increases the MIC’s of H. influenzae between four- and eight-fold [Lofland et al 2003]. The postantibiotic effect (PAE) duration of LBM-415 ranged from 0.3 to 1.4 hours for S. pneumoniae. The PAE duration of LBM-415 for S. pneumoniae was not affected by strain susceptibility to β-lactams or macrolides [Kosowska-Shick et al 2007].
BB-81384, BB-83698, NVP LBM-415, and PDF-611 exhibit primarily bacteriostatic activity against pneumococci, staphylococci, mycobacteria, and M. catarrhalis, even at medium concentrations up to and exceeding tenfold the MIC [Credito et al 2004; Ednie et al 2004; Gross et al 2004; Lofland et al 2004; Fritsche et al 2005; Teo et al 2006]. However, bactericidal activity has been noted against C. pneumoniae (with NVP PDF 386) [Roblin & Hammerschlag 2003], H. influenzae (with BB-83698) [Lofland et al 2004] and S. pneumoniae (in 5/16 [31%] strains with BB-83698) [Lofland et al 2003]. NVP LBM-415 is active both intra- and extra-cellularly against a variety of Legionella species but such activity is bacteriostatic in nature [Edelstein et al 2005]. Preliminary data from combination studies with NVP LBM-415 demonstrate rare synergy with the vast majority of combinations resulting in indifference [Fritsche et al 2005].
The in vitro growth curves of S. pneumoniae after exposure to linezolid and NVP LBM-415 reveal marked differences in the time to onset of effect. The inhibitory effect of linezolid was already apparent at the 1 hour sampling timepoint while that of the PDI was delayed until the 2 hour timepoint [Azoulay-Dupuis et al 2004]. Dry-form broth microdilution panels for susceptibility testing in NVP LBM-415 clinical trials have been validated (99.2% of dry-form MIC results were within ±1 log2 dilution of the reference standards with between-day and within-day reproducibility being 96.7% and 98.9%, respectively) [Fritsche et al 2004a]. Quality control guidelines have also been established for this same PDI for disk diffusion and broth microdilution testing, each using 4 strains of American Type Culture Collection (ATCC) microorganisms [Anderegg et al 2003, 2004].
Provisional MIC and disk diffusion zone size criteria have been published for NVP LBM-415. Sensitive corresponds to an MIC value of 4 mg/L or less (zone size of 20 mm or greater) while resistance is denoted by an MIC value of 16 mg/L or greater (zone size of 16 mm or less) [Fritsche et al 2004]. In a collection of 2625 isolates, no strains of staphylococci, S. pneumoniae, other streptococci, or enterococci had MIC/zone size values corresponding to resistant. The authors thus felt that until resistant isolates of streptococci and staphylococci are seen, the only interpretive category for these microorganisms should be sensitive. For enterococci and H. influenzae, all three interpretive criteria should be developed, based on pharmacokinetic/pharmacodynamic and MIC population considerations [Fritsche et al 2004].
The development of resistance to PDIs may be a worrisome issue as these agents enter clinical trials and, eventually, the marketplace. Resistant mutants of S. pneumoniae and S. aureus were selected at medium concentrations of 2 to 32 times the MIC and their frequencies were 10−8 to 10−10 and 2 × 10−6 to 2 × 10−8, respectively [Leeds et al 2004]. Multistep resistance selection testing yielded resistant clones with fourfold or greater increases in MIC in 11/12 (92%) strains of S. pneumoniae after 14–50 daily passages. MICs rose from 0.125–1.0 mg/L (parents) to 2–>16.0 mg/L (mutants) [Kosowska-Shick et al 2007]. For H. influenzae, two distinct mutants were selected, one at medium concentrations of 8 to 32 times the MIC (frequency of 10−8 to 10−9) and the other at medium concentrations of 2 to 4 times the MIC (frequency of 6 × 10−8) [Leeds et al 2004]. At a medium concentration of 10 times the MIC, resistant mutants of M. bovis BCG arose at frequencies of 5 × 10−7 or less [Teo et al 2006]. In a global surveillance study conducted between 2002 and 2004, one S. aureus isolate was isolated with high-level resistance (MIC ≥ 1024 mg/L) to NVP LBM-415. Resistance was due to multiple sequence changes in resistance phenotype genes (defB [1 nucleotide change] and fmt [3 nucleotide changes]). At the time of collection of this isolate, PDIs had not been used in humans at all and in vitro and in vivo (animal) exposures had been limited to a small number of clinical laboratories [Watters et al 2006].
Mutations in the formyl transferase gene (fmt) reduce the susceptibilities of S. aureus, H. influenzae, M. bovis BCG and E. coli for PDIs. These mutations also produce cross-resistance to all PDIs [Margolis et al 2000, Apfel et al 2001; Leeds et al 2004; Teo et al 2006]. These mutations also put these organisms at a disadvantage as their growth rates fall, morphologic changes occur (only with H. influenzae), and their virulence attenuates compared to wild-type strains [Margolis et al 2000; Apfel et al 2001; Leeds et al 2004; Teo et al 2006]. Mutations in the defB gene reduce the susceptibility of S. pneumoniae to PDIs [Kosowska-Shick et al 2007]. In contrast to the fmt gene, mutations of the defB gene may not produce cross-resistance across all PDIs [Margolis et al 2001]. Another potential mechanism of resistance is overexpression of the target enzyme due to the def gene amplification by the microorganism-of-interest and/or surrounding microorganisms (“bystanders”) [Apfel et al 2001]. This has been noted as a resistance mechanism for H. influenzae [Dean et al 2007].
