

Abstract
CM101, an antiangiogenic polysaccharide derived from group B streptococcus, was administered by i.v. injection 1 hr post-spinal-cord crush injury in an effort to prevent inflammatory angiogenesis and gliosis (scarring) in a mouse model. We postulated that gliosis would sterically prevent the reestablishment of neuronal connectivity; thus, treatment with CM101 was repeated every other day for five more infusions for the purpose of facilitating regeneration of neuronal function. Twenty-five of 26 mice treated with CM101 survived 28 days after surgery, and 24 of 26 recovered walking ability within 2–12 days. Only 6 of 14 mice in the control groups survived 24 hr after spinal cord injury, and none recovered function in paralyzed limbs. MRI analysis of injured untreated and treated animals showed that CM101 reduced the area of damage at the site of spinal cord compression, which was corroborated by histological analysis of spinal cord sections from treated and control animals. Electrophysiologic measurements on isolated central nervous system and neurons in culture showed that CM101 protected axons from Wallerian degeneration; reversed γ-aminobutyrate-mediated depolarization occurring in traumatized neurons; and improved recovery of neuronal conductivity of isolated central nervous system in culture.
Functional recovery from spinal cord injury (SCI) in humans still constitutes a major clinical challenge. Traumatic insults to the spinal cord induce both immediate mechanical damage and subsequent tissue degeneration (1). Tissue damage progresses in a setting of ischemia, hemorrhage, and edema leading to neuronal necrosis (2). Additional evidence suggests that both global and focal ischemic cell loss, in part, may reflect apoptosis in neurons as a late consequence of the injury (1, 3). Experimental observations in cell culture and animal models suggest that the mammalian central nervous system (CNS) neurons may be capable of axonal regrowth and reconnection of injured processes under some conditions (4). Cheng et al. (5) devised a method of peripheral nerve grafts for spinal cord repair in paralyzed rats that created great interest (6). Additional approaches to achieve axonal regeneration in the adult CNS include grafts of cultured rat olfactory bulb cells (7), genetically modified fibroblasts (8), peripheral nerve grafts and cotreatment with selected growth factors (9), intercostal nerves and cauda equina anastomoses (10), neurite growth inhibitor-neutralizing antibody IN-1 (11, 12), and brain-derived neurotrophic factors (13, 14). There is, however, still no cure for the catastrophic consequence of SCI in paralyzed patients. Gendelman and Tardieu (15) have shown that trauma of the mature CNS leads to hypoxia-induced angiogenesis and infiltration of activated brain macrophages and neutrophils, all of which leads to scarring (16, 17). Gliosis, the formation of a scar in injured CNS, involves hypoxia-driven inflammatory angiogenesis (18, 19). Gliosis includes proliferation of astrocytes and microglia, and inflammatory infiltration of peripheral macrophages (15–17). CM101 is an antipathoangiogenic polysaccharide (20) derived from group B streptococcus (GBS) (21) that inhibits angiogenesis and subsequent infiltration of inflammatory cells and thereby formation of granulation tissue, which produces scarring (M. Neeman, R. Abramowitch, B.D.W., and C.G.H., unpublished data).
In this study we explored the hypothesis that gliosis of the spinal cord, which depends on hypoxia-induced angiogenesis, could be inhibited by i.v. administration of CM101, and that the anticipated reduced gliosis would facilitate neuronal reconnectivity after injury. We used a direct spinal cord compression/crush method (22), which resulted in SCI similar to those observed with other mechanical injury models (23–27) and demonstrate here, in a randomized study, a dramatic survival and recovery of walking ability in paralyzed mice after treatment with CM101. In support of our in vivo observation, we demonstrate prevention by CM101 of Wallerian degeneration of injured axons in cultured spinal cord neurons and that γ-aminobutyrate (GABA)-mediated depolarization occurring in traumatized spinal cord neurons in culture (28, 29) was reversed by CM101. Furthermore, we show that CM101 improved recovery of neuronal conductivity in traumatized cultured CNS from mice embryos.
METHODS
CM101 Preparation.
CM101 is a bacterial polysaccharide produced by GBS from several serotypes (H. Sundell, H.-P. Yan, B.D.W., C. E. Carter, and C.G.H., unpublished work) with an average molecular mass of 300 kDa by gel filtration when compared with dextran standards and is composed of GalNAc/GlcNAc/Gal/Glc/Man in an approximate ratio of 1:1:3:1:1 (30). CM101 was produced for clinical trials by culturing GBS in Todd Hewitt Broth (Becton Dickinson) to late-logarithmic phase. The culture was autoclaved and the bacteria were removed by centrifugation and filtration. The supernatant was concentrated by recovering the retentate from a 10-K nominal molecular weight limit (NMWL) polysulfone cassette filter (Millipore). CM101 was recovered from an alcohol precipitate by phenol-water extraction and DEAE-Sephadex and Sepharose column chromatography. Final purification was achieved by lentil lectin chromatography (30). The resultant polysaccharide solution was filter-sterilized, aliquoted into sterile vials at a concentration of 150 μg/ml, and then lyophilized by an Food and Drug Administration-approved contractor for clinical use. Samples of CM101 from this lot were provided for use in this study. CM101 was dissolved in PBS and administered at 30 or 60 μg/kg in 100-μl doses for i.v. injections of injured mice.
