

Abstract
Episodic hypoxia causes repetitive inspiratory activation that induces a form of respiratory plasticity termed long-term facilitation (LTF). While LTF is a function of the hypoxic exposures and inspiratory activation, their relative importance in evoking LTF is unknown. The aims of this study were to: (1) dissociate the relative roles played by episodic hypoxia and respiratory activation in LTF; and (2) determine whether the magnitude of LTF varies as a function of hypoxic intensity. We did this by examining the effects of episodic hypoxia in postnatal rats (15–25 days old), which unlike adult rats exhibit a prominent hypoxia-induced respiratory depression. We quantified inspiratory phrenic nerve activity generated by the in situ working-heart brainstem before, during and for 60 min after episodic hypoxia. We demonstrate that episodic hypoxia evokes LTF despite the fact that it potently suppresses inspiratory activity during individual hypoxic exposures (P < 0.05). Specifically, we show that after episodic hypoxia (three 5 min periods of 10% O2) respiratory frequency increased to 40 ± 3.3% above baseline values over the next 60 min (P < 0.001). Continuous hypoxia (15 min of 10% O2) had no lasting effects on respiratory frequency (P > 0.05). To determine if LTF magnitude was affected by hypoxic intensity, the episodic hypoxia protocol was repeated under three different O2 tensions. We demonstrate that the magnitude and time course of LTF depend on hypoxic severity, with more intense hypoxia inducing a more potent degree of LTF. We conclude that inspiratory activation is not required for LTF induction, and that hypoxia per se is the physiological stimulus for eliciting hypoxia-induced respiratory LTF.
The respiratory motor control system exhibits marked neuroplasticity when challenged with repeated perturbations in O2 levels (Bach & Mitchell, 1996; Mitchell et al. 2001a; Mahamed et al. 2005; Beecroft et al. 2006). One form of respiratory plasticity is long-term facilitation (LTF), a prolonged increase in respiratory motor output that persists (> 1 h) following exposure to episodic hypoxia (Powell et al. 1998; Baker-Herman & Mitchell, 2002; Baker-Herman et al. 2004). LTF is characterized by a persistent increase in the amplitude and/or frequency of inspiratory efforts of the upper airway and diaphragm muscles following episodic but not continuous hypoxia. It is observed in anaesthetized and freely behaving rats (Fuller et al. 2000; Olson et al. 2001; McGuire et al. 2002; Feldman et al. 2003; McGuire et al. 2003) as well as in awake and sleeping humans (Babcock et al. 2003; Harris et al. 2006). LTF has also been evoked in awake goats (Turner & Mitchell, 1997), ducks (Mitchell et al. 2001b) and dogs (Cao et al. 1992); however, results are variable because it has not been found in anaesthetized, spontaneously breathing rats (Janssen & Fregosi, 2000) or in some healthy awake humans (Jordan et al. 2002).
LTF can be elicited in anaesthetized rats without intact peripheral chemoreceptors, suggesting that it is mediated by a central neural mechanism (Bavis & Mitchell, 2003). It is hypothesized that the serotonergic and noradrenergic systems (e.g. medullary raphe and locus coeruleus) underlie hypoxia-induced LTF because blockade of either 5-HT2A (Fuller et al. 2001; McGuire et al. 2004) or α1 adrenergic (Neverova et al. 2007) receptors abolish this phenomenon.