Another potential mechanism of resistance to the PDIs involves efflux pumps located in the cell membrane/wall which act to actively pump antimicrobials out of the cell. Inactivation of the AcrAB-TolC efflux pump of H. influenzae enhances susceptibility to NVP LBM-415 [Dean et al 2005; Fritsche et al 2005]. In strains with MICs of 16 mg/L or greater, genetic deletion of AcrAB produces hypersusceptible strains and inactivation of AcrAB or TolC significantly enhances susceptibility [Dean et al 2005; Fritsche et al 2005]. In contrast to the effect of mutations in the fmt gene, the presence of AcrAB or TolC does not alter the growth rate or virulence of H. influenzae [Neckerman 2005]. Perhaps the presence of this efflux pump may account for the modest-marginal intrinsic activities of PDIs against H. influenzae (Table 1 and 2). In addition, the presence of efflux pumps also result in reduced susceptibility to the macrolides [Leeds et al 2004].
Lastly, mutations in/near the folD gene have a similar effect to that of mutations in the fmt gene since both genes are necessary to encode the proteins needed for the formylation of methionyl initiator transfer RNA. The folD gene encodes the bifunctional enzyme methylene tetrahydrofolate-dehydrogenase and -cyclohydrolase. Again, bacterial fitness suffers in acquiring this form of resistance, leading to greatly reduced growth rates and virulence [Nilsson et al 2006].
Concerns regarding resistance issues played a significant role in suspension of further development of Ro 66-0376 and Ro 66-6976 (Roche).
In vivo (animal) pharmacodynamics
BB-81384 has been evaluated in three animal infection models: murine systemic infection (sepsis) model, neutropenic mouse thigh infection model, and murine lung infection model [Gross et al 2004]. In the murine sepsis model, an inoculum of S. pneumoniae was injected into the peritoneal cavity followed one hour later by a single oral dose of antimicrobial. Five day survival rates (endpoint) were 0, 50, 100, 100, and 100 percent following BB-81384 10, 30, and 90 mg/kg, amoxicillin/clavulanate 10 mg/kg, and azithromycin 10 mg/kg, respectively. When BB-81384 was administered twice on the day of inoculation (ie, 1 and 5 hours post-inoculation), the ED50 (dose effective at prolonging survival to at least 5 days in 50% of the group) fell from 30 to 20 mg/kg. In the neutropenic mouse thigh infection model, an inoculum of S. pneumoniae was injected IM into mice rendered neutropenic by intraperitoneal injections of cyclophosphamide followed 2 hours later by a single oral dose of antimicrobial. BB-81384 30 and 60 mg/kg and amoxicillin/clavulanate 10 mg/kg produced log10 thigh CFU reductions of 3.8, not done and 5.5 at 5 hours post-inoculation, respectively and 2.6, 3.3, and 5.0 at 24 hours post-inoculation, respectively. In the murine lung infection model, an inoculum of S. pneumoniae was instilled intranasally followed by 3 days of antimicrobials. BB-81384 100 mg/kg once daily for 3 days produced 100 percent survival at 3 days but none at 6 days. Three-day regimens of 50 mg/kg twice daily and 100 mg/kg twice daily both produced 100 percent survival at 3 days and 40 and 67 percent survival at 6 days, respectively. In addition, the latter two regimens reduced the 22 hr log10 lung CFU counts by 4.3 and 5.6 and the 50 hr log10 CFU counts by 4.5 and 6.0, respectively [Gross et al 2004].
Oral and subcutaneous BB-83698 has also been evaluated in the neutropenic (and normal) mouse thigh infection model. Eight strains of S. pneumoniae were investigated, 5 being penicillin-resistant and 3 being multi-resistant. Regimens evaluated included single 20 and 80 mg/kg doses and 24 hour treatment with 5 to 640 mg/kg/day given at dosing intervals of 3, 6, 12, and 24 hours. Based on thigh CFU values, BB-83698 was bactericidal and exhibited in vivo post-antibiotic effect (PAE) values of 6 to 13 hours. The pharmacodynamic parameter which best correlated with efficacy in neutropenic mice was the ratio of 24 hour area under the plasma concentration versus time curve (AUC)/MIC (r2 = 0.91). Efficacy was predicted when AUC/MIC was 133 or higher. The ratio of peak plasma concentration (Cmax)/MIC and proportion of the dosing interval where plasma concentration exceeded the MIC had lower r2 values of 0.50 and 0.60, respectively. The dosing interval had no effect on the total dose needed to exert a bacteriostatic effect in neutropenic mice. These data supported evaluation of a once-daily dosing regimen for the treatment of pneumococcal infections [Craig 2001].
In a murine model of pneumococcal pneumonia (MICs of the 4 test strains were 0.06 to 0.25 mg/L), BB-83698 80 mg/kg twice daily SC or 160 mg/kg once daily SC protected 70 to 100 percent of animals at 10 days post-inoculation. Bacterial burden in the bloodstream and lungs both fell in a dose-dependent fashion [Azoulay-Dupuis et al 2004]. After a single 80 mg/kg SC dose, mean serum and lung tissue Cmax/MIC ratios were 238 and 1032 to 1, respectively. Corresponding mean 24 hour AUC/MIC ratios were 957 and 3823 to 1 [Azoulay-Dupuis et al 2004].
BB-3497 (IV, oral) was evaluated in the murine sepsis model using S. aureus Smith. The ED50’s were 7 mg/kg (IV) and 8 mg/kg (oral). With a methicillin-resistant S. aureus test strain, the ED50 for oral drug rose to 14 mg/kg [Clements et al 2001]. NVP LBM-415 was evaluated in a M. pneumoniae pneumonia model in mice. Drug was dosed as 50 mg/kg once daily SC. As compared to untreated mice, drug-treated mice exhibited significant reductions in bronchoalveolar lavage (BAL) fluid M. pneumoniae CFU counts on days 6 and 13 on treatment and lung histopathology scores on days 3, 6, and 13 on treatment and 7 days after treatment. Airway obstruction fell significantly in active-treated mice on days 1, 3, and 6 on treatment and 7 days after treatment while airway hyperresponsiveness fell only on day 3 on treatment. Significant reductions were found in the BAL fluid concentrations of most, but not all, cytokines and chemokines (eg, tumor necrosis factor alpha, interferon gamma, interleukins −6 and −12, functional interleukin −8, monocyte chemotactic protein −1, macrophage inflammatory protein 1±, monokine induced by interferon-gamma, and interferon-inducible protein 10) in active-treated mice [Fonseca-Aten et al 2005].
