

Abstract
Highly active antiretroviral therapy (HAART) has dramatically improved the prognosis of patients with HIV. Low adherence and toxicity among HIV-positive patients starting HAART, however, can lead to discontinuation of therapy and limit long-term treatment success. Moreover, increasing prevalence of primary resistance (>10%) as well as the accumulation of mutations resulting from continued selection pressure exerted by ongoing antiretroviral treatment in patients failing virologically, mean that new compounds are needed that retain antiretroviral activity against resistant strains. Tipranavir (Aptivus®) is a novel protease inhibitor (NPPI), which is characterized by a unique genetic resistance profile that allows it to remain active against HIV strains resistant to currently licensed protease inhibitors (PIs). Tipranavir was approved and licensed in the US and Europe in 2005 for treatment-experienced patients. This review summarizes the currently available data and studies on tipranavir and discusses the possible position of tipranavir in the currently available armamentarium of antiretroviral drugs.
Keywords: tipranavir, resistance, salvage, AIDS
Introduction
Substantial improvements in HIV/AIDS-associated morbidity and mortality has been achieved, since highly active antiretroviral therapy (HAART) was introduced in the mid-1990s (Carpenter et al 2000; Jones et al 1999; Palella et al 1998). However, despite these achievements, an increasing population of patients harbour HIV strains resistant against at least one class of antiretroviral agents (Little et al 2002). Of a representative sample from 83,475 patients in the US taking antiretroviral (ARV) therapy, 76% had plasma viral loads greater than 500 copies/mL, and were resistant to at least one drug; 48% were infected with HIV that was resistant to two ARV drug classes (Richman et al 2004). Furthermore, in a further study up to 50% of patients were reported to have failed their initial antiretroviral regimen after a median duration of only 1.6 years (Bartlett et al 2001; Chen et al 2003). However, recent data suggest that with the availability of boosted protease inhibitors (PIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs), with improved pharmacokinetic profiles and increasing forgiveness of the regimens, the percentage of patients developing virological failure under first-line ARV therapy is declining substantially (Lampe et al 2005).
Moreover, another important issue in resistance development is the increasing number of new primary multi-drug resistant HIV infections which in some areas like New York (USA) already exceeds 3.8% (Clavel and Hance 2004; Markowitz et al 2005; Boden et al 1999).
The emergence of ARV drug resistance reduces the ability of current agents to control viral replication and to construct effective ARV regimens for patients who are failing their treatment regimens. Evidently, with each drug failure the number of alternative agents that are active against the resistant virus becomes more limited (Montaner 2003; Yeni et al 2004) and therefore the need for new potent drugs is obviously growing with the development of (multidrug-)resistant strains and it is necessary to provide new treatment options for patients with resistant viral isolates (Montaner 2003).
Tipranavir (Aptivus®, Boehringer Ingelheim) (TPV) was licensed and approved in June and October 2005 in the US and Europe, respectively. TPV is a novel protease inhibitor which is highly selective for therapeutic intervention in the viral life cycle by blocking the HIV-1 and HIV-2 protease (Thaisrivongs and Strohbach 1999). Importantly, TPV has a unique resistance profile which is characterized by sufficient antiviral activity in vitro against viral strains cross-resistant to other marketed protease inhibitors (Larder et al 2000). This property makes the drug interesting for treatment of patients who are PI-experienced or who are infected with PI-resistant viral isolates.
Dosage of TPV
A self-emulsifying drug delivery system (SEDDS), contains 250 mg TPV free acid in a soft gel TPV capsule. Dosage for treatment-experienced patients is recommended at 500 mg twice daily taken with a meal, as defined in the pre-clinical Phase IIb dose optimization study (BI 1182.52) performed in 216 treatment-experienced patients (Gathe et al 2003). TPV, as other PIs, must be co-administered with ritonavir (RTV) to enhance plasma levels; a RTV dose of 200 mg bid is recommended. In study 1182.52, patients replaced their failing PI for TPV/r administered at 3 different doses for 2 weeks and subsequently changed their background regimens and were followed for a total of 24 weeks. Patients who were taking 500/200 mg TPV/r, showed similar antiviral activity compared with patients treated with 750/200 mg TPV/r, although the latter group experienced 10% more frequent (severe) adverse events after 24 weeks. In addition, one arm was treated with 500 mg TPV and a boosting dosage of RTV 100 mg bid, as commonly used for other PIs. Even though patients achieved a pVL reduction of more than 0.5 log10 after 2 weeks, the viral load reduction could not be sustained after 24 weeks. Thus, the daily pill burden for TPV/r is eight pills, which is higher than for the other currently licensed PIs. In addition the use of TPV/r in a once-daily regimen has not been investigated and is thus not available at present.
Resistance profile of tipranavir
In vitro data has shown, that primary TPV resistance develops slowly and involves the acquisition of several specific protease gene mutations (Doyon et al 2005). These results demonstrate that in vitro up to 10 mutations are involved in the development of resistance to TPV: L10F, I13V, V32I, L33F, M36I, K45I, I54V, A71V, V82L, and I84V. All of these mutations have previously been described in the presence of other PIs except for the active site mutation V82L which seems to be unique to TPV (Johnson et al 2005). HIV protease mutation of Val82 to Ala, Phe, Thr, or Ser has previously been implicated in resistance to PIs. However, this is the first description of a Leu substitution at the V82 position (Johnson et al 2005). Interestingly, an initial TPV resistance study identified V82L in the protease of emerging viruses, but investigators were unable to reconstitute a resistance phenotype by reintroducing this mutation into a wild type background (Kemp et al 2000). This study confirmed that V82L alone does not confer resistance to TPV, but contributes to a 2.4-fold increase in resistance when selected for on a background of five pre-existing protease mutations. These results also highlight the requirement for the presence of six specific mutations in the protease (I13V, V32I, L33F, K45I, V82L, I84V), and three in the active site of the enzyme to confer >10-fold resistance to TPV. Therefore, it suggests that although the majority of mutations selected by this PI do not significantly differ from those selected by other PIs, the genetic barrier for the development of resistance to TPV is higher than for most other PIs. This could explain why TPV has such a broad activity against PI resistant clinical isolates.
To determine the relationship between protease mutations detected in genotype to phenotypic susceptibility and virologic response to TPV a series of regression analyses were performed using the TPV program Phase II data. Validation included analyses of Phase III study datasets to determine if the same mutations would be selected and to assess how these mutations contribute to multiple regression models of tipranavir phenotype and of virologic response. A string of 16 protease positions and 21 mutations were identified : 10V, 13V, 20M/R/V, 33F, 35G, 36I, 43T, 46L, 47V, 54A/M/V, 58E, 69K, 74P, 82L/T, 83D, and 84V. An increasing number of mutations (>8 TPV associated mutations) at these positions were associated with reduced phenotypic susceptibility and blunted virologic responses to TPV. Lower TPV scores and the use of active drugs, especially of a new class, were associated with improved responses to a TPV-containing regimen.