Experimental Animals.
CD-1 mice were obtained from Charles River Breeding Laboratories and maintained in cages with free access to water and food in the Animal Care Facility. All experimental procedures were in accordance with protocols approved by the Vanderbilt University Animal Care Committee.
SCI.
The animals, 8–10 weeks old, were anesthetized with ketamine (87 mg/kg) and xylazine (15 mg/kg) (80:20, stock solution; i.p.) and positioned in a spinal stereotactic apparatus with fixation at the ears under a stereomicroscope. The skin above the vertebral column was shaved, decontaminated, and treated with antiseptic. A 15- to 20-mm skin incision was made and the vertebral column was exposed. After the spinal thoracic region was exposed by separation of dorsal muscles to the sides, the spinous processes of vertebrae T8–T13 were exposed. A small incision made with microscissors separated posterior processes of T10 and T11, and the dura mater was exposed intact. Animals with intact dura then were prepared for injury by applying additional fixation at the T8 spinal process, using surgical forceps. Spinal cord in the vertebral canal between T10 and T11 was compressed gradually to reduce a segment diameter from 2 mm to 0.1–0.2 mm. This procedure was performed under 4×–30× magnification and resulted in crush damage to all corticospinal tracts. Each mouse was operated on once. Either a bilateral mechanical compression or a lateral compression of the spinal cord to involve the whole segment, or one side of the segment between T10 and T11, was performed. Animals from the 2 groups of 15 monoplegic and 25 paraplegic mice were randomly assigned to control and treatment groups. All inside layers were closed with surgical sutures. Skin was closed with a tissue glue, and animals were randomized to control and treatment groups. Each operated animal was allowed to recover from surgery with unlimited access to food and water. Care was taken to minimize disruption of the vertebral column.
Clinical Treatment.
Randomized mono- or paraplegic mice were treated with i.v. infusions of CM101. The first dose of CM101, either 30 or 60 μg/kg, was infused into a tail vein 1 hr after surgery, and this treatment was repeated five times every other day.
Open-Field Walking Scoring.
We used the open-field walking scoring system adopted from Cheng et al. (5), which measures each mouse’s gross locomotor ability during a 3- to 5-min observation period. One animal at a time was allowed to move freely inside a plastic tray [8 inches × 11 inches × 3 inches (1 inch = 2.54 cm)]. Locomotor activity was videotaped and animals were scored according to a modified Tarlov scale (5), which ranged from 0 (flaccid or spastic paralysis) to 5 (normal walking). This scale accurately reflected a pattern of recovery of locomotor function as follows: 1, no spontaneous movement but recovery of sensory response; 2, uncoordinated movement of spontaneous groups of muscles; 3, movement of two or all three major joints in hind limbs and active support and uncoordinated gait or short bouts of coordinated gait (limply walk); 4, coordination of walking function with small deficit in hind limbs and gait (this score also included walking on knuckles or the medial surface of the foot or some toe drags or walking on wide base); and 5, full recovery of normal walking function. Scores were generated from the paralyzed limb in monoplegic animals. In paraplegic mice, animal scores were calculated by averaging scores from each hind limb.
CNS Cultures.
Isolated CNS cultures were prepared by the methods devised by Saunders et al. (31) and Nicholls et al. (32). Intact brain and spinal cord from 20–21 gestational day mouse embryos was dissected at 4°C in Eagle’s minimum essential medium (MEM, GIBCO). The CNS preparations were maintained in MEM, pH 7.4, supplemented with 0.2% fetal calf serum (GIBCO), 7S nerve growth factor (30 ng/ml; Sigma), insulin (10 μg/ml), and gentamicin sulfate (0.1 mg/ml) at 25°C in an incubator with 5% CO2/95% air. Medium with or without CM101 (0.3 μg/ml) was changed daily, and cultures were maintained for up to 16 days.
Neuronal Cell Cultures.