VRC 3375 was evaluated in the murine sepsis model (S. aureus Smith). The ED50 values were 32 mg/kg (IV), 17 mg/kg (SC), and 21 mg/kg (oral) [Chen et al 2004]. In this same model, the ED50 for VRC 4307 was 17.9 mg/kg (SC) (no efficacy was seen with oral VRC 4307, even at 30 mg/kg) [Hackbarth et al 2002]. For VRC 4232, the ED50 was 29.7 mg/kg (SC) [Hackbarth et al 2002].
The acute toxicology has been described for one PDI, VRC 3375. The LD50 (lethal dose for 50% of mice) was 447 mg/kg (IV), with 3 deaths occurring immediately post-injection. No deaths were reported after SC and oral dosing. Day 8 necropsy results were unremarkable except in 3 mice in the 500 mg/kg SC groups (all had dark red skin plaques at the injection site) [Chen et al 2004].
Animal pharmacokinetics
The BB series of PDIs are quantified in biological samples using high performance liquid chromatography (HPLC) with tandem mass spectrometry (MS) [Clements et al 2001; Azoulay-Dupuis et al 2004; Gross et al 2004; Ramanathan-Girish et al 2004]. The VRC series of PDIs are also quantitated using HPLC with tandem MS as well as HPLC with ultraviolet detection [Hackbarth et al 2002, Chen et al 2004a;Teo et al 2006]. Table 5 illustrates the mean pharmacokinetic parameters of a variety of peptide deformylase inhibitors in mice and rats [Clements et al 2001; Craig 2001; Hackbarth et al 2002; Azoulay-Dupuis et al 2004; Chen et al 2004a; Gross et al 2004; Ramanathan-Girish et al 2004; Teo et al 2006].
Table 5.
Mean pharmacokinetic parameters for selected peptide deformylase inhibitors in mice and rats
| Reference/species | Compound | Regimen | Cmax (mg/L) | Tmax (min.) | AUC (mg/L•h) | CL (mL/min/kg) | t 1/2 (h) | Vss(L/Kg) | F (%) |
|---|---|---|---|---|---|---|---|---|---|
| [Azoulay-Dupuiset al 2004]/mice | BB-83698 | 80 mg/kg SC × 1 dose | 14.3a | 60 | 57.4a,c | – | 2.5a | – | – |
| 61.9b | 60 | 229.4b,c | – | 2.6b | – | – | |||
| [Ramanathan-Girish et al 2004]/ mice | BB-83698 | 10 mg/kg IV × 1 dose | 6.3 | – | 3.8 | 44.4 | 1.0 | 2.0 | – |
| 50 mg/kg IV × 1 dose | 44.6 | – | 55.1 | 15.1 | 2.6 | 2.6 | – | ||
| [Craig 2001]/ neutropenic mice | BB-83698 | 20 and 80 mg/kg SC and PO × 1 dose each | – | – | – | – | 1.6–1.7 | – | 50 |
| [Gross et al 2004]/ mice | BB-81384 | 10 mg/kg IV × 1 dose | 15.1 | – | 6.5d | 25 | 2.2 | 1.6 | – |
| 10 mg/kg oral × 1 dose | 1.6 | – | 3.6d | – | 3.1 | – | 55 | ||
| 50 mg/kg oral × 1 dose | 21.3 | – | 28.6d | – | 3.7 | – | 88 | ||
| [Clements et al 2001]/rats | BB-3497 | 100 mg/kg oral × 1 dose | 24.0 | – | 34e | – | – | – | – |
| [Chen et al 2004a]/ mice | VRC 3375 | 100 mg/kg IV × 1 dose | – | – | – | – | 0.25 | – | – |
| 100 mg/kg SC × 1 dose | 66 | 20 | – | – | 0.25 | – | – | ||
| 100 mg/kg PO × 1 dose | 43 | 10 | – | – | 0.25 | – | 64 | ||
| [Hackbarth et al 2002]/mice | VRC 4232 | 13 mg/kg IV × 1 dose | 5580 | – | – | – | 1.1 | – | – |
| 13 mg/kg oral × 1 dose | 167 | – | – | – | – | – | 3.2 | ||
| VRC 4307 | 3.7 mg/kg IV × 1 dose | 1720 | – | – | – | 0.1 | – | – | |
| 3.7 mg/kg oral × 1 dose | 1.5 | 15 | – | – | – | – | 0.1 | ||
| [Teo et al 2006]/ mice | PDF-611 | 1 mg/kg IV × 1 dose | 604f | – | 254g | 65.8 | 0.5 | 3.0 | – |
| 5 mg/kg oral × 1 dose | 194f | 15 | 566g | – | 3.7 | – | 45 |
Abbreviations:Cmax = peak plasma concentration, Tmax = time to Cmax, AUC = area under the plasma concentration-versus-time curve, CL = total body clearance, t 1/2 = terminal disposition half-life, Vss = volume of distribution at steady-state, F = oral bioavailability.
serum.
lung.
Over 10 hrs. post-dose.
Over 5 hrs. post-dose.
Over 24 hrs. post-dose.
ng/mL.
ng/mL•h.