In addition, TPV shows considerable efficacy in the presence of universal protease inhibitor resistance associated mutations (PRAMs) like L33I/V/F, V82A/F/L/T, I84V and L90M, which are mutations conferring resistance against all currently available PIs. This robust activity against protease inhibitor (PI-) resistant strains still remains unexplained, although some authors have speculated that TPV has a broader molecular flexibility, which allows it to fit into the active pocket of the protease enzyme (Larder et al 2000). Other mutations, outside the active site (secondary mutations), can increase resistance in the presence of major primary mutations like L10I/V, K20M/L/T, and I54V, as well as additional primary mutations such as M46I, and V82A/F/L/T. However, even the activity of TPV is not unlimited; despite good short term activity and a >1.0 log viral load reduction after 2 weeks in patients whose isolates carried 3 or more PRAMs, the susceptibility to TPV begins to decrease at this level of protease resistance, probably in part, due to the lack of active drugs to support TPV’s activity.
It is important to point out that the resistant strains which can emerge under a “classic” PI-based regimen differ slightly from those that emerge under a TPV based regimen. This means that a failure of a TPV-containing regimen does not necessarily imply that the virus has become resistant to all other PIs. However, in vitro data suggests that TPV/r resistant viral strains, which developed from wild-type HIV-1, exhibited a 2 to 154 fold decrease in susceptibility to the PIs amprenavir, indinavir, lopinavir, nelfinavir and ritonavir, but still remained sensitive to saquinavir (Doyon et al 2005).
This is most probably attributable to the fact that TPV failed to select for the SQV primary mutations G48V and L90M in these studies (Johnson et al 2005; Jacobsen et al 1995). Characterization of human cross-resistance was most significant for ATV and RTV, and in vitro resistance for these was previously reported to be associated with mutations at positions V32I/L33F/A71V/I84V (ATV) (Gong et al 2000) and V82F/I84V (RTV) (Markowitz et al 1995).
Efficacy and antiviral activity
TPV appears to have antiviral activity against most viral isolates with PI resistance (Larder et al 2000; Back et al 2000; Poppe et al 1997; Rusconi et al 2000; Schwartz et al 2002). In a highly active antiretroviral therapy regimen, especially in chronic HIV-1 infected patients with multiple resistant isolates, it is often difficult to discern the effects of a single drug. To be able to draw direct conclusions for the efficacy of a single drug, it is therefore important to obtain monotherapy data, which can be extracted from several studies. In the above cited dose finding study BI 1182.52 (n = 216) the failing PI was discontinued and replaced by TPV/r for two weeks with no adjustment changes in the backbone regimen. In this context TPV/r was considered a functional monotherapy. After two weeks, an optimized backbone regimen (OBR) was added based on genotypic testing and patients were switched to the OBR + TPV/r. In the intent to treat analysis (last observation carried forward [LOCF]) the median plasma viral load (pVLs) decreased by 0.87–1.18 log10 copies/mL during the first two weeks of TPV/r functional monotherapy (Gathe et al 2003). Similar reductions were obtained when treatment-naïve patients (n = 31) received TPV or TPV/r monotherapy in different dosages for 14 days (McCallister et al 2004). At day 15 the median decreases in pVL were significantly greater in the two RTV-boosted arms with a decline of −1.43 log10 copies/mL and −1.64 log10 copies/mL compared to the unboosted TPV arm treated with 1200 mg BID (old formulation of TPV), in which the patients had only a pVL reduction of 0.77 log10 copies/mL. This reduction was too low to keep a sustainable plasma viral load reduction as it has been shown for different ARVs that a decrease of pVL of less than 0.75 log10 copies/mL already at day 6 indicates a poor long-term response in 99% of the patients (Polis et al 2001). Whether lower doses of TPV/r than the one currently approved can exert significant antiviral activity in patients with less resistant isolates remains to be determined.
Efficacy data of the Phase III trials: RESIST-1 and RESIST-2
The TPV Phase III program studied treatment-experienced patients with proven viral resistance in two large, multi-national, controlled, pivotal trials (RESIST-1 and RESIST-2 -Randomized Evaluation of Strategic Intervention in Multi-Drug ReSistant Patient with Tipranavir) (Hicks et al 2006). RESIST-1, performed in the United States, Canada and Australia and RESIST-2, performed in 19 countries of Europe and Latin America, were Phase III safety and efficacy trials in 620 (RESIST-1) and 863 (RESIST-2) HIV- positive adults with three-class ARV experience, who had failed at least two PI-based regimens, and whose isolates had no more than two protease mutations at position 33, 82, 84 or 90. This outline was employed based on the assumption that patients with three or more mutations at these key positions were less likely to respond to any PI-based regimen, including TPV/r.
Both trials examined the treatment response up to 48 weeks of TPV/r versus a comparator (control) group (CPI) in which patients received one of several marketed RTV-boosted PIs. Before randomization, investigators selected a comparator PI (among lopinavir, indinavir, saquinavir, and amprenavir) that offered patients the best opportunity for treatment response and an optimized background regimen based on treatment history and resistance testing. The use of enfuvirtide (ENF) was permitted in both arms, but had to be chosen before randomization. Following the 1:1 randomization, patients could be treated with any of four RTV-boosted comparator PIs or TPV/r along with an optimized non-PI ARV backbone regimen. Patients in these trials were highly treatment-experienced (median use of 12 ARV before randomization) and the majority were most likely resistant to the comparator PI chosen. The median baseline viral load and CD4+ count were 4.82 log10 copies/mL and 155 cells/μl, respectively.
A total of 1483 patients were randomized and treated in RESIST-1 and -2. At week 24, data of 1159 patients was available for analysis. The two treatment arms were well matched in demographics and disease baseline characteristics (see Table 1 for baseline characteristics). Patients had taken a median of 12 ARV drugs before enrolment: six NRTIs, one NNRTI and four PIs. Twelve percent of the patients had taken enfuvirtide previously. In RESIST-1 the most frequently chosen comparator PI was lopinavir/r but was amprenavir/r in RESIST-2. However based on genotypic data, the virus was already resistant to the pre-selected PI in 66.7% of the patients in the comparator group.
Table 1.