Embryonic mouse spinal cords (gestational day 13–14) were minced and dispersed by trituration to single cells and small clumps (33, 34). The neurons were plated 4–20 weeks before experimentation in collagen-coated dishes in medium composed of 80% (vol/vol) MEM, 10% fetal calf serum, and 10% heat-inactivated horse serum, supplemented with 10 ng/ml 7S nerve growth factor and 1 ml/liter Mito Serum Extender (Collaborative Research). Growth of fast-dividing nonneuronal cells was suppressed by brief treatment with 5-fluoro-2′-deoxyuridine after 1 week. Continued cultures were maintained in MEM in the above medium minus the fetal calf serum but supplemented with 5.5 g of glucose/1.5 g of sodium bicarbonate per liter diluted to 300–325 mOsm with distilled water. After equilibration with incubator atmosphere containing 10% CO2, the pH of the culture medium was 7.4. Medium was changed two or three times a week.
Electrophysiological Measurements on Isolated CNS.
Isolated CNSs were placed in culture at room temperature for 5–7 days (31, 32, 35) and subjected to electrophysiological measurements before and after mechanical crush injury. Two microelectrodes were placed on each side of the crush, one to record changes in membrane/field potential induced by pulses fired by the other electrode (36). Stimulation pulses of 2–8 nA (250- to 500-ms duration) repeated after 30–45 s were used to examine translesion conductivity. Measurements of conductivity illustrating axon-to-axon transmission were performed on days 1 and 5 with and without CM101 present in the cultures and with and without trauma. Control CNSs were subjected to 0.5- to 4-nA pulses lasting 100–300 ms, which produced repetitive firing of field potentials or postsynaptic action potentials (28, 33, 34, 36).
Electrophysiological Measurements on Isolated Neurons.
Electrophysiological function was studied in cultured spinal cord neurons by measuring GABA-induced responses in normal and traumatized neurons (34–36). A microelectrode was placed into the cell by overcompensation of the microelectrode capacitance to induce oscillation of the tip. A bridge circuit in the Axoclamp-2B amplifier (Axon Instruments) allowed simultaneous measurement of membrane potential (Em) and transmission of polarizing current through the recording microelectrode for phasic or steady-state changes of potential. The amplified somatic membrane potential was recorded on a storage oscilloscope for on-line monitoring and photographing. Trauma to individual axons was induced by axonal compression for 15–60 s by using the heat-polished patch–clamp electrode tip. PBS used during all experiments was supplemented with 5–7 mM MgCl2 to suppress spontaneous firing of action potentials (33, 34).
RESULTS
In Vivo Procedures.
Our objective was to create a mouse model of spinal cord trauma that would resemble the clinical scenario of mechanical SCI (22) and to study the effects of i.v. administration of CM101, an antipathoangiogenic agent (20) that inhibits scarring. After an initial small pilot experiment to refine procedures and demonstrate probable success, 15 animals underwent a neurosurgical procedure resulting in single hind-limb paralysis. Whole-segment compression in 25 mice resulted in loss of function of both hind limbs mimicking severe spinal cord damage observed in accident victims.
Recovery of Walking Ability.
Animals were observed and recovery from paralysis was recorded on videotape and scored daily for 21 days and weekly for up to 110 days. Scores were given by two “blinded” observers using the open-field walking score test (5). Within the first 24 hr after SCI on day 0 (Fig. 1 A and B, arrowhead), 8 of 14 control animals had died and all 26 treated mice survived. Mice in all study groups were dragging their hind limb(s) (Fig. 2 A and B, Day 1). By day 5, 2 more controls and 2 of 26 treated animals had died. On day 28, 1 additional control animal died (Table 1). Restoration of partial walking ability (minimum “limply walk”—score 3; Fig. 1 A and B) was observed early and within 48 hr post-SCI in 17 of 26 treated (7 of 10 monoplegic; 10 of 16 paraplegic) mice. All treated mice had reached a score of at least 3 by day 12 (last treatment was administered on day 10). Fig. 2 shows still frames of a representative paraplegic mouse recovering walking function (Fig. 2B) on days 7 and 14 compared with the placebo-treated control (Fig. 2A). Recovery of walking function was observed in four of four monoplegic mice treated with CM101 (30 μg/kg), and in six of six monoplegic mice treated with CM101 (60 μg/kg). Among the paraplegic mice, seven of eight mice receiving CM101 (30 μg/kg) and seven of eight mice receiving CM101 (60 μg/kg) showed improvement of walking ability (scores between 3 and 5) (Fig. 1). Survival in the treatment groups averaged 92.3% versus 28.6% in controls on day 6 (Table 1). Two mice from each paraplegic group were sacrificed on day 6 for histology. Of the remaining mice, all treated and one of the two remaining controls survived through day 35. Seventeen CM101-treated animals were kept past 110 days (Table 1) and retained a score of 5, normal walking, and over this time period regained full control of their bladders and tails. Wounds healed rapidly and no scars were found at the site of surgical incision in CM101-treated mice compared with controls.
Figure 1.