As LTF is elicited by not only episodic hypoxia, but also by repetitive carotid sinus nerve stimulation in vivo (Millhorn et al. 1980; Ling et al. 1997) or episodic application of exogenous serotonergic or noradrenergic receptor agonists in vitro (Bocchiaro & Feldman, 2004; Feldman et al. 2005), all of which act to transiently augment inspiratory activity, it is possible that this phenomenon is the by-product of a non-specific activity-dependent respiratory activation. In terms of hypoxia-induced LTF, it is currently unknown whether it can be evoked solely by episodic hypoxia or whether it requires both hypoxia and respiratory activation. It is therefore necessary to determine whether episodic hypoxia requires concomitant inspiratory activation for the induction of LTF. In adult rodents, it is not possible to dissociate these variables from one another, and therefore document their independent roles in LTF, because respiratory activity is potently stimulated by hypoxia (Bach & Mitchell, 1996; Baker & Mitchell, 2000; Olson et al. 2001; Fuller et al. 2002). The present study circumvents this limitation by studying hypoxia-induced LTF in postnatal rats, which unlike adult rats, exhibit a prominent ventilatory depression when exposed to hypoxia (Eden & Hanson, 1987; Hilaire & Duron, 1999; Mortola, 1999). We therefore take advantage of the postnatal hypoxic ventilatory response to test the hypothesis that hypoxia-induced LTF is caused by episodic hypoxic stimuli and not by activity-dependent inspiratory activation.
Accordingly, this study was designed to determine whether: (1) episodic hypoxia could evoke LTF in the absence of inspiratory activation; and (2) the magnitude of LTF varies as a function of hypoxic intensity. We demonstrate that episodic hypoxia-induced ventilatory suppression induces long-term facilitation of phrenic motor output in postnatal (15–25 days old) rats, and that the magnitude and time course of LTF is affected by the severity of the episodic hypoxia. These data demonstrate, for the first time, that episodic hypoxia and not inspiratory activation, is the physiological stimulus that underlies the induction of hypoxia-induced LTF in neonatal rats.
Methods
Experimental preparation
All experiment were performed in the in situ working-heart brainstem preparation, which is an established model for studying the neural mechanisms of respiratory control in postnatal rats (Paton, 1996; Wilson et al. 2001; Potts et al. 2005; Day & Wilson, 2006). The in situ preparation is arterially perfused with artificial cerebral spinal fluid (ASCF) that permits oxygenation of the respiratory network so that it generates a normal breathing rhythm on the phrenic and hypoglossal nerves innervating the diaphragm and genioglossus muscles, respectively. We used the in situ preparation for two reasons. First, the respiratory response to hypoxia has been previously described in this model system (St-John & Paton, 2000) and, like the hypoxic response of postnatal rats in vivo, it is characterized by a potent decrease in respiratory motor output. Second, because the in situ preparation is arterially perfused, changes in neural ventilation, such as in response to hypoxia, do not lead to subsequent changes in CO2 or O2 levels because lung ventilation is absent. This ensures that long-term changes in neural ventilation after episodic hypoxia are not due to ventilatory-induced fluctuations in O2 and CO2 levels.
Experiments were performed on postnatal male Sprague–Dawley rats (20.5 ± 0.7 days old, range 15–25 days; 46.7 ± 2.8 g, range 30–70 g; Charles River Laboratories). Rat pups were housed (12 h light–dark cycle) with their mothers and had unlimited access to both food and water. Male rats were randomly selected and exposed to one of three experimental protocols (see below, Experimental protocols), which were performed between 12.00 and 18.00 h. All procedures were approved by the Animal Care and Use Committee at the University of Toronto.
Rats were anaesthetized in a chamber ventilated with 5% halothane (in 50% O2). After complete abolition of the foot-pinch withdrawal reflex, they were transected subdiaphragmatically and immersed in cold, oxygenated (95% O2 and 5% CO2) ACSF (125 mm NaCl, 5.0 mm KCl, 1.25 mm KH2PO4, 24 mm NaHCO3, 2.5 mm CaCl, 1.25 mm MgSO4 and 20 mm dextrose). Both parietal bones were removed and all brain structures rostral to the superior colliculus were removed by aspiration.