Baseline demographic data of the RESIST studies I and II
| Total treated | 582 | 577 | ||
|---|---|---|---|---|
| Median Age (years [range]) | 43.0 | [17-80] | 43.0 | [21-72] |
| Gender (male, N[%]) | 503 | [86.4] | 516 | [89.4] |
| Race (Ewhite, N [%]) | 430 | [73,9] | 414 | [71.8] |
| Median baseline HIV-1 RNA (log10 copies/ml [range]) | 4.83 | [2.34–6.52] | 4.82 | [2.01–6.76] |
| Patients stratified by baseline viral load (N [%]) | ||||
| ≤10,000 copies/ml | 91 | [15.6] | 90 | [15.6] |
| >10,000–100,000 copies/ml | 259 | [44.5] | 253 | [43.8] |
| >100,000 copies/ml | 232 | [39.9] | 234 | [40.6] |
| Median baseline | 155 | [1–1893] | 158 | [1–1184] |
| CD4 + cell count (cells/μl [range]) | ||||
| Patients stratified by baseline CD4 + count (N [%]) | ||||
| ≥350 cells/μl | 87 | [14.9] | 99 | [17.2] |
| 201–350 cells/μl | 133 | [22.9] | 134 | [23.2] |
| 50–200 cells/μl | 226 | [38.8] | 193 | [33.4] |
| <50 cells/μl | 133 | [22.9] | 148 | [25.6] |
In the combined analysis of these two studies, the mean plasma viral load reduction was 1.2 log10 copies/ml in the TPV/r arm and 0.6 log10 copies/mL in the comparator arm at week 24. This response was maintained at week 48 and the mean change in VL was a reduction of −1.14 log10 copies/mL in patients taking TPV/r and only −0.54 log10 copies/mL in the control arm at this time point. The maximal pVL reduction occurred at week 4 in the TPV/r arm and at week 2 in the control group. Achieving an undetectable viral load (<50 cop/mL) in these treatment-experienced populations was difficult, taking into account the extensive antiretroviral pre-treatment of the study subjects. Only 23.9% of the patients in the TPV/r and 9.4% in the control group were fully suppressed after being administered the prescribed chemotherapeutic regimens at week 24 (p < 0.001). Similar to what has been seen in other studies, patients with lower pVL and higher CD4+ counts at baseline responded better to therapy than those with more advanced disease (van Leth et al 2004; Clotet et al 2004). Patients with 2 or 3 previously used PIs, who were more likely to have additional ARV drugs to use as background regimens, had a greater decrease in viral load than more experienced patients and therefore were more likely to achieve a fully suppressed viral load. Also the additional use of enfuvirtide (ENF) was associated with a better outcome for both groups; 58.2% of the patients taking TPV/r plus ENF achieved a treatment response, defined as VL reduction ≥1.0 log10 copies/mL, compared to 25.8% of patients in the control group + ENF (Figure 1) (Cooper et al 2005). This observation was similar to other studies, that patients taking regimens containing additional, active ARV drugs, experienced a substantially higher decrease in viral load and increase in CD4+ counts compared to those who had no more additional active drugs available.
Figure 1.
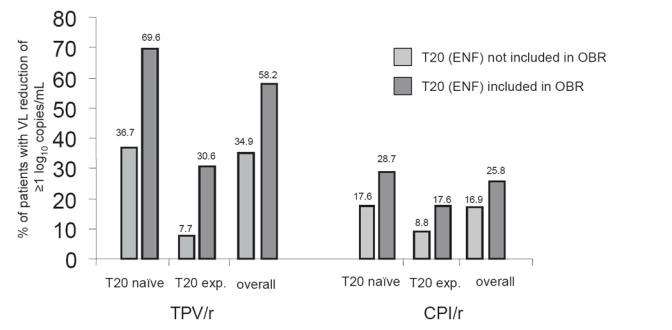
Effect of T20 (ENF) on Treatment Response in the TPV/r compared to the CPI/r Arms.
At week 48 patients taking TPV/r were twice as likely to experience a treatment response as those in the control group. The treatment response (TR) rate was 33.6% (251/746) in the TPV/r arm and 15.3% (113/737) in the comparator arm (p < 0.001). More than 80% of patients who achieved a treatment response had week 48 plasma viral loads below 400 c/mL. The TR was 48.5% (82/169) in patients who took TPV/r plus an optimized background regimen (OBR) that contained at least one active ARV, eg, ENF, but it was 20.0% (27/135) in patients who took CPI/r plus ENF. Patients taking TPV/r were more likely to achieve a durable response than control patients.
In one sub-analysis the treatment response rate of each single comparator PI was stratified and compared to the treatment response rate of TPV/r at week 24. This analysis showed that the treatment response rates for each single comparator PIs were lower than the response rate for TPV/r. In detail, the treatment response rates were 39.6% for TPV/r, 21.4% for LPV/r, 15.3% for SQV/r and 18.8% for APV/r. However, comparing the TPV/r group to a LPV/r inexperienced group in a sub-interim analysis, the difference in virologic response in both groups was not statistically significant (Cooper et al 2005) (see Figure 2).
Figure 2.
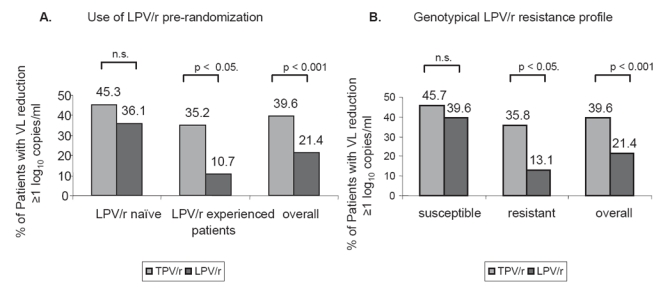
Comparison of Efficacy of TPV/r and LPV/r in LPV stratum (data from the RESIST interim analysis).
In a 48-week analysis, the median time to treatment failure (TTF) was 113 days in the overall TPV/r group and 0 days in the CPI/r group (p < 0.0001). The median TTF was 0 days in the control group because <50% of the control patients achieved a virologic response in the RESIST studies, even though they were taking a standard of care PI/r plus an OBR. Patients randomized to the TPV/r arm had a 37% lower probability of treatment failure than those randomized to a CPI/r (HR = 0.63, p < 0.0001).
Including an active ARV in the OBR, such as ENF, increased the median TTF to 337 days in the TPV/r arm but the TTF was unchanged in the CPI/r arm in patients who took ENF (0 days) (p < 0.0001) (Cahn et al 2005).