Recovery of hind-limb function in the six different study groups of monoplegic (A) or paraplegic (B) mice randomly assigned to groups. Scores improved significantly by day 3 in the treated groups when monoplegic (A) and paraplegic (B) mice were compared with controls (P < 0.001 Kruskal—Wallis test from day 3 on, in paraplegic animals).
Figure 2.

Sequential video frames demonstrating paralysis and recovery in experimental animals. (A) Representative control paraplegic mouse videotaped on day 1, score 0; on day 7, score 0; and on day 14, score 0. (B) A representative paraplegic mouse treated with 60 μg/kg CM101 and videotaped on day 1, score 0; on day 7, score 5; and on day 14, score 5.
Table 1.
Animal survival after experimental SCI
| No. surviving on day
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Animals | 0 | 1 | 2 | 3 | 5 | 6 | 7 | 21 | 35 | 110 |
| Monoplegic control | 5 | 2 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 0 |
| Mono + CM101 (30 μg/kg) | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| Mono + CM101 (60 μg/kg) | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 4 |
| Paraplegic control | 9 | 4 | 4 | 4 | 3 | 3* | 1 | 1 | 0 | 0 |
| Para + CM101 (30 μg/kg) | 8 | 8 | 8 | 8 | 7 | 7* | 5 | 5 | 5 | 5 |
| Para + CM101 (60 μg/kg) | 8 | 8 | 7 | 7 | 7 | 7* | 5 | 5 | 5 | 4 |
*Two animals were sacrificed for histological examination.
Evaluation by MRI.
The area of spine damage and the process of healing including observation for late degenerative changes were documented by MRI on day 21 after injury. MRI showed localized lesions in the spinal cord in treated mice compared with control mice. A large area of damage at the site of compression involving the surrounding tissue and the spinal cord is obvious in the control animal (Fig. 3A). CM101 significantly reduced the compression-induced scarring as shown in the representative treated animal (Fig. 3B).
Figure 3.

MRI of spinal cord from control (A) and CM101-treated animals (B) is visualized by using a mouse-sized coil 1 inch in diameter, 4.6-T magnetic field at 200 MHz, and 300-μm slices. Images were obtained 21 days after SCI. Image quality was variable as a result of a coil size and slice geometry. A large area of damage to the spinal cord, indicated by bar for size and by arrow for location, is obvious in the untreated animal, whereas in the treated animal (B), CM101 appears to have minimized the damage resulting in a limited lesional area indicated by the bar and arrow.
Histopathologic Examination.
The spinal cords from treated and untreated animals were processed for histopathological examination to determine the level of gliosis and overall recovery of tissue structure (Fig. 4). The data corroborated the MRI analysis and showed a significantly reduced area of gliosis and fibrosis in treated mice (Fig. 4B) compared with control (Fig. 4A).
Figure 4.
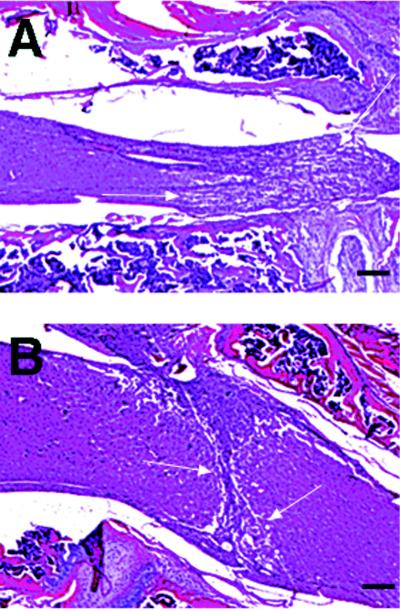
Representative micrographs illustrating histopathological staining with hematoxylin and eosin. Sagittal section of the spinal cord from a paraplegic control (A) and a representative CM101-treated (B) mouse sacrificed on day 6 after injury after four treatments with 30 μg/kg CM101. Amount of hemorrhage, fibrosis, and gliosis formed in the spinal cord (area between arrows) in the CM101-treated mice is reduced compared with control. (Bars = 100 μm.)
In Vitro Studies.
Studies on isolated CNS. All CNSs maintained in culture showed viability on day 5–7, during which time electrophysiological measurements were performed. Figs. 5 A and B shows representative data from several observations made with or without CM101 in nontraumatized spinal cords on days 1 and 5. Likewise, Fig. 5 C–F shows representative measurements with traumatized neurons in CNS without (Fig. 5 C and D) and with (Fig. 5 E and F) 0.3 μg/ml CM101 in the culture media.
Figure 5.