The preparation was then placed into a recording chamber and the descending aorta cannulated using single lumen catheters (18 and 20 gauge; Harvard Apparatus). The preparation was continuously perfused with warm, oxygenated (95% O2 and 5% CO2) ACSF to which oncotic (2.5% Ficoll 70; Sigma), paralytic (2 μg ml−1 pancuronium bromide; Sabex), and anti-inflammatory agents (8 μg ml−1 dexamethasone; Vetoquinol) had been added. The lungs were ligated and removed. Temperature of the preparation was measured via a thermistor (WPI Inc.) and maintained at 29.9 ± 0.2°C by heating the ASCF in a water-bath (Thermoneslab, Model RTE7). Arterial pressure was measured by placing a catheter into the ascending aorta and connecting it to a blood pressure transducer (Harvard Apparatus). Aortic pressure was maintained at levels approximating that of intact rats (i.e. 80–100 mmHg; Kasparov & Paton, 1997) by adjusting the flow rate of the peristaltic pump (Watson-Marlow 502S).
Experimental recording
The electrical activity recorded from left or right phrenic and hypoglossal nerves served as an index of central respiratory rhythm. Nerves were dissected and isolated from surrounding connective tissue; their most distal ends were cut and drawn into bi-polar suction electrodes. Nerve activity was amplified (Neurolog, NL104), filtered (band pass 500–5000 Hz; Neurolog, NL126), and integrated (time constant, 100 ms). Nerve activity was digitized (1 kHz; Cambridge Electronic Design, Micro1401) and stored on a computer (Dell) using Spike 2 software (Cambridge Electronic Design). Specially written software (National Instruments, Labview) processed integrated nerve signals so that both the frequency and amplitude of nerve signals were automatically calculated and stored in 60 s time bins.
Aortic pressure and cranial temperature were concurrently recorded with nerve activities and were digitized (40 Hz; Cambridge Electronic Design, Micro1401) and stored on a computer (Dell) using Spike 2 software (Cambridge Electronic Design).
Experimental protocols
Preparations were left to stabilize for 60 min before recordings were made. Following this stabilization period, baseline phrenic and hypoglossal nerve activities were recorded for 10–15 min before performing one of three experimental protocols. Each in situ preparation was exposed to only one protocol and one level of hypoxia.
Protocol 1 – Episodic hypoxia
These experiments were designed to determine whether: (1) episodic hypoxia could evoke LTF in the absence of inspiratory activation; and (2) the magnitude of LTF varies as a function of hypoxic intensity. Respiratory motor output was recorded under baseline conditions (95% O2–5% CO2) for 10–15 min; then each preparation was exposed to one of three levels of episodic hypoxia: (i) severe – 10% O2–5% CO2–85% N2; (ii) moderate – 20% O2–5% CO2–75% N2; or (iii) mild – 40% O2–5% CO2–55% N2. The episodic hypoxia protocol consisted of three cycles of 5 min of hypoxia (severe, moderate or mild) that alternated with 5 min of baseline conditions. Following episodic hypoxia, baseline conditions were reinstated and phrenic nerve activity was recorded for another 60 min. The mean ages of the rats used to make in situ preparations for the severe, moderate and mild hypoxia groups were: 21 ± 1.9, 21 ± 1.4 and 21 ± 0.8 days old, respectively. There was no significant difference between these age groups (one-way ANOVA, P < 0.05).
Although tissue 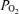 was not measured in these experiments, previous data demonstrate that gassing the perfusate of the in situ preparation with 95% O2 produces adequate oxygenation of the respiratory network (Paton, 1996; Day & Wilson, 2006) therefore gassing the perfusate with less than 95% O2 should cause CNS hypoxaemia. Indeed, we observed that each of the three levels of hypoxia (severe: 10% O2; moderate: 20% O2; and, mild: 40% O2) produced tissue hypoxaemia because respiratory motor output was potently and rapidly affected by each level of hypoxia (see Fig. 2).
was not measured in these experiments, previous data demonstrate that gassing the perfusate of the in situ preparation with 95% O2 produces adequate oxygenation of the respiratory network (Paton, 1996; Day & Wilson, 2006) therefore gassing the perfusate with less than 95% O2 should cause CNS hypoxaemia. Indeed, we observed that each of the three levels of hypoxia (severe: 10% O2; moderate: 20% O2; and, mild: 40% O2) produced tissue hypoxaemia because respiratory motor output was potently and rapidly affected by each level of hypoxia (see Fig. 2).