As for the viral load reduction the CD4+ count increase was significantly different in the TPV/r arm and the comparator arm. By week 24, the median CD4+ cell count had risen by 34 cells/μl in the TPV/r and only by 4 cells/μl in the control group. At 48 weeks, only the mean CD4+ cell count is published and therefore unrivalled to the 24 week data. The mean CD4+ T cell count rose by 45 cells/μl in the TPV/r arm and only by 21 cells/μl in the control group by week 48 (Cooper et al 2005; Cahn et al 2005).
As the participation in the RESIST-1 and -2 studies was restricted for those patients infected with virus that carried three or more mutations at codons 33, 82, 84 and 90 (PRAMs), they were eligible to be enrolled in the study BI 1182.51. Enrolled were 315 HIV-1 infected, triple ARV-class experienced patients, who had at least received two PI based ARV regimens and a pVL of above 1000 log10 copies/mL at the study entry. The patients were randomized to receive one of the bid regimens containing either LPV/r + OBR, APV/r + OBR, SQV/r + OBR or TPV/r + OBR. During the first two weeks of administration of TPV/r the median pVL reduction from baseline was −1.06 log10 copies/mL (Figure 3). By contrast, the median reduction in the other treatment arms was lower and ranged from a decline of −0.15 to −0.38 log10 copies/mL (Leith et al 2004). When TPV/r was added to the other single PI regimen, there was a further reduction in median pVL in all three PI arms by −0.96–1.19 log10 copies/mL. Subsequently, however, virological rebound was noticed in all study arms (Boehringer Ingelheim GmbH International 2005). This trend towards baseline pVLs in all treatment arms may be accounted for a lack of a sufficient OBR, which suggests that not enough additional, active drugs were part of the new regimen to support the antiviral activity of TPV/r. This data underlines the need to construct a TPV/r-based HAART regimen similar to the other PIs, with as many active drugs as possible in order to achieve a durable response.
Figure 3.
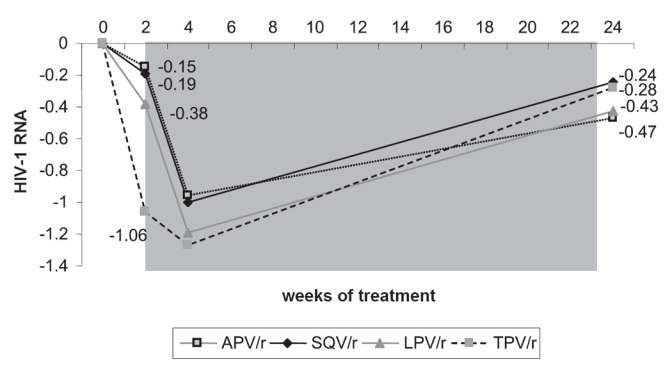
BI 1182.51 study.
Side effects
At 24 weeks, adverse events and side effects associated with TPV/r are commonly gastrointestinal, such as diarrhoea (10.9%), nausea (6.7%), vomiting (3.4%), and abdominal pain (2.8%) (Cahn et al 2005; Pierone et al 2005; Hicks 2004). Other adverse events reported in the studies were pyrexia (4.6%), headache (3.1%), bronchitis (2.9%), depression (2%), rash (2%) asthenia and fatigue (4%). As tipranavir is a sulfa-containing (2-Pyridinesulfonamide) drug, it should be used with caution in patients with a known sulfa allergy, although a final association has not been proven. Mild to moderate rashes including urticarial rash, maculopapular rash, and possible photosensitivity have been reported in subjects receiving TPV. In Phase 2 and 3 trials, rash was observed in 14% of females and in 8–10% of males receiving TPV/r (GmbH B.I. 2004). Overall the majority of the reported side effects were mild or moderately intense and comparable with the other boosted PIs. The most common side effects observed in RESIST-1 and RESIST-2 are summarized in Table 2.
Table 2.
Adverse events (AEs) in RESIST and RESIST 2 trials
| Adverse events | TPV/r + OBR (n = 746) |
CPI/r + OBR (n = 737) [%] |
|---|---|---|
| Gastrointestinal Disorders | 46.8 | 42.6 |
| Diarrhoea | 13.4 | 11.1 |
| Nausea | 11.7 | 7.9 |
| Vomiting | 3.8 | 2.8 |
| Flatulence | 2.9 | 1.9 |
| Abdominal disentsion | 2.5 | 1.8 |
| Abdominal pain | 2.4 | 2.7 |
| Loose stools | 1.6 | 1.2 |
| Dyspepsia | 1.1 | 0.7 |
| Metabolism and Nutrition Disorders | ||
| Anorexia | 1.1 | 0.9 |
| Hypertriglyeridaemia | 1.9 | 0.8 |
| Hyperlipidaemia | 1.2 | 0.4 |
| General Disorders | ||
| Fatigue | 4.4 | 2.6 |
| Nervous System Disorders | ||
| Headache | 3.5 | 1.2 |
| Skin Disorders | ||
| Rash | 1.6 | 0.9 |
| Puritus | 1.1 | 0.4 |
The long-term effects of tipranavir are clearly presently unknown. In a 4-year follow-up study of treatment experienced patients, TPV/r therapy was well tolerated, adverse events were not associated with treatment discontinuation, and no new adverse event emerged after long-term follow-up (Pierone et al 2005). Recently, however, Boehringer Ingelheim has distributed information on 14 reports of fatal and non-fatal intracranial hemorrhage in the APTIVUS clinical development programme. These have occurred in 13 out of 6,840 HIV-1 infected individuals receiving APTIVUS/r in clinical trials, which corresponds to a rate of 0.2/100 patient exposure years (PEY). A literature review of ICH in HIV-infected individuals found that the rate of ICH observed in AIDS patients not receiving combination antiretroviral therapy is the same (0.2/100 PEY) as reported in the APTIVUS clinical trials. Notably this rate is approximately 25-fold higher than the rate in non-HIV-infected persons. Therefore, the clinical impact of these findings still needs to be determined.
Some laboratory abnormalities are potentially important and should be monitored closely. In patients enrolled in the RESIST studies taking ritonavir boosted tipranavir, 45.1% showed elevated triglycerides, 17.5% elevated liver enzymes (esp. γ-GT and AST) and 14.6% elevated cholesterol at week 24 (% figures refer to all severity Grades {1–4}). Table 3 summarises the grade 3 and 4 laboratory abnormalities recorded in the RESIST trials. The proportion of patients who experienced a Grade 3 or 4 adverse event in elevated liver enzymes were 6.9% in RESIST-1 versus 1.3% in the CPI arm and 5.2% in RESIST-2 vs 2.2% in the CPI group. Similarly, the Grade 3/4 cholesterol elevations were higher in the TPV/r arm with 3.3% vs 0.3%, as well as the Grade 3/4 triglyceride levels with 21% vs 11.3% in the control group. Other reported laboratory abnormalities, more frequently observed, were decreased white blood cell count (3.6%) and elevated amylase (2.9%) during this study, which however were lower than in the comparator PI-arm.