Effects of CM101 on isolated CNS from mouse embryos. Control recording was made by using two intracellular microelectrodes placed on each side of the spinal cord lesion on day 1 (A) and day 5 (B) in vitro. Compression and crush of isolated tissue at the level of T10 of the spinal cord produced significant reduction of conduction of electric stimuli on day 1 (C). On day 5 (D), in the absence of CM101, no electric stimuli were detected after multiple and increased stimulation. In spinal cord tissue cultured with CM101 (0.3 μg/ml), a slightly longer latency of transmitted excitation was seen on day 1 (E) and significant improvement in conductivity was observed on day 5 (F). B-1, D-1, and F-1 are expansions of the time axis of day 5 recordings. Action potentials (B-1 and F-1) suggest neuronal components of pulse transmission that are absent in untreated CNS (D-1).
Excitory stimulation in injured isolated CNS produced a brief depolarizing response (1–8 mV) with sporadic action potentials on day 1 (Fig. 5C) and day 5 (Fig. 5D) (n = 8) compared with control (Fig. 5A) (n = 4) or control with CM101 (Fig. 5B) (n = 4), where a 100- to 300-ms pulse gave repetitive field and action potentials of a maximum of 60 mV from −60 to 0 mV. Traumatized CNSs with CM101 (0.3 μg/ml) in the media and in the superfusate during the electrophysiological experiments (Fig. 5E) showed minimal response on day 1. Recording performed on day 2 showed improved current transmission through the lesion site (data not shown). Measurements on day 5 after injury in the presence of CM101 produced intensive bidirectional change of the field potential (Fig. 5F). Detailed analysis of the field action and postsynaptic action potential tracings suggests neuronal routing of stimuli between the electrodes in control neurons (Fig. 5B-1) and in traumatized neurons treated with CM101 (Fig. 5F-1). Lack of action potentials on day 5 in injured untreated spinal cords suggests lack of axonal connectivity (Fig. 5D-1). The presence of action potentials in CM101-treated CNS (Fig. 5F-1) provides evidence for axonal reconnection and regeneration in central neurons of the isolated traumatized CNS.
Effect of CM101 on GABA-induced depolarization in traumatized neuron.
Inhibitory neurotransmitter, GABA (10 μM), applied for 3 sec by pressure ejection from a blunt-tipped patch–clamp pipette (marked “X” in Fig. 6) produced hyperpolarization from −60 to −68 mV in normal mouse spinal cord neurons (n = 20) (Fig. 7 A–C). Trauma was induced by compression of axons using the patch–clamp microelectrode. The response to the induced trauma in untreated neurons was dramatic. Rapid swelling was followed by axonal autoamputation and cell death in 95% of the cells (Fig. 6 B and C). However, only a 5% loss of cells occurred with CM101 and no axonal amputation or Wallerian degeneration in the surviving 95% of neurons was seen (Fig. 6 E and F). GABA then was applied 5–30 min after injury for 3 sec and produced action potential from −65 mV to +20 mV followed by depolarization from −65 to −30 mV ± 10 mV (n = 10) (Fig. 7D). GABA (10 μM) applied on day 2 after injury in cultured untreated neurons produced less pronounced but still depolarizing responses (Fig. 7E). Additional firing of action potentials indicative of cell injury was observed in about 30% of the untreated neurons. Depolarization (from −67 to −41 mV) was still present on day 5 after the injury (Fig. 7F). CM101 treatment produced a time-dependent modification of the GABA-mediated depolarization in the mechanically injured neurons (Fig. 7 G and I). GABA (10 μM) applied on day 2 after injury in CM101-treated neurons produced less-pronounced depolarization and less firing of action potentials (compare Fig. 7 H with E), and, by day 5, GABA response returned to near normal with no depolarizing response and no action potential in the CM101-treated spinal cord neurons (n = 10), indicating partial recovery from trauma.
Figure 6.

Mechanical injury of the spinal cord neurons cultured in a monolayer network. (A–C) Control. (D—F) Cultured with CM101 (0.3 μg/ml) 48 hr before injury and for the duration of the experiment. The patch–clamp pipette (X) was used to produce mechanical crush injury to single neurons. After obtaining control image (A), mild compression was applied to axons (arrowhead) to produce cell damage. Axon and soma swelled within 1–3 min (B), and axon autoamputation and autolysis was observed in 95% of neurons within 5–30 min. Within 1–4 hr, swelling was reduced (C) in the remaining injured untreated neurons. CM101 treatment (D) prevented cell death (95% survival) and axonal amputation (two injured neurons shown: arrowhead in E); however, CM101 did not protect axons from swelling (E), which occurred within 2–3 min after crush injury and reversed within 6 hr (F). Z, intracellular microelectrode used to record from a single neuron. (Bar = 50 μm.)
Figure 7.