Figure 2. Typical respiratory responses to hypoxia.

Representative examples of integrated (in arbitrary units) phrenic and hypoglossal nerve activity before (baseline conditions 95% O2) and after exposure to severe (10% O2; top trace), moderate (20% O2; centre trace) and mild (40% O2; bottom trace) hypoxia. All levels of hypoxia caused a potent suppression of both respiratory frequency and inspiratory amplitude. Baseline traces are typical examples of nerve activity in the minute preceding hypoxia and hypoxic traces are representative examples of respiratory activity in the 5th minute of the first hypoxic exposure.
Protocol 2 – Continuous hypoxia
This protocol was designed to demonstrate that continuous hypoxia-induced ventilatory depression does not induce LTF. Respiratory motor output was recorded under baseline conditions and then the preparation was exposed to 15 min of continuous severe, moderate or mild hypoxia (see Protocol 1 for O2 concentrations). Following the 15 min period of hypoxia, baseline conditions were reinstated and respiratory motor output was recorded for another 60 min. The mean age of the rats used to make in situ preparations for this treatment group was 20 ± 0.5 days old.
Protocol 3 – Time control
This protocol was designed to demonstrate that respiratory rhythm remained stable across the experimental recording period. Respiratory motor output was recorded for a 150 min period under baseline conditions. It was necessary to demonstrate respiratory stability over a 150 min period since this is the time required to complete the episodic and continuous hypoxia protocols (see Protocols 1 and 2). The mean age of the rats used to make in situ preparations for this experimental group was 20 ± 2.5 days old.
To further demonstrate maintained respiratory responsiveness, rats from each protocol were exposed to 5 min of 10% CO2 at the beginning (0 min) and end (150 min) of the experiment. These hypercapnic responses were used to confirm that the respiratory control system consistently responded to respiratory challenges across the recording period, and they were also used to demonstrate that inspiratory amplitude could be elevated above baseline values. It was important to confirm the latter because inspiratory amplitude (60 min after the last hypoxic episode) was unaffected by exposure to episodic hypoxia, and so this approach enabled us to demonstrate that inspiratory amplitude could indeed have been elevated as part of an LTF response.
Analyses
Phrenic motor output
Although hypoglossal motor output was recorded, its activity was not quantified because there was no observable difference between either the short-term or long-term responses to hypoxia as compared to phrenic nerve activity. Therefore, integrated phrenic nerve activity was used to determine both respiratory frequency and inspiratory amplitude. Respiratory frequency was calculated by averaging the number of inspiratory nerve discharges that occurred in 60 s and is expressed as bursts per minute. Inspiratory amplitude was calculated by measuring the difference between the expiratory phase and peak inspiratory amplitude for each integrated burst. These values are presented in arbitrary units (a.u.) and were averaged over the same 60 s period that was used to determine mean respiratory frequency. Data are also expressed as neural minute ventilation (n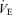 ) and were calculated as the product of respiratory frequency and inspiratory amplitude. Values expressed as per cent change were calculated as measured value minus baseline value divided by baseline value multiplied by 100%.
) and were calculated as the product of respiratory frequency and inspiratory amplitude. Values expressed as per cent change were calculated as measured value minus baseline value divided by baseline value multiplied by 100%.
Measurements
All data presented were based on 60 s averages. Respiratory frequency, phrenic inspiratory amplitude and minute neural ventilation were measured for the minute proceeding (baseline) and during each minute of both episodic and continuous hypoxia (severe, moderate and mild). These variables were also analysed at 15, 30, 45 and 60 min post-hypoxia. In the time control experiments, respiratory frequency and inspiratory amplitude were analysed every 30 min over a 150 min period. The respiratory responses to 10% CO2 were determined by comparing respiratory frequency and inspiratory amplitude in the minute preceding CO2 exposure (baseline) to those in the 5th minute of the exposure.