Table 3.
Grade 3 and 4 laboratory events in Resist-1 and Resist-2 (week 24 data)*
| TPV/r + OBR (n = 732) [%] |
CPI/r + OBR (n = 726) [%] |
|
|---|---|---|
| Haematology | ||
| WBC count decrease | 3.5 | 5.5 |
| Chemistry | ||
| ALT | 5.9 | 1.8 |
| AST | 4.0 | 1.7 |
| Amylase | 4.5 | 5.9 |
| Cholesterol | 3.3 | 0.3 |
| Triglycerides | 21.0 | 11.3 |
reported in = 2% of patients
Tipranavir is contraindicated in patients with moderate and severe hepatic impairment (Child-Pugh Class B and C) and should be used with caution in patients with known hepatitis B or C. TPV has been associated with hepatitis and significant liver damage (hepatic decompensation), including some fatal cases (Pierone et al 2005). Therefore liver function tests should be performed prior to starting treatment with TPV and rechecked regularly throughout the duration of treatment. Further subanalyses in the RESIST studies were performed in patients with hepatitis coinfection upon inclusion in the study. Overall 49 patients in the TPV/r arms and 68 patients in the control arms had an HCV infection in the RESIST-1/2 studies; 26 (TPV/r) and 42 (CPI) were infected with HBV. The types and frequencies of AEs were comparable in hepatitis negative and hepatitis co-infected patients, whereas no hepatic events occurred in ≥5% of either hepatitis negative or hepatitis co-infected TPV/r patients. The frequencies of hepatobiliary events were similar in hepatitis negative and hepatitis co-infected patients. The antiviral activity of TPV was not affected by HCV or HBV co-infection. Patients with HCV or HBV co-infection however, are at an increased risk for developing transaminase elevations and, therefore a close monitoring of liver parameters is necessary in these patients (Rockstroh et al 2005).
A total of 3/75 (4%) patients who died while on or after taking TPV/r were considered treatment-related in the RESIST studies. Two of these three patients had severely complicated end-stage AIDS and a definite reason for death could not be stated by the investigator. One of these patients died as a result of multi-system organ failure including hepatic failure with hyperbilirubinemia. The third patient did not have end-stage AIDS and was taking TPV/r with ddI, d4T, NVP as a backbone. He was hospitalized with severe lactic acidosis and elevated hepatic transaminases, subsequently developing respiratory failure and brain stem infarction (Boehringer Ingelheim GmbH International 2005; 2004 unpublished data, Boehringer Ingelheim GmbH). This case of hepatotoxicity was most likely due to mitochondrial toxicity deriving from the d4T/ddI combination inducing hepatic steatosis and lactic acidosis.
Drug-drug interactions
As tipranavir is being metabolized through the CYP3A4 system in the endoplasmatic reticulum of the liver, the concurrent use of medications using the same metabolism pathway may induce significant drug-drug interactions. Drugs such as HMG-CoA reductase inhibitors or warfarin can lead to substantial increases in plasma concentrations (2005 Boehringer Ingelheim International GmbH; Boehringer Ingelheim Pharmaceuticals Inc 2005; Boffito 2006). Indeed the product label states that co-administration of TPV and 200 mg of RTV with lovastatin or simvastatin is not recommended. Pravastatin and atorvastatin however can be co-administered. With regard to atorvastatin the recommendation is given to start with the lowest dose possible and to perform careful monitoring. Patients receiving concomitant warfarin therapy should receive frequent INR (international normalized ratio) monitoring upon initiation of TPV/r therapy. Rifampin, St. John’s Wort (Hypericum perforIatum) and delavirdine have been demonstrated to lower the levels of TPV by 80% with the risk of diminished plasma concentration and consequent reduced antiviral effects (Boehringer Ingelheim GmbH International 2005; 2005 Boehringer Ingelheim International GmbH; Boehringer Ingelheim Pharmaceuticals Inc 2005). Additionally, as it has been shown for atazanavir or fos-amprenavir, antacids partially lower the plasma levels of TPV with an AUC reduction of 25–30% and Cmax by 25–30% (Boffito 2006; Van Heeswijk et al 2004). Table 4 summarises some of the main drugs which should not be co-administered with TPV/r or have significant potential for drug-drug interactions.
Table 4.
Drugs that should not be coadministered with TPV/r
| Potential for serious and/or life theatening reactions | |
| Antiarrhythmics | Amiodarone |
| Bepridil | |
| Flecainide | |
| Propafenone | |
| Qunidine | |
| Antihistamines | Astemizole |
| Terfenadine | |
| Ergot derviates | Dihydroergotamine |
| Ergonovine | |
| Ergotamine | |
| Methylergonovine | |
| GI motility agent | Cisapride |
| Neuroleptic | Piomizide |
| Sertindole | |
| Sedatives | Midazolam |
| Triazolam | |
| Risk of sub-therapeutic TPV/r levels | |
| Antibiotics | Rifampicin |
| Herbal preparations | St John’s Wort |
| Increased risk of myopathy, including rhabdomyolysis | |
| HMG-CoA reductase inhibitors | Lovastatin |
| Simvastatin | |
It is important to highlight that TPV may have an effect on the plasma levels of other antiretroviral drugs and dose adjustment has not been established yet (2005 Boehringer Ingelheim International GmbH; Boehringer Ingelheim Pharmaceuticals Inc 2005; Boffito 2006). The addition of TPV/r did not change the pharmacokinetics of stavudine (d4T), lamivudine (3TC), efavirenz (EFV), enfuvirtide (ENF) or nevirapine (NVP) and therefore no adjustment in dosage was required when they were co-administered with TPV/r. However, as seen with other RTV boosted PIs, the exposures to abacavir (ABC) and zidovudine (AZT) were both significantly decreased by 35% and 40% of the recommended area under the curve (AUC) dosage at steady state, although the clinical significance of these findings still remains undetermined. Similarly, levels of didanosine (ddI) were significantly reduced by 20%, and ddI administration several hours prior to TPV/r dosing has been advised. Tenofovir levels were reduced by 30% which, however, was not significant and therefore no dose adjustment is recommended.