Absorption
BB-83698 is reasonably well-absorbed by the oral route, with a mean bioavailability of 50 percent in neutropenic mice [Craig 2001]. Absorption proceeds rapidly after oral administration, with a mean time to peak plasma concentration (Tmax) of about 1 hour [Azoulay-Dupuis et al 2004]. BB-81384 is also reasonably well-absorbed after oral administration, with mean bioavailabilities of 55 percent (10 mg/kg orally) and 88 percent (50 mg/kg orally) [Gross et al 2004]. Of the VRC series of compounds, only VRC 3375 is reasonably well-absorbed, with a mean bioavailability of 64 percent and Tmax of 0.167 to 0.33 hours [Chen et al 2004a]. The mean oral bioavailabilities of VRC 4232 and VRC 4307 are 3.2 and 0.1 percent, respectively [Hackbarth et al 2002]. Absorption proceeds rapidly with VRC 4307, the mean Tmax being 0.25 hours [Hackbarth et al 2002]. The antimycobacterial PDF-611 is also reasonably well-absorbed, with a mean bioavailability of 45 percent and Tmax of 0.25 h [Teo et al 2006].
For NVP LBM-415, the mean Tmax after oral dosing was the same in rats and mice (0.5 hr) [Chen et al 2004a]. Oral bioavailability averaged 65% in mice but ranged from 21 to 94% in rats over a dose range of 12 to 436 mg/kg (latter being an effect of saturable metabolism, not altered bioavailability) [Chen et al 2004a].
Distribution
For VRC 3375 in mice, concentrations in heart, kidney, and lung tissue exceeded those in serum while those in muscle and serum were comparable at 3 minutes following IV dosing of 100 mg/kg [Chen et al 2004a].
For BB-81384, after a single 10 mg/kg oral dose, lung tissue and plasma concentrations were approximately equivalent over a five hour period following dosing while thigh muscle concentrations were approximately one-third those in plasma over the same time period. Peak concentrations were achieved within 20 minutes in all 3 compartments [Gross et al 2004]. For BB-83698, after an 80 mg/kg SC dose, lung tissue concentrations exceeded those of serum by approximately four-fold, whether measured by Cmax or AUC (Table 5) [Azoulay-Dupuis et al 2004]. For NVP LBM-415, lung tissue concentrations were double those in plasma after both oral and IV dosing in mice [Chen et al 2004a].
Metabolism and excretion
VRC 4232/4307 are rapidly metabolized in mouse and rat hepatic microsomes, in contrast to their metabolism in human microsomes [Hackbarth et al 2002]. For VRC 4232, metabolism by mouse, rat, and human microsomes is extensive, with hydroxylation predominating but also oxidation, hydrolysis, and reduction, all followed by glucuronidation [Hackbarth et al 2002]. As all metabolites exhibit an altered hydroxamic acid moiety, all should be microbiologically inactive [Hackbarth et al 2002]. The pattern of metabolites varied with species and with the model used (ie, in vivo animal versus in vitro liver microsomes). Thus, the patterns of the three major metabolites were different for the in vivo mouse, mouse liver microsome, rat liver microsome and human liver microsome models. In human liver microsomes, the three major metabolic products (in descending order of amount) were the product of proline hydroxylation and hydrolysis, the product of hydrolysis alone, and the glucuronide conjugate of the parent compound [Hackbarth et al 2002].
Multiple-dose studies with BB-83698 have been conducted in rats and dogs at 3 dose levels (10, 22, and 50 mg/kg IV once daily × 28 days). In rats, mean day 1 peak plasma concentrations rose 2.5-fold (expected 2.2-fold from 10 to 22 mg/kg) and 4.9-fold (expected 5-fold from 10 to 50 mg/kg) in males with corresponding values in females of 2.6- and 6.6-fold. Mean day 1 AUC rose 2.2-fold and 6.6-fold in males and 3.1-fold and 8.8-fold in females. Mean day 28 peak plasma concentrations rose 3.4-fold and 3.3-fold in males and 2.6-fold and 4.2-fold in females. Mean day 28 AUC rose 3.4-fold and 14.4-fold in males and 2.7-fold and 9.4-fold in females. In rats, BB-83698 exhibited non-linear pharmacokinetics in both genders, especially at the higher 50 mg/kg dose and when quantitated using AUC data (ie, the rise in AUC was disproportionately high compared to the rise in dose). In the dog, non-linear pharmacokinetics were also noted in both genders. However, this was apparent with both Cmax and AUC data and with the 22 and 50 mg/kg dose. In the case of the dog, the rises in Cmax and AUC were disproportionately low compared to the rises in dose (dog data not shown) [Ramanathan-Girish et al 2004].
From day 1 to day 28, the mean degrees of accumulation as measured by change in Cmax were 1.2-, 1.6-, and 0.8-fold in males and 1.5-, 1.6-, and 1.0-fold in females (with doses of 10, 22, and 50 mg/kg, respectively). The corresponding degrees of accumulation as measured by change in AUC were 0.8-, 1.4-, and 1.9-fold in males and 0.8-, 0.7-, and 0.8- fold in females. In both rats and dogs, accumulation over 28 days occurred to a minor extent, if at all, whether quantitated using Cmax or AUC data (dog data not shown) [Ramanathan-Girish et al 2004].
After IV administration of NVP LBM-415, the mean total body clearances in mice and rats were 5.19 and 1.27 L/h/kg, respectively [Chen et al 2004]. Peak plasma concentration and AUC did not rise in proportion to dose in rats due to (presumed) saturable metabolism [Chen et al 2004]. In the first 24 hours after IV drug administration, mean urinary excretion of parent compound was 19 percent of the dose in mice and 48 percent of the dose in rats [Chen et al 2004a]. Mean biliary excretion in the first 7 hours after IV dosing was 23 percent in rats [Chen et al 2004a].