Effects of CM101 on cultured mouse spinal cord neurons traumatized by axonal mechanical compression and stimulated with 3-s pulses of 10 μM GABA (horizontal lines below readout). CM101 in vitro reversed GABA-mediated depolarizing responses in injured mammalian central neurons in the culture (G and I). The oscilloscope traces A, B, and C represent recordings of hyperpolarization from the control uninjured neurons. The oscilloscope traces D, E, and F represent recordings of depolarization from the mechanically injured neurons on days 1, 2, and 5 after trauma, respectively. The oscilloscope traces G, H, and I represent recordings from the mechanically injured neurons on days 1, 2, and 5 after trauma, treated with CM101-supplemented (0.3 μg/ml) culture medium. Calibration at the right refers to respective row(s).
DISCUSSION
Electrophysiological studies using intracellular microelectrode recordings from single neurons in culture showed that CM101 has a protective effect on the survival of neurons in culture subjected to trauma, presumably through a cellular lectin carbohydrate interaction that will warrant further studies at the molecular level. The effect on survival, however, is reflected in the recovery in injured neurons of GABA-induced hyperpolarization, characteristic of the normal neuron, over a 5-day period. Thus, CM101 inhibited axonal amputation and facilitated reestablishment of neuronal conductivity in vitro.
The isolated CNSs incubated with CM101 after crush show a restored conductivity by day 5, and it may be assumed that CM101 had diffused through the crushed area to be able to inhibit the inflammatory angiogenesis and scarring event, which otherwise would prevent recovery of connectivity. Restored conductivity in isolated CNS would be the equivalent to recovery of walking in adult animals.
Microelectrode field potential recordings from the isolated and cultured CNS showed neuronal characteristics of pulse transmission across the spinal lesions. The neuronal component consisted of action potentials and the glial component consisted of the oscillatory wave of the recorded field potential. Action potentials were observed in nontraumatic controls and in CM101-treated injured spinal cords but not in traumatized untreated controls. Our in vitro data indicate that CM101 facilitated the return of neuron-type firing across the lesion, demonstrating reconnection of axons in the traumatized neurons. The oscillatory low wave potential observed in injured spinal cords not treated with CM101 may, in fact, be a result of glial syncytium.
Ramón y Cajal has described the formation of growth cones and sprouts shortly after the lesions in the spinal cord (37). He observed that neuronal regeneration was present but aborted and that axonal elongation did not occur. His quantitative assessment of regeneration in the transected corticospinal tract was confirmed and refined by Snell and Schwab (38).
Observations with CM101 as an antiangiogenic (20) and antineovascularization agent in cancer patients (39, 40) support that CM101 inhibits hypoxia-induced pathologic angiogenesis. When present in circulation in vivo, CM101 appears to act as an antiangiogenic agent by effectively inhibiting inflammatory angiogenesis induced by hypoxia in a tumor (20) or wounded tissue (M. Neeman, personal communication). Similarly, the mice in this study treated with CM101 experienced rapid healing of their wounds from the surgical incisions without a scar.
We demonstrate by pathologic examination and MRI analysis that CM101 treatment reduces the extent of expected gliosis in the spinal cord after spinal cord crushes and that paraplegic mice treated with CM101 recovered the ability to walk compared with the few survivors not treated with CM101 who were unable to use their rear limbs.
We postulate that antipathoangiogenic CM101 caused the observed inhibition of gliosis, which facilitated axonal reconnection and subsequent recovery of function. The MRI and histology data support that CM101 minimized the acute and chronic inflammation in the CNS leading to recovery of walking ability in 24 of 26 mice within 2–12 days. Furthermore, treatment with CM101 after spinal cord crush leading to paralysis had a dramatic effect on survival since 25 of 26 CM101-treated animals survived the trauma, whereas only 6 of 14 animals in the control group survived for 48 hr; 6 animals died within 24 hr.
Application of CM101 in acute SCI in humans is suggested by the apparent mechanism of action and clinical experience with CM101 in cancer (39, 40). Potential application of CM101 in chronic SCI, however, would require a resection of the scar that would have formed after the original trauma. Regeneration of function then possibly could be obtained by inhibiting rescarring with CM101 treatment and promoting neuronal regrowth with specific growth factors, which may include insulin-like growth factor I, basic fibroblast growth factor, or transforming growth factor-βS (9), or brain-derived neurotrophic factor NT-3 (14), ciliary neurotrophic factor (13, 14), transplantation of neural progenitor cells (41), or by treatment with macrophages (42).
SCI affects a large number of physically active individuals, with tragic consequences. Our data demonstrated that i.v. administration of CM101 after injury was capable of producing both a remarkably rapid regeneration of spinal cord function in acute SCI, as evidenced by recovery of walking ability within days of treatment, and a quite significant protection from early fatalities among the treated animals.