As the NRTIs therapeutic windows are not well-established, the clinical relevance of these changes is unknown. Moreover the packaging labels for ABC, AZT and ddI do not advise alterations in dosage when co-administered with level reducing drugs. Therefore a co-administration with ABC or AZT is thus so far not recommended unless no other NRTIs are available (2005 Boehringer Ingelheim International GmbH; Boehringer Ingelheim Pharmaceuticals Inc 2005; Boffito, Maitland and Pozniak 2006). Similar appropriate doses for the combination of TPV/r with APV/r, LPV/r or SQV/r have not been established as TPV/r reduces the AUC at steady state for APV/r (44%) LPV/r (55%) and SQV/r (76%) (Leith et al 2004). These findings make a double PI use with TPV/r inadvisable. In addition, there is an increased risk of Grade 3/4 elevations in hepatic transaminase enzymes if TPV/r is co-administered with APV/r. No data are currently available on interactions between TPV/r and indinavir, atazanavir, fos-amprenavir or nelfinavir. Therefore co-administration of any PI with TPV/r is not recommended at this time.
In one drug interaction trial in healthy female volunteers, who were administered a single dose of ethinyl estradiol followed by TPV/r, more than 50% (n = 29) of subjects (n = 52) developed a rash. Rash accompanied by joint pain or stiffness, throat tightness, or generalized itching led to discontinuation of TPV/r in 20 of the patients. The symptoms were resolved after treatment discontinuation. Women taking estrogen-containing medications with TPV/r have an increased risk of developing a mild to moderate rash and it is associated with a 50% reduction in ethinyl estradiol levels. This large reduction is below the therapeutic window for use of a contraceptive (Boehringer Ingelheim GmbH International 2005; 2004 unpublished data Boehringer Ingelheim GmbH; Boffito 2006).
Discussion
TPV is a protease inhibitor with a unique resistance profile compared with other protease inhibitors. Based on the existing data TPV is best used in patients who have failed currently available PIs. In RESIST-1 and RESIST-2 tipranavir has evidently demonstrated its virological efficacy in multiple-PI resistant patients. Clearly, overall long-term efficacy depends on further active components within combination treatment including TPV. Enfuvirtide or other active drugs according to genotypic resistance testing are ideal partners for treatment of patients with PI mutations in whom prior treatment with lopinavir or other RTV boosted PIs has failed. Indeed TPV/R may have limited efficacy when used without enfuvirtide in highly drug-experienced patients.
However, the use of TPV in the current HIV treatment armamentarium will probably be limited. The RESIST studies have shown that tipranavir treatment results in a better outcome in patients who had used 2 or 3 PIs previously than in more experienced patients (Rockstroh, Villacian, Quinson et al 2005). But for patients exhibiting less resistance, compared with lopinavir, TPV may offer limited advantage in lopinavir sensitive patients, as lopinavir’s side effect profile seems to be slightly better than for TPV/r and their virological efficacies were almost identical in this specific patient group. In the more advanced treatment population on the other hand, several other new protease inhibitors such as darunavir (TMC-114) (so far only licensed in the US) and brecanavir will become available soon, which are currently in phase II-III of development with encouraging in vitro data showing good susceptibility as well as results from first clinical trials demonstrating good virological efficacy against PI-resistant HIV strains (King et al 2004; Koh et al 2003; Katlama et al 2005; Wilkin et al 2005). Indeed, in the POWER-1 trial in patients who were enfurvirtide-naive and received darunavir in combination with enfurvirtide 63% of patients achieved less than 50 copies/ml at week 24 (n = 19) highlighting the antiretroviral potency of the new compound. Moreover, the safety profile was favorable with no significant difference to the comparator PI arm. However, no studies have yet directly compared TMC-114 or brecanavir to tipranavir. In addition, large differences in the number of treated patients (in the clinical trials) as well as different inclusion criteria and baseline resistance profiles limit the comparison between tipranavir and TMC-114 or brecanavir at this time.
As a future scenario it appeared possible that tipranavir might also be chosen for treatment of naïve HIV-1 infected patients. A study in treatment naïve patients (BI trial 1188.33) receiving TPV/r in different dosages (one arm looked at lower ritonavir doses for boosting ie, 100 mg RTV bid) however, was recently stopped because of increased liver toxicity in the TPV-arm, making the earlier use of tipranavir unlikely. Preliminary data from another trial for 14 days TPV therapy in treatment inexperienced patients had initially demonstrated high efficacy, good safety and good tolerability by the patients (McCallister et al 2004).
Under consideration of the current trend towards a more simplified and better tolerable first line regimen, the daily pill burden and the tolerability profile of TPV make this agent an improbable first choice at present. TPV in the licensed dosing is associated with more adverse events and discontinuations mostly due to Grade 3 and 4 ALT or triglyceride elevations than other boosted PIs. However, the health-related quality of life measured by the Medical Outcomes Study-HIV Health Survey (MOS-HIV) in patients enrolled in RESIST 1 and 2, showed no significant statistical difference between TPV/r and a boosted comparator PI (Woo et al 2005), suggesting that most patients adapt to boosted PI/r adverse events.
In conclusion, tipranavir is a new and very potent antiretroviral drug, which is an excellent choice for patients, who have experience with 2 to 3 PIs; most of these patients’ isolates remain sensitive to TPV/r. Preferably, additional, active drugs (especially a drug from a class the patient has never received before) should be used in a TPV/r containing regimen, which as with all other ARV agents, would increase the durability of response to TPV/r significantly. Also TPV/r should be considered in salvage therapy as studies showed higher viral load suppression even though the genetic resistance profile showed several PI primary mutations. Nevertheless, TPV/r should be used with caution in patients with underlying hepatic impairment under close laboratory observation as the risk for development of hepatotoxicity is increased.