Statistical analysis
Two-way RM ANOVAs were used to determine whether individual hypoxic episodes (e.g. 5 min severe hypoxia) suppressed respiratory output, and whether there was a difference between the magnitude of respiratory suppression between the severe, moderate and mild hypoxic groups. The two factors were experimental treatment (i.e. severe, moderate or mild hypoxia) and time (i.e. each minute of baseline versus each minute of hypoxia). Two-way RM ANOVAs were also used to determine whether episodic hypoxia induced long-term changes in respiratory motor output, and whether the degree of hypoxia was a significant determinant of the response. The two factors were experimental treatment (i.e. episodic hypoxia, continuous hypoxia and time control) and time (i.e. baseline and 15, 30, 45, 60 min post-hypoxia). Statistical differences were determined using the Student–Newman–Keuls method post hoc test and were considered significant when P < 0.05. A paired t test (parametric) was used to determine the affects of 10% CO2 on respiratory motor output at the beginning (0 min) and end of experiments (150 min). All statistical tests, sample sizes and P values are listed in parentheses within the results. Analyses were performed using Sigmastat (Jandel Scientific Inc., San Rafael, CA, USA). Data are expressed as means ± standard errors (mean ± s.e.m.) and this symbol (*) indicates statistical significance compared to baseline.
Results
Episodic hypoxia
Individual hypoxic episodes suppress respiratory activity
Figure 1A depicts a typical response from one in situ preparation; it illustrates that phrenic motor output was depressed by each of the three 5 min episodes of severe (10% O2) hypoxia and returned to pre-hypoxic levels during control conditions (i.e. 95% O2). Hypoglossal motor output was also suppressed during each hypoxic episode and returned to baseline levels during control conditions (Fig. 2).
Figure 1. Representative examples demonstrating how respiratory activity is affected by episodic hypoxia, continuous hypoxia or time (control).

A, episodic hypoxia: a typical example demonstrating that respiratory activity of the phrenic nerve (integrated and in arbitrary units) is suppressed by each 5 min cycle of hypoxia and that respiratory frequency (Rf) progressively increases above baseline levels over the 60 min period post-hypoxia (i.e. frequency LTF). At the end of the 60 min period, inspiratory amplitude was unaffected by episodic hypoxia. B, continuous hypoxia: a representative example showing that while respiratory activity is depressed during the 15 min period of continuous hypoxia, this did not result in long-term changes in respiratory frequency (i.e. no LTF). Inspiratory amplitude did not return to baseline levels after continuous hypoxia. C, time control: a typical example showing that respiratory frequency and inspiratory amplitude remain constant over the 150 min recording period. Bpm, bursts per minute.
Group data demonstrate that neural minute ventilation (phrenic) was significantly suppressed (two-way RM ANOVA, P < 0.05) below baseline conditions by all levels of hypoxia (e.g. severe group: n = 8; moderate group: n = 6; and mild group: n = 10; Fig. 3). Although neural minute ventilation was suppressed during each hypoxic episode, it returned to baseline levels when control conditions were reinstated (P > 0.05; Fig. 3). Hypoxia-induced suppression of both phrenic and hypoglossal motor output was attributable to declines in both respiratory frequency and inspiratory amplitude (Fig. 2), suggesting that inspiratory drive to both motor pools was potently reduced during individual hypoxic episodes.
Figure 3. Group data showing how respiratory activity is affected by episodes of severe, moderate or mild hypoxia.