Acknowledgments
We dedicate this paper to Mr. Christopher Reeve. We thank Prof. Guy M. McKhann of Krieger Mind and Brain Institute at The Johns Hopkins University and Profs. Michael Waterman, Department of Biochemistry, James V. Staros, Department of Molecular Biology, and David M. Lovinger, Department of Molecular Physiology and Biophysics, Vanderbilt University, and Prof. Stephen Lloyd, University of Texas Medical Branch–Center for Molecular Science, Galveston, for reviewing this manuscript. We thank Vicki Janson for technical assistance, Drs. David L. Page and Mahlon Johnson for the histopathological examinations, Dr. Lee Hakil for MRI study support, George Holburn for MRI technical assistance, Dr. Kevin Standard for MRI evaluation, Pam Chunn for secretarial assistance, and RobMarc Production, Inc., for videotaping and video editing services. This research was supported by a grant from CarboMed, Inc., in which the authors have a financial interest.
ABBREVIATIONS
- GBS
group B streptococcus
- CNS
central nervous system
- SCI
spinal cord injury
- GABA
γ-aminobutyrate
References
- 1. Liu X Z, Xu X M, Hu R, Du C, Zhang S X, McDonald J W, Dong H X, Wu Y J, Fan G S, Jacquin M F, et al. J Neurosci. 1997;17:5395–5406. doi: 10.1523/JNEUROSCI.17-14-05395.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olsson Y, Sharma H S, Pettersson A, Cervos-Navarro J. Prog Brain Res. 1992;91:197–203. doi: 10.1016/s0079-6123(08)62335-3. [DOI] [PubMed] [Google Scholar]
- 3.Crowe M J, Bresnahan J C, Shuman S L, Masters J N, Beattie M S. Nat Med. 1997;3:73–76. doi: 10.1038/nm0197-73. [DOI] [PubMed] [Google Scholar]
- 4.Nicholls J, Saunders N. Trends Neurosci. 1996;19:229–234. doi: 10.1016/0166-2236(96)10021-7. [DOI] [PubMed] [Google Scholar]
- 5.Cheng H, Cao Y, Olson L. Science. 1996;273:510–513. doi: 10.1126/science.273.5274.510. [DOI] [PubMed] [Google Scholar]
- 6.Young W. Science. 1996;273:451. doi: 10.1126/science.273.5274.451. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Field P M, Raisman G. Science. 1997;277:2000–2002. doi: 10.1126/science.277.5334.2000. [DOI] [PubMed] [Google Scholar]
- 8.Tuszynski M H, Peterson D A, Ray J, Baird A, Nakahara Y, Gage F H. Exp Neurol. 1994;126:1–14. doi: 10.1006/exnr.1994.1037. [DOI] [PubMed] [Google Scholar]
- 9.Houle J, Ye J-H, Kane C J M. Restorative Neurol Neurosci. 1996;10(4):205–215. doi: 10.3233/RNN-1996-10403. [DOI] [PubMed] [Google Scholar]
- 10.Zhao S R, Beuerman R W, Kline D G. J Reconstr Microsurg. 1997;13(1):39–45. doi: 10.1055/s-2008-1063939. [DOI] [PubMed] [Google Scholar]
- 11.Bregman B S, Kunkel-Bagden E, Schnell L, Dai H N, Gao D, Schwab M E. Nature (London) 1995;378:498–501. doi: 10.1038/378498a0. [DOI] [PubMed] [Google Scholar]
- 12.Schwab M E. Int J Dev Neurosci. 1996;14:379–385. [PubMed] [Google Scholar]
- 13.Ye J-H, Houle J. Exp Neurol. 1997;143:70–81. doi: 10.1006/exnr.1996.6353. [DOI] [PubMed] [Google Scholar]
- 14.Grill R, Murai K, Blesch A, Gage F H, Tuszynski M H. J Neurosci. 1997;17:5560–5572. doi: 10.1523/JNEUROSCI.17-14-05560.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gendelman M, Tardieu M. J Leukocyte Biol. 1994;56:389–398. doi: 10.1002/jlb.56.3.389. [DOI] [PubMed] [Google Scholar]
- 16.Mallat M, Chamak B. J Leukocyte Biol. 1994;56:416–422. doi: 10.1002/jlb.56.3.416. [DOI] [PubMed] [Google Scholar]
- 17.Gehrmann J, Matsumoto Y, Dreutzberg G W. Brain Res Rev. 1995;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- 18.Tatagiba M, Brosamle C, Schwab M E. Neurosurgery. 1997;40:541–547. doi: 10.1097/00006123-199703000-00023. [DOI] [PubMed] [Google Scholar]