Human pharmacokinetics
Few published data exist regarding the pharmacokinetics of PDIs in humans. One published phase I clinical trial evaluated the pharmacokinetics and tolerability of ascending single intravenous doses (10, 25, 50, 100, 200, 325, 400, 475 mg) of BB-83698 in healthy male volunteers. All doses were administered over 15 minutes. No significant adverse events (AEs) occurred at any dose level. The overall mean ± SD clearance was 238 ± 130 mL/min, with CL’s ranging from 189 mL/min (475 mg) to 521 mL/min (10 mg). Linearity was evident with Cmax and AUC data, although these data did appear to deviate somewhat at the lowest and highest doses. Further studies are necessary to resolve this issue. However, Vdss and t 1/2 did vary significantly with dose (ranging from 57–106 L and 4.8–16.7 h, respectively). Plasma protein binding ranged from 78 to 82 percent [Ramanathan-Girish et al 2004].
Similar single and multiple dose pharmacokinetic studies have also been conducted with oral NVP LBM-415. After single oral doses of 100, 250, 500, 1000, 2000, and 3000 mg, the median Tmax was 1 hr or less in all dosing groups. Linearity was noted for dose-normalized 12-h AUC data. The mean t 1/2 ranged from 2 to 3 h except in the 2000 mg group (where the value was 4.2 h). When a 1000 mg dose was taken immediately after finishing breakfast, as compared to ingestion in the fasting state, the Tmax was prolonged (median rose from 0.5 to 2 h) and Cmax fell (mean fell from 15.5 to 6.7 mg/L) but the AUC was statistically unchanged. After multiple oral dosing (250, 500, and 1000 mg twice daily for 11 d), no accumulation was noted and steady-state was achieved after one day of dosing. These data supported the evaluation of a twice daily regimen in clinical trials [Jain et al 2005].
Timelines for development of BB-83698 and NVP LBM-415
BB-83698
In October 2002, BB-83698 (developed by British Biotech in collaboration with Genesoft) entered phase I studies in humans. Although dose-limiting central nervous system adverse events (AEs) such as trauma, unsteady gait and seizures had occurred in dogs, no significant AEs were seen in humans. The probable therapeutic dose was judged to be 475 mg, based on an AUC/MIC value of 184 (assuming, in turn, an MIC for S. pneumoniae of 0.25 mg/L). Development was terminated for unknown reasons when British Biotech became Vernalis. Oscient has now purchased this compound and one must wait to see the drug’s ultimate fate.
NVP LBM-415
In October 2003, NVP LBM-415 (developed by Vicuron in collaboration with Novartis) entered phase I studies in humans. Multiple dose pharmacokinetic studies used dosage regimens of 250 and 500 mg twice daily. No significant AEs were seen after administration of single doses up to and including 3g. However, Novartis and Vicuron discontinued its development in April 2004. Vicuron was purchased by Pfizer in June 2005. However, Novartis appears to be still pursuing PDI’s, most recently the antimycobacterial NVP PDF-611 (LBK611).
Conclusion
The relatively rapid development of microbial resistance after the entry of every new antimicrobial into the marketplace necessitates a constant supply of new agents to maintain effective pharmacotherapy. Despite extensive efforts to identify novel lead compounds from molecular targets, only the peptide deformylase inhibitors (PDIs) have shown any real promise, with some advancing to phase I human trials. Bacterial peptide deformylase, which catalyzes the removal of the N-formyl group from N-terminal methionine following translation, is essential for bacterial protein synthesis, growth, and survival. The majority of PDIs are pseudopeptide hydroxamic acids and two of these (IV BB-83698 and oral NVP LBM-415) entered phase I human trials. However, agents to the present have suffered from major potential liabilities. Their in vitro activity has been limited to gram-positive aerobes and some anaerobes and has been quite modest against the majority of such species (MIC90 values ranging from 1–8 mg/L). They have exerted bacteriostatic, not bacteriocidal, activity, thus reducing their potential usefulness in the management of serious infections in the immunocompromised. The relative ease with which microorganisms have been able to develop resistance and the multiple available mechanisms of resistance (mutations in fmt, defB, folD genes; AcrAB/TolC efflux pump; overexpression of peptide deformylase) are worrisome. These could portend a short timespan of efficacy after marketing. Despite these current liabilities, further pursuit of more potent and broader spectrum PDIs which are less susceptible to bacterial mechanisms of resistance is still warranted.
Note in proof
The recent discovery of a peptide deformylase homologue (mitochondrial PDF or mPDF) in humans has raised major objections to using PDF as a target of antimicrobial drugs. Indeed, mPDF is functional, displaying PDF activity in the human mitochondrion; is involved in the same essential pathway as in bacteria; is inhibited in vitro and in vivo by actinonin (see above); and, when inhibited, produces an antiproliferative effect triggered by mitochondrial dysfunction leading to cell death. Fortunately, PDF exists in 3 forms: PDF1B and PDF2 (both are bacterial PDFs) and PDF1A (human mPDF). Thus, the search is now on for compounds that selectively inhibit PDFs 1B and 2 and have no effect on PDF1A. At least one compound with this spectrum of enzyme activity is now known: 2-(5-bromo-1H-indol-3-yl)-N-hydroxyacetamide [Boularot et al 2007].