References
- Back NK, van Wijk A, Remmerswaal D, et al. In vitro tipranavir susceptibility of HIV-1 isolates with reduced susceptibility to other protease inhibitors. Aids. 2000;14:101–2. doi: 10.1097/00002030-200001070-00019. [DOI] [PubMed] [Google Scholar]
- Bartlett JA, DeMasi R, Quinn J, et al. Overview of the effectiveness of triple combination therapy in antiretroviral-naive HIV-1 infected adults. Aids. 2001;15:1369–77. doi: 10.1097/00002030-200107270-00006. [DOI] [PubMed] [Google Scholar]
- Boden D, Hurley A, Zhang L, et al. HIV-1 drug resistance in newly infected individuals. JAMA. 1999;282:1135–41. doi: 10.1001/jama.282.12.1135. [DOI] [PubMed] [Google Scholar]
- Boehringer Ingelheim.2004. Unpublished data. Boehringer Ingelheim GmbH Investigator’s Brochure
- Boehringer Ingelheim GmbH International. Aptivus (tipranavir) Capsules -SPC. 2005 [Google Scholar]
- Boehringer Ingelheim GmbH International. Product Monograph, Aptivus (tipranavir) 2005 [Google Scholar]
- Boehringer Ingelheim Pharmaceuticals Inc. Aptivus (tipranavir) Capsules -Prescribing Information. 2005 [Google Scholar]
- Boffito M, Maitland D, Pozniak A. Practical perspectives on the use of tipranavir in combinations with other medications: Lessons learned from pharmacokinetic studies. J Clin Pharmacol. 2006;46:130–9. doi: 10.1177/0091270005283279. [DOI] [PubMed] [Google Scholar]
- Cahn P, Hicks C, Teams f.t.R.-a.R.-S. 2005. RESIST-1 (R-1) and RESIST-2 (R-2) 48 week meta-analyses demonstrate superiority of protease inhibitor (PI) tipranavir+ritonavir (TPV/r) over an optimized comparator PI (CPI/r) regimen in antiretroviral (ARV) experienced patients. 10th European AIDS Conference (EACS); 17–20 November 2005; Dublin, Ireland. Abs. # PS3/8. [Google Scholar]
- Carpenter CC, Cooper DA, Fischl MA, et al. Antiretroviral therapy in adults: Updated recommendations of the international AIDS society-U S A Panel. JAMA. 2000;283:381–90. doi: 10.1001/jama.283.3.381. [DOI] [PubMed] [Google Scholar]
- Chen RY, Westfall AO, Mugavero MJ, et al. Duration of highly active antiretroviral therapy regimens. Clin Infect Dis. 2003;37:714–22. doi: 10.1086/377271. [DOI] [PubMed] [Google Scholar]
- Clavel F, Hance AJ. HIV drug resistance. N Engl J Med. 2004;350:1023–35. doi: 10.1056/NEJMra025195. [DOI] [PubMed] [Google Scholar]
- Clotet B, Raffi F, Cooper D, et al. Clinical management of treatment-experienced, HIV-infected patients with the fusion inhibitor enfuvirtide: consensus recommendations. Aids. 2004;18:1137–46. doi: 10.1097/00002030-200405210-00007. [DOI] [PubMed] [Google Scholar]
- Cooper D, Hicks C, Cahn P, et al. 24-week RESIST study analyses: The efficacy of tipranavir/ritonavir is superior to lopinavir/ritonavir, and the TPV/r treatment response is enhanced by inclusion of genotypically active antiretrovirals in the optimized background regimen. Conference on Retroviral and Opportunistic Infections (CROI); 2005; Boston, MA. 2005. Abs # 560. [Google Scholar]
- Doyon L, Tremblay S, Bourgon L, et al. Selection and characterization of HIV-1 showing reduced susceptibility to the non-peptidic protease inhibitor tipranavir. Antiviral Res. 2005;68:27–35. doi: 10.1016/j.antiviral.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Gathe J, Kohlbrenner VM, Pierone G, et al. Tipranavir/ritonavir demonstrates potent efficacy in multiple protease inhibitor experienced patients: BI 1182.52. 10th Conference on Retroviral an Opportunistic Infections (CROI); Boston. 2003. Abs. # 179. [Google Scholar]
- GmbH BI. Investigator’s Brochure. 2004 [Google Scholar]
- Gong YF, Robinson BS, Rose RE, et al. In vitro resistance profile of the human immunodeficiency virus type 1 protease inhibitor BMS-232632. Antimicrob Agents Chemother. 2000;44:2319–26. doi: 10.1128/aac.44.9.2319-2326.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks C. RESIST-1: A phase 3, randomized, controlled, open-label, multicenter trial comparing tipranavir/ritonavir (TPV/r) to an optimized comparator protease inhibitor/r (CPI/r) regimen in antiretroviral (ARV) experienced patients: 24-week trial. 44th Interscience Conference on Antimicrobial Agents and Chemotherapy; October 30 – November 2; Washington, DC; 2004. 2004. Abstract H-1137a. [Google Scholar]
- Hicks C, Cahn CP, Cooper DA, et al. for the RESIST Investigator Group. Durable efficacy of tipranavir-ritonavir in combination with an optimised background regimen of antiretroviral drugs for treatment-experienced HIV-1-infected patients at 48 weeks in the Randomized Evaluation of Strategic Intervention in multi-drug reSistant patients with Tipranavir (RESIST) studies: an analysis of combined data from two randomised open-label trials. Lancet. 2006;368:466–75. doi: 10.1016/S0140-6736(06)69154-X. [DOI] [PubMed] [Google Scholar]
- Jacobsen H, Yasargil K, Winslow DL, et al. Characterization of human immunodeficiency virus type 1 mutants with decreased sensitivity to proteinase inhibitor Ro 31-8959. Virology. 1995;206:527–34. doi: 10.1016/s0042-6822(95)80069-7. [DOI] [PubMed] [Google Scholar]
- Johnson VA, Brun-Vezinet F, Clotet B, et al. Update of the drug resistance mutations in HIV-1: 2005. Top HIV Med. 2005;13:51–7. [PubMed] [Google Scholar]
- Jones JL, Hanson DL, Dworkin MS, et al. Surveillance for AIDS-defining opportunistic illnesses, 1992–1997. MMWR CDC Surveill Summ. 1999;48:1–22. [PubMed] [Google Scholar]
- Katlama C, Berger D, Bellos N, et al. Efficacy of TMC114/r in 3-class experienced patients with limited treatment options: 24-week planned interim analysis of 2 96-week multinational dose-finding Trials. Conference on Retroviral and Opportunistic Infections (CROI); 2005; Boston, MA. 2005. 164LB. [Google Scholar]
- Kemp SD, Salim M, Field N, et al. Site-directed mutagenesis and in vitro drug selection studies have failed to reveal a consistent genotypic resistance pattern for tipranavir. Interscience Conference on Antimicrobial Agents and Chemotherapy. 2000 Abstract # 2113. [Google Scholar]
- King NM, Prabu-Jeyabalan M, Nalivaika EA, et al. Structural and thermodynamic basis for the binding of TMC114, a next-generation human immunodeficiency virus type 1 protease inhibitor. J Virol. 2004;78:12012–21. doi: 10.1128/JVI.78.21.12012-12021.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh Y, Nakata H, Maeda K, et al. Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob Agents Chemother. 2003;47:3123–9. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe F, Gatell J, Staszewski S, et al. Trends over time in initial virological failure of first HAART: 1996 to 2002. A joint cohort Analysis of 4143 subjects conference on retroviral and opportunistic infections (CROI); 2005; Boston, MA. 2005. Abstr. # 593. [Google Scholar]