- 19.Ferrara N. Breast Cancer Res Treatment. 1995;36:127–137. doi: 10.1007/BF00666035. [DOI] [PubMed] [Google Scholar]
- 20.Yan, H.-P., Carter, C. E., Wang, E.-Z., Page, D. L., Washington, K., Wamil, B. D., Yakes, F. M., Thurman, G. B. & Hellerqvist, C. G. (1998) Angiogenesis, in press. [DOI] [PubMed]
- 21.Hellerqvist C G, Rojas J, Green R S, Sell S, Sundell H W, Stahlman M T. Pediatr Res. 1981;15:892–898. doi: 10.1203/00006450-198106000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Z, Guth L. Exp Neurol. 1997;147:159–171. doi: 10.1006/exnr.1997.6590. [DOI] [PubMed] [Google Scholar]
- 23.Guth L, Albuquerque E X, Deshpande S S, Barrett C P, Donati E J, Warnick J E. J Neurosurg. 1980;52:73–86. doi: 10.3171/jns.1980.52.1.0073. [DOI] [PubMed] [Google Scholar]
- 24.Blight A, Young U. J Neurol Sci. 1989;91:15–34. doi: 10.1016/0022-510x(89)90073-7. [DOI] [PubMed] [Google Scholar]
- 25.Nobel L, Wrathall J. Exp Neurol. 1989;103:34–40. doi: 10.1016/0014-4886(89)90182-9. [DOI] [PubMed] [Google Scholar]
- 26.Behrmann D L, Bresnahan J C, Beattie M S, Shah B R. J Neurotrauma. 1992;9:197–217. doi: 10.1089/neu.1992.9.197. [DOI] [PubMed] [Google Scholar]
- 27.Basso D M, Beattie M S, Bresnaham J C. Exp Neurol. 1996;139:244–256. doi: 10.1006/exnr.1996.0098. [DOI] [PubMed] [Google Scholar]
- 28.Wamil A W, Parris W C V. Curr Rev Pain. 1997;1:251–264. [Google Scholar]
- 29.Van den Pol A N, Obrietan K, Chen G. J Neurosci. 1996;16:4283–4292. doi: 10.1523/JNEUROSCI.16-13-04283.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hellerqvist, C. G. (1991) Therapeutic Agent and Method of Inhibiting Vascularization of Tumors, U.S. Patent 5,010,062 (patents have been issued in Europe, Canada, Australia, and Japan).
- 31.Saunders N R, Deal A, Knott G W, Vaiga Z M, Nicholls J G. Clin Exp Pharmacol Physiol. 1995;22:518–526. doi: 10.1111/j.1440-1681.1995.tb02060.x. [DOI] [PubMed] [Google Scholar]
- 32.Nicholls J G, Stewart R R, Erulkar S D, Saunders N R. J Exp Biol. 1990;152:1–15. doi: 10.1242/jeb.152.1.1. [DOI] [PubMed] [Google Scholar]
- 33.Wamil A W, Schmutz M, Portet C H, Feldmann K F, McLean M J. Eur J Pharmacol. 1994;271:301–308. doi: 10.1016/0014-2999(94)90787-0. [DOI] [PubMed] [Google Scholar]
- 34.Wamil A W, Loscher W, McLean M J. J Pharmacol Exp Ther. 1997;280:1349–1356. [PubMed] [Google Scholar]
- 35.Mollgard K, Balslev Y, Janas M S, Treherne J M, Saunders N R, Nichols J G. J Neurocytol. 1994;23:151–165. doi: 10.1007/BF01181557. [DOI] [PubMed] [Google Scholar]
- 36.Sotelo C, Alvardo-Mallart R M. Trends Neurosci. 1991;14:350–355. doi: 10.1016/0166-2236(91)90161-m. [DOI] [PubMed] [Google Scholar]
- 37.Ramón y Cajal S. Degeneration and Regeneration of the Nervous System. London: Oxford Univ. Press; 1928. [Google Scholar]
- 38.Snell L, Schwab M E. Eur J Neurosci. 1993;5:1156–1171. doi: 10.1111/j.1460-9568.1993.tb00970.x. [DOI] [PubMed] [Google Scholar]
- 39.Wamil B D, Thurman G B, Sundell H W, DeVore R F, Wakefield G B, Johnson D H, Wang Y-F, Hellerqvist C G. J Cancer Res Clin Oncol. 1997;123:173–179. doi: 10.1007/BF01214670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeVore R F, Hellerqvist C G, Wakefield G B, Wamil B D, Thurman G B, Minton P A, Sundell H W, Yan H-P, Carter C E, Wang Y-F, et al. J Clin Cancer Res. 1997;3:365–372. [PubMed] [Google Scholar]
- 41.Lundberg C, Winkler C, Whittemore S R, Bjorklund A. Brain Res. 1996;737:295–300. doi: 10.1016/0006-8993(96)00923-7. [DOI] [PubMed] [Google Scholar]
- 42.Rapalino O, Lazarov-Spiegler O, Agranov E, Velan G J, Yoles E, Fraidakis M, Solomon A, Gepstein R, Katz A, Belkin M, et al. Nat Med. 1998;4:814–821. doi: 10.1038/nm0798-814. [DOI] [PubMed] [Google Scholar]