Group data demonstrating that neural minute ventilation (n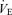 ) decreases during each 5 min hypoxic episode (filled bars) and that it returns to pre-hypoxia levels when baseline conditions (open bars) are reinstated. The top, middle and bottom graphs represent how n
) decreases during each 5 min hypoxic episode (filled bars) and that it returns to pre-hypoxia levels when baseline conditions (open bars) are reinstated. The top, middle and bottom graphs represent how n changes in response to each 5 min episode of severe (10% O2), moderate (20% O2) and mild (40% O2) hypoxia, respectively. The first open bar represents the baseline condition (95% O2), which was calculated by averaging n
changes in response to each 5 min episode of severe (10% O2), moderate (20% O2) and mild (40% O2) hypoxia, respectively. The first open bar represents the baseline condition (95% O2), which was calculated by averaging n in the minute preceding the first hypoxic exposure. Each bar thereafter represents the average group value per minute expressed as a percentage of the baseline. The upper dotted line in each graph represents baseline and the lower dotted line represents the average level of suppression during the 5th minute of the hypoxic episodes.
in the minute preceding the first hypoxic exposure. Each bar thereafter represents the average group value per minute expressed as a percentage of the baseline. The upper dotted line in each graph represents baseline and the lower dotted line represents the average level of suppression during the 5th minute of the hypoxic episodes.
Figure 4 demonstrates that respiratory motor output was suppressed by the same degree during each of the three episodes of hypoxia (whether severe, moderate or mild). During severe hypoxia, neural minute ventilation was significantly depressed by 42 ± 18, 40 ± 16 and 39 ± 16% (P < 0.001 for all three hypoxic episodes) during the 5th minute of the first, second and third hypoxic exposures, respectively. During moderate hypoxia, neural minute ventilation was reduced by 65 ± 9, 63 ± 11 and 58 ± 14% (P < 0.01 for all three hypoxic episodes) during the 5th minute of each of the three episodes; and during mild hypoxia, it was depressed by 61 ± 18, 62 ± 16 and 59 ± 16% (P < 0.05 for all three hypoxic episodes). However, neural minute ventilation was suppressed by the same degree in the severe, moderate and mild hypoxic groups (Fig. 4), that is, there was no statistically significant difference in the magnitude of respiratory depression among treatment groups (two-way RM ANOVA, P = 0.876).
Figure 4. Group data showing that neural minute ventilation is suppressed by each 5 min cycle of severe, moderate and mild hypoxia.

Mean group data demonstrating the magnitude of respiratory suppression during the first, second and third exposures to severe (10% O2), moderate (20% O2) and mild (40% O2) hypoxia. Neural minute ventilation (n ) was significantly suppressed (P < 0.05) during the 5th minute of each hypoxic cycle in all three groups (i.e. severe, moderate and mild hypoxia); however, there was no significant difference in the degree of suppression between the groups (2-way RM ANOVA, P = 0.876). Each bar in the graph represents a 1 min average. Baseline values of n
) was significantly suppressed (P < 0.05) during the 5th minute of each hypoxic cycle in all three groups (i.e. severe, moderate and mild hypoxia); however, there was no significant difference in the degree of suppression between the groups (2-way RM ANOVA, P = 0.876). Each bar in the graph represents a 1 min average. Baseline values of n were calculated in the 1 min preceding the first hypoxic exposure. n
were calculated in the 1 min preceding the first hypoxic exposure. n during hypoxia represents the percentage change from baseline during the 5th minute of each hypoxic exposure. *Significant decrease (P < 0.05) from baseline conditions.
during hypoxia represents the percentage change from baseline during the 5th minute of each hypoxic exposure. *Significant decrease (P < 0.05) from baseline conditions.
There was no sign of gasping (i.e. increased inspiratory amplitude and decrementing inspiratory burst pattern) at any hypoxic level (see Fig. 2), which agrees with previously reported findings that exposure to 5% O2 (in 8% CO2) is required to induce gasping in the in situ model system (Paton et al. 2006).
Episodic hypoxia induces respiratory long-term facilitation
All levels of episodic hypoxia, whether severe, moderate or mild, depressed inspiratory activity during each hypoxic episode; however, they also induced a persistent augmentation of respiratory motor output during the following 60 min post-hypoxia (i.e. LTF). These long-term changes were manifested by progressive increases in respiratory frequency; they were not due to changes in inspiratory amplitude (Figs 5 and 6). These effects are illustrated in Fig. 1A, which depicts: (a) respiratory suppression during each hypoxic episode; and (b) a progressive increase in respiratory frequency during the 60 min post-hypoxia.