References
- Anderegg TR, Biedenbach DJ, Jones RN Quality Control Working Group. Quality control guidelines for MIC susceptibility testing of NVP PDF-713: a novel peptide deformylase inhibitor. Int J Antimicrob Agents. 2003;22:84–6. doi: 10.1016/s0924-8579(03)00114-6. letter. [DOI] [PubMed] [Google Scholar]
- Anderegg TR, Jones RN Quality Control Working Group. Disk diffusion quality control guidelines for NVP-PDF-713: a novel peptide deformylase inhibitor. Diagn Microbiol Infect Dis. 2004;48:55–7. doi: 10.1016/S0732-8893(03)00162-7. [DOI] [PubMed] [Google Scholar]
- Apfel C, Banner DW, Bur D, et al. Hydroxamic acid derivatives as potent peptide deformylase inhibitors and antibacterial agents. J Med Chem. 2000;43:2324–31. doi: 10.1021/jm000018k. [DOI] [PubMed] [Google Scholar]
- Apfel CM, Locker H, Evers S, et al. Peptide deformylase as an antibacterial drug target: target validation and resistance development. Antimicrob Agents Chemother. 2001;45:1058–64. doi: 10.1128/AAC.45.4.1058-1064.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azoulay-Dupuis E, Mohler J, Bedos JP. Efficacy of BB-83698, a novel peptide deformylase inhibitor, in a mouse model of pneumococcal pneumonia. Antimicrob Agents Chemother. 2004;48:80–5. doi: 10.1128/AAC.48.1.80-85.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JM, Turnidge JD, Inoue M, et al. Activity of a peptide deformylase inhibitor LBM415 (NVP PDF-713) tested against recent clinical isolates from Japan. J Antimicrob Chemother. 2005;55:276–8. doi: 10.1093/jac/dkh547. letter. [DOI] [PubMed] [Google Scholar]
- Boularot A, Giglione C, Petit S, et al. Discovery and refinement of a new structural class of potent peptide deformylase inhibitors. J Med Chem. 2007;50:10–20. doi: 10.1021/jm060910c. [DOI] [PubMed] [Google Scholar]
- Bowker KE, Noel AR, MacGowan AP. In vitro activities of nine peptide deformylase inhibitors and five comparator agents against respiratory and skin pathogens. Int J Antimicrob Agents. 2003;22:557–61. doi: 10.1016/s0924-8579(03)00246-2. [DOI] [PubMed] [Google Scholar]
- Butler MS, Buss AD. Natural products–the future scaffolds for novel antibiotics? Biochem Pharmacol. 2006;71:919–29. doi: 10.1016/j.bcp.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Chan PF, O’Dwyor KM, Palmer LM, et al. Characterization of a novel fructose-regulated promoter (PfcsK) suitable for gene essentiality and antibacterial mode-of-action studies in Streptococcus pneumoniae. J Bacteriol. 2003;185:2051–8. doi: 10.1128/JB.185.6.2051-2058.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DZ, Patel DV, Hackbarth CJ, et al. Actinonin a naturally occurring antibacterial agent is a potent deformylase inhibitor. Biochemistry. 2000;39:1256–62. doi: 10.1021/bi992245y. [DOI] [PubMed] [Google Scholar]
- Chen D, Hackbarth C, Ni ZJ, et al. Peptide deformylase inhibitors as antibacterial agents: identification of VRC3375 a proline-3-alkylsuccinyl hydroxamate derivative by using an integrated combinatorial and medicinal chemistry approach. Antimicrob Agents Chemother. 2004;48:250–61. doi: 10.1128/AAC.48.1.250-261.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Tembe V, Cramer J, et al. LBM415 (VIC-104959), a novel peptide deformylase inhibitor with favorable pharmacokinetic profile in rodents. Presented at the 44th Annual Meeting of the Interscience Conference on Antimicrobial Agents and Chemotherapy; November 2004a; Washington, D.C.. abstract F-1965–2004. [Google Scholar]
- Clements JM, Beckett RP, Brown A, et al. Antibiotic activity and characterization of BB-3497 a novel peptide deformylase inhibitor. Antimicrobial Agents Chemother. 2001;45:563–70. doi: 10.1128/AAC.45.2.563-570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig WA. In vivo pharmacodynamics of BB-83698, a deformylase inhibitor. Presented at the 41st Interscience Conference on Antimicrobial Agents and Chemotherapy; December 2001; Chicago, IL. abstract F-355. [Google Scholar]
- Credito K, Lin G, Ednie LM, Appelbaum PC. Antistaphylococcal activity of LBM415, a new peptide deformylase inhibitor compared with those of other agents. Antimicrob Agents Chemother. 2004;48:4033–6. doi: 10.1128/AAC.48.10.4033-4036.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cynamon MH, Alvirez-Freites E, Yeo AET. BB-3497 a peptide deformylase inhibitor, is active against Mycobacterium tuberculosis. J Antimicrob Chemother. 2004;53:403–5. doi: 10.1093/jac/dkh054. letter. [DOI] [PubMed] [Google Scholar]
- Dean CR, Narayan S, Daigle DM, et al. Role of the AcrAB-TolC efflux pump in determining susceptibility of Haemophilus influenzae to the novel peptide deformylase inhibitor LBM415. Antimicrob Agents Chemother. 2005;49:3129–35. doi: 10.1128/AAC.49.8.3129-3135.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean CR, Narayan S, Richards J, et al. Reduced susceptibility of Haemophilus influenzae to the peptide deformylase inhibitor LBM415 can result from target protein overexpression due to amplified chromosomal def gene copy number. Antimicrob Agents Chemother. 2007;51:1004–10. doi: 10.1128/AAC.01103-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein PH, Hu B, Edelstein MAC. In vitro and intracellular activities of LBM415 (NVP PDF-713) against Legionella pneumophila. Antimicrob Agents Chemother. 2005;49:2533–5. doi: 10.1128/AAC.49.6.2533-2535.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ednie LM, Pankuch G, Appelbaum PC. Antipneumococcal activity of LBM415, a new peptide deformylase inhibitor, compared with those of other agents. Antimicrob Agents Chemother. 2004;48:4027–32. doi: 10.1128/AAC.48.10.4027-4032.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca-Aten M, Salvatore CM, Mejias A, et al. Evaluation of LBM415 (NVP PDF-713), a novel peptide deformylase inhibitor, for treatment of experimental Mycoplasma pneumoniae pneumonia. Antimicrob Agents Chemother. 2005;49:4128–36. doi: 10.1128/AAC.49.10.4128-4136.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsche TR, Moet GJ, Jones RN. Commerical broth microdilution panel validation and reproducibility trials for NVP PDF-713 (LBM415), a novel inhibitor of bacterial peptide deformylase. Clin Microbiol Infect. 2004;10:857–60. doi: 10.1111/j.1198-743X.2004.00946.x. [DOI] [PubMed] [Google Scholar]