- Larder BA, Hertogs K, Bloor S, et al. Tipranavir inhibits broadly protease inhibitor-resistant HIV-1 clinical samples. Aids. 2000;14:1943–8. doi: 10.1097/00002030-200009080-00009. [DOI] [PubMed] [Google Scholar]
- Leith J, Walmsley S, Katlama C, et al. Pharmacokinetics and safety of Tipranavir/Ritonavir (TPV/r) Alone or in Combination with Saquinavir (SQV), Amprenavir (APV), or Lopinavir (LPV): Interim analysis of BI1182.51. 5th International Workshop on Clinical Pharmacology of HIV Therapy, Rome, Italy. 2004 session 5 Abs. 5.1. [Google Scholar]
- Little SJ, Holte S, Routy JP, et al. Antiretroviral-drug resistance among patients recently infected with HIV. N Engl J Med. 2002;347:385–94. doi: 10.1056/NEJMoa013552. [DOI] [PubMed] [Google Scholar]
- Markowitz M, Mo H, Kempf DJ, et al. Selection and analysis of human immunodeficiency virus type 1 variants with increased resistance to ABT-538, a novel protease inhibitor. J Virol. 1995;69:701–6. doi: 10.1128/jvi.69.2.701-706.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz M, Mohri H, Mehandru S, et al. Infection with multidrug resistant, dual-tropic HIV-1 and rapid progression to AIDS: a case report. Lancet. 2005;365:1031–8. doi: 10.1016/S0140-6736(05)71139-9. [DOI] [PubMed] [Google Scholar]
- McCallister S, Valdez H, Curry K, et al. A 14-day dose-response study of the efficacy, safety, and pharmacokinetics of the nonpeptidic protease inhibitor tipranavir in treatment-naive HIV-1-infected patients. J Acquir Immune Defic Syndr. 2004;35:376–82. doi: 10.1097/00126334-200404010-00007. [DOI] [PubMed] [Google Scholar]
- Montaner JS. Challenges of the unmet needs in the treatment-experienced patient. AIDS Read. 2003;13:S5–9. [PubMed] [Google Scholar]
- Palella FJ, Jr, Delaney KM, Moorman AC, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med. 1998;338:853–60. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- Pierone G, Drulak M, Arasteh K, et al. A long-term open-label rollover trial assessing the safety and tolerability of combination tipranavir and ritonavir (TPV/r) Use in HIV-1–infected patients. The 3rd IAS Conference on HIV Pathogenesis and Treatment (IAS 2005); July 24–27, 2005; Rio de Janeiro, Brazil Poster WePe6.2C05. 2005. [Google Scholar]
- Polis MA, Sidorov IA, Yoder C, et al. Correlation between reduction in plasma HIV-1 RNA concentration 1 week after start of antiretroviral treatment and longer-term efficacy. Lancet. 2001;358:1760–5. doi: 10.1016/s0140-6736(01)06802-7. [DOI] [PubMed] [Google Scholar]
- Poppe SM, Slade DE, Chong KT, et al. Antiviral activity of the dihydropyrone PNU-140690, a new nonpeptidic human immunodeficiency virus protease inhibitor. Antimicrob Agents Chemother. 1997;41:1058–63. doi: 10.1128/aac.41.5.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman DD, Morton SC, Wrin T, et al. The prevalence of antiretroviral drug resistance in the United States. Aids. 2004;18:1393–1401. doi: 10.1097/01.aids.0000131310.52526.c7. [DOI] [PubMed] [Google Scholar]
- Rockstroh J, Sulkowski M, Neubacher D, et al. 24-Week Efficacy of tipranavir boosted with ritonavir (TPV/r) in Hepatitis B (HBV) or Hepatitis C (HCV) Co-Infected Patients. 45th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy; December 16–19th, 2005; Washington D.C.. 2005. H-525. [Google Scholar]
- Rockstroh J, Villacian J, Quinson A, et al. 24-week analysis of the efficacy of tipranavir boosted with ritonavir (TPV/r) in HIV patients stratified by previous protease inhibitor (PI) Use. 10th European AIDS Conference (EACS); November 17–20, 2005; Dublin, Ireland. 2005. PE7.9/11. [Google Scholar]
- Rusconi S, La Seta Catamancio S, Citterio P, et al. Susceptibility to PNU-140690 (Tipranavir) of human immunodeficiency virus type 1 isolates derived from patients with multidrug resistance to other protease inhibitors. Antimicrob Agents Chemother. 2000;44:1328–32. doi: 10.1128/aac.44.5.1328-1332.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz R, Kazanjian P, Slater L, et al. Resistance to tipranavir is uncommon in a randomized trial of tipranavir/ritonavir (TPV/RTV) in Multiple PI-Failure Patients (BI 1182.2). 9th Conference on Retroviruses and Opportunistic Infections; 2002; Seattle, WA. 2002. Session 75, Poster Session 562-T. [Google Scholar]
- Thaisrivongs S, Strohbach JW. Structure-based discovery of Tipranavir disodium (PNU-140690E): a potent, orally bioavailable, nonpeptidic HIV protease inhibitor. Biopolymers. 1999;51:51–8. doi: 10.1002/(SICI)1097-0282(1999)51:1<51::AID-BIP6>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Van Heeswijk R, Sabo J, Cooper C, et al. The pharmacokinetic interaction between tipranavir/ritonavir 500/200 mg bid (TPV/r) and atorvastatin, antacid, and CYP3A4 in healthy adult volunteers. 5th International Workshop on Clinical Pharmacology of HIV Therapy, Rome, Italy. 2004 Abstract #35. [Google Scholar]
- van Leth F, Phanuphak P, Ruxrungtham K, et al. Comparison of first-line antiretroviral therapy with regimens including nevirapine, efavirenz, or both drugs, plus stavudine and lamivudine: a randomised open-label trial, the 2NN Study. Lancet. 2004;363:1253–63. doi: 10.1016/S0140-6736(04)15997-7. [DOI] [PubMed] [Google Scholar]
- Wilkin T, Haubrich R, Steinhart C, et al. TMC114/r Superior to Standard of Care in 3-Class-Experienced Patients: 24-Wks Primary Analysis of the Power 2 Study (C202). 45th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy; December 16–19th, 2005; Washington D.C.. 2005. H-413. [Google Scholar]
- Wu A, Huang I, Lobo F, et al. Health-related quality of life with tipranavir in combination with an optimized background regimen in 2 randomized clinical trials. 12th Conference on Retroviruses and Opportunistic Infections; February 22–25, 2005; Boston, Massachusetts. 2005. Poster #654. [Google Scholar]
- Yeni PG, Hammer SM, Hirsch MS, et al. Treatment for adult HIV infection: 2004 recommendations of the International AIDS Society-USA Panel. JAMA. 2004;292:251–65. doi: 10.1001/jama.292.2.251. [DOI] [PubMed] [Google Scholar]