- Fritsche TR, Rhomberg PF, Jones RN. Comparisons of the inter-method susceptibility testing accuracy for LBM415 (NVP PDF-713) using 2,625 recent clinical isolates. Presented at the 44th Annual Meeting of the Interscience Conference on Antimicrobial Agents and Chemotherapy; November 2004a; Washington, D.C.. abstract D-1919–2004. [Google Scholar]
- Fritsche TR, Sader HS, Cleeland R, Jones RN. Comparative antimicrobial characterization of LBM415 (NVP PDF-713), a new peptide deformylase inhibitor of clinical importance. Antimicrob Agents Chemother. 2005;49:1468–76. doi: 10.1128/AAC.49.4.1468-1476.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Dahlgren C, Bylund J. Subinhibitory concentrations of the deformylase inhibitor actinonin increase bacterial release of neutrophil-activating peptides: a new approach to antimicrobial chemotherapy. Antimicrob Agents Chemother. 2003;47:2545–50. doi: 10.1128/AAC.47.8.2545-2550.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross M, Clements J, Beckett RP, et al. Oral anti-pneumococcal activity and pharmacokinetic profiling of a novel peptide deformylase inhibitor. J Antimicrob Chemother. 2004;53:487–93. doi: 10.1093/jac/dkh108. [DOI] [PubMed] [Google Scholar]
- Guilloteau J-P, Mathieu M, Giglione C, et al. The crystal structures of four peptide deformylases bound to the antibiotic actinonin reveal two distinct types: a platform for structure-based design of antibacterial agents. J Mol Biol. 2002;320:951–62. doi: 10.1016/s0022-2836(02)00549-1. [DOI] [PubMed] [Google Scholar]
- Hackbarth CJ, Chen DZ, Lewis JG, et al. N-alkyl urea hydroxamic acids as a new class of peptide deformylase inhibitors with antibacterial activity. Antimicrob Agents Chemother. 2002;46:2752–64. doi: 10.1128/AAC.46.9.2752-2764.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackbarth CJ, Lopez S, Wu C, et al. Activity of peptide deformylase (PDF) inhibitors against gram negative bacteria. Presented at the 43rd Annual Meeting of the Interscience Conference on Antimicrobial Agents and Chemotherapy; September 2003; Chicago, IL. abstract F-1479–2003. [Google Scholar]
- Hu x, Nguyen KT, Verlinde CLMJ, Hol WGJ, Pei D. Structure-based design of a macrocyclic inhibitor for peptide deformylase. J Med Chem. 2003;46:3771–4. doi: 10.1021/jm034113f. letter. [DOI] [PubMed] [Google Scholar]
- Hu X, Nguyen KT, Jiang VC, Lofland D, Moser HE, Pei P. Macrocyclic inhibitors for peptide deformylase: a structure-activity relationship study of the ring size. J Med Chem. 2004;47:4941–9. doi: 10.1021/jm049592c. [DOI] [PubMed] [Google Scholar]
- Huo B, Gong W, Rajagopalan PTR, Zhou Y, Pei D, Chen MK. Structural basis for the design of antibiotics targeting peptide deformylase. Biochemistry. 1999;38:4712–9. doi: 10.1021/bi982594c. [DOI] [PubMed] [Google Scholar]
- Jain R, Sundram A, Lopez S, et al. α-substituted hydroxamic acids as novel bacterial deformylase inhibitor-based antibacterial agents. Bioorg Med Chem Lett. 2003;13:4223–8. doi: 10.1016/j.bmcl.2003.07.020. [DOI] [PubMed] [Google Scholar]
- Jain R, Chen D, White RJ, Patel DV, Yuan Z. Bacterial peptide deformylase inhibitors: a new class of antimicrobials. Curr Med Chem. 2005;12:1607–21. doi: 10.2174/0929867054367194. [DOI] [PubMed] [Google Scholar]
- Jones RN, Rhomberg PR. Comparative spectum and activity of NVP-PDF386 (VRC4887), a new peptide deformylase inhibitor. J Antimicrob Chemother. 2003;51:157–61. doi: 10.1093/jac/dkg055. [DOI] [PubMed] [Google Scholar]
- Jones RN, Fritsche TR, Sader HS. Antimicrobial spectrum and activity of NVP PDF-716, a novel peptide deformylase inhibitor, tested against 1,837 recent gram-positive clinical isolates. Diagn Microbiol Infect Dis. 2004;49:63–5. doi: 10.1016/j.diagmicrobio.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Jones RN, Moet GL, Sader HS, Fritsche TR. Potential utility of a peptide deformylase inhibitor (NVP PDF-713) against oxazolidinone-resistant or streptogramin-resistant gram-positive organism isolates. J Antimicrob Chemother. 2004a;53:804–7. doi: 10.1093/jac/dkh184. [DOI] [PubMed] [Google Scholar]
- Jones RN, Sader HS, Fritsche TR. Antimicrobial activity of LBM415 (NVP PDF-713) tested against pathogenic Neisseria spp. (Neisseria gonorrhoeae and Neisseria meningitides) Diagn Microbiol Infect Dis. 2005;51:139–41. doi: 10.1016/j.diagmicrobio.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Kosowska-Shick K, Credito KL, Pankuch GA, Dewasse B, McGhee P, Appelbaum PC. Multistep resistance selection and PAE studies on antipneumococcal activity of LBM415 compared to other agents. Antimicrob Agents Chemother. 2007;51:770–3. doi: 10.1128/AAC.01150-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeds JA, Dean CR, Favre B, et al. In vitro selection of decreased susceptibility to the novel peptide deformylase inhibitor LB415 in three pathogens. Presented at the 44th Annual Meeting of the Interscience Conference on Antimicrobial Agents and Chemotherapy; November 2004; Washington, D.C.. abstract C1-1880–2004. [Google Scholar]
- Lofland D, Difuntorum S, Waller A, Clements J, Johnson K. Antibacterial spectrum, bactericidal activity, and susceptibility testing of the peptide deformylase inhibitor GBB-83698. Presented at the 43rd Annual Meeting of the Interscience Conference on Antimicrobial Agents and Chemotherapy; September 2003; Chicago, IL. abstract E-1712–2003. [Google Scholar]
- Lofland D, Difuntorum S, Waller A, et al. In vitro antibacterial activity of the peptide deformylase inhibitor BB-83698. J Antimicrob Chemother. 2004;53:664–8. doi: 10.1093/jac/dkh129. [DOI] [PubMed] [Google Scholar]
- Margolis PS, Hackbarth CJ, Young DC, et al. Peptide deformylase in Staphylococcus aureus: resistance to inhibition is mediated by mutations in the formyltransferase gene. Antimicrob Agents Chemother. 2000;44:1825–31. doi: 10.1128/aac.44.7.1825-1831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis P, Hackbarth C, Lopez S, et al. Resistance of Streptococcus pneumoniae to deformylase inhibitors is due to mutations in defB. Antimicrob Agents Chemother. 2001;45:2432–5. doi: 10.1128/AAC.45.9.2432-2435.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazel D, Pochet S, Marliere P. Genetic characterization of polypeptide deformylase, a distinctive enzyme of eubacterial translation. EMBO J. 1994;13:914–23. doi: 10.1002/j.1460-2075.1994.tb06335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckerman G, Dean CR, Yu D, Manni K, Narayan S, Dzink-Fox J. Role of AcrAB-TolC efflux in reducing susceptibility to LBM-415, azithromycin, and telithromycin in a Haemophilus influenzae murine lung infection model. Presented at the 45th Annual Meeting of the Interscience Conference on Antimicrobial Agents and Chemotherapy; December 2005; Washington, DC. abstract C1-1033-2005. [Google Scholar]
- Nilsson AI, Zorzet A, Kanth A, Dahlstrom S, Berg OG, Andersson DI. Reducing the fitness costs of antibiotic resistance by the amplification of initiator tRNA genes. PNAS. 2006;103:6976–81. doi: 10.1073/pnas.0602171103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan-Girish S, McColm J, Clements JM, et al. Pharmacokinetics in animals and humans of a first-in-class peptide deformylase inhibitor. Antimicrob Agents Chemother. 2004;48:4835–42. doi: 10.1128/AAC.48.12.4835-4842.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roblin PM, Hammerschlag MR. In vitro activity of a new antibiotic, NVP-PDF386 (VRC4887) Antimicrob Agents Chemother. 2003;47:1447–8. doi: 10.1128/AAC.47.4.1447-1448.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snydman DR, Jacobus NV, McDermott LA. Evaluation of the in vitro activity of NVP-LBM415 against clinical anaerobic isolates with emphasis on the Bacteroides fragilis group. J Antimicrob Chemother. 2005;55:1024–8. doi: 10.1093/jac/dki107. [DOI] [PubMed] [Google Scholar]
- Teo JWP, Thayalan P, Beer D, et al. Peptide deformylase inhibitors as potent antimycobacterial agents. Antimicrob Agents Chemother. 2006;50:3665–73. doi: 10.1128/AAC.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Aller GS, Nandigama R, Petit CM, et al. Mechanism of time-dependent inhibition of polypeptide deformylase by actinonin. Biochemistry. 2005;44:253–60. doi: 10.1021/bi048632b. [DOI] [PubMed] [Google Scholar]
- Waites KB, Reddy NB, Crabb DM, Duffy LB. Comparative in vitro activities of investigational peptide deformylase inhibitor NVP LBM-415 and other agents against human mycoplasmas and ureaplasmas. Antimicrob Agents Chemother. 2005;49:2541–2. doi: 10.1128/AAC.49.6.2541-2542.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, White R, Yuan Z. Proteomic study of peptide deformylase inhibition in Streptococcus pneumoniae and Staphylococcus aureus. Antimicrob Agents Chemother. 2006;50:1656–63. doi: 10.1128/AAC.50.5.1656-1663.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watters AA, Jones RN, Leeds JA, Denys G, Sader HS, Fritsche TR. Antimicrobial activity of a novel peptide deformylase inhibitor, LBM415, tested against respiratory tract and cutaneous infection pathogens: a global surveillance report (2003–2004) J Antimicrob Chemother. 2006;57:914–23. doi: 10.1093/jac/dkl093. [DOI] [PubMed] [Google Scholar]
- Wise R, Andrews JM, Ashby J. In vitro activities of peptide deformylase inhibitors against gram-positive pathogens. Antimicrob Agents Chemother. 2002;46:1117–18. doi: 10.1128/AAC.46.4.1117-1118.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootton M, Howe RA, MacGowan AP, Walsh TR, Bennett PM. In vitro activity of BB83698 and two other peptide deformylase inhibitors compared to ciprofloxacin, moxifloxacin, gentamicin and linezolid against heterogeneous glycopeptide intermediate Staphylococcus aureus (hGISA) and GISA. Presented at the 41st Interscience Conference on Antimicrobial Agents and Chemotherapy; December 2001; Chicago, IL. abstract F-352. [Google Scholar]
- Yeo J-S, Zheng C-J, Lee S, Kwak J-H, Kim W-G. Macrolactin N, a new peptide deformylase inhibitor produced by Bacillus subtilis. Bioorg Med Chem Lett. 2006;16:4889–92. doi: 10.1016/j.bmcl.2006.06.058. [DOI] [PubMed] [Google Scholar]