Figure 5. Typical examples demonstrating how inspiratory phrenic nerve activity is affected by episodic hypoxia, continuous hypoxia or time (control).

Episodic and continuous hypoxia: representative examples of integrated phrenic nerve activity before (baseline), and 15, 30, 45 and 60 min after exposure to episodic hypoxia (10% O2; top trace) or continuous hypoxia (10% O2; middle trace). Episodic hypoxia induced a progressive increase in respiratory frequency over time; however, continuous hypoxia had no such effect. Time control: a typical example demonstrating the stability of inspiratory phrenic nerve activity across the 150 min experimental recording period. Integrated traces are in arbitrary units; horizontal dotted lines represent the average amplitude of inspiratory bursts during baseline conditions.
Figure 6. Group data demonstrating how respiratory frequency, inspiratory amplitude and neural minute ventilation are affected by episodic hypoxia, continuous hypoxia or time.

A, respiratory frequency (Rf): mean group data demonstrating that Rf is: (1) significantly increased at 15, 30, 45 and 60 min after severe episodic hypoxia (10% O2; n = 8; continuous line); (2) unaffected by severe continuous hypoxia (10% O2; n = 8; dashed line); and (3) stable during the 150 min recording period (n = 9; dotted line). B, inspiratory amplitude (Iamp): mean group data showing Iamp at 15, 30, 45 and 60 min after exposure to episodic or continuous hypoxia. Iamp remained stable across the 150 min baseline control period (dotted line) and is unaffected by episodic hypoxia (continuous line); however, it was suppressed by continuous hypoxic (dashed line). C, neural minute ventilation (n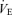 ): group data demonstrating n
): group data demonstrating n before and after episodic or continuous hypoxia. Episodic hypoxia non-significantly facilitated n
before and after episodic or continuous hypoxia. Episodic hypoxia non-significantly facilitated n  (continuous line), while continuous hypoxia suppressed it (dashed line). n
(continuous line), while continuous hypoxia suppressed it (dashed line). n  remained stable in the time control group (dotted line). Data are expressed as mean percentage changes from baseline (± s.e.m.); *Significant difference (P < 0.05) compared to baseline values.
remained stable in the time control group (dotted line). Data are expressed as mean percentage changes from baseline (± s.e.m.); *Significant difference (P < 0.05) compared to baseline values.
Mean data for the severe hypoxia group demonstrate that respiratory frequency returned to baseline levels after the third hypoxic episode (P = 0.09), but over the next 60 min progressively increased to 40 ± 3.3% above baseline values (P < 0.001 at 60 min), with respiratory frequency remaining significantly above baseline values at all time points post-hypoxia (n = 8, two-way RM ANOVA, P < 0.05; Figs 5 and 6A). While severe episodic hypoxia induced long-term increases in respiratory frequency (Table 1), there were no time-dependent effects on inspiratory amplitude (P < 0.05; Figs 5 and 6B). Although neural minute ventilation increased after episodic hypoxia, this trend did not reach statistical significance (P > 0.05; Fig. 6C).
Table 1.
Measurements of respiratory frequency before, and for 60 min after episodic or continuous hypoxia
| Protocol | Baseline | 15 min | 30 min | 45 min | 60 min |
|---|---|---|---|---|---|
| Control (n = 9) | 23 ± 1.8 | 23 ± 2.3 | 22 ± 2.4 | 24 ± 2.2 | 21 ± 2.9 |
| EH (n = 8) | 24 ± 2.3 | 28 ± 3.6* | 30 ± 3.6* | 32 ± 3.2* | 34 ± 3.3* |
| CH (n = 8) | 24 ± 1.4 | 20 ± 1.5 | 23 ± 1.4 | 25 ± 0.8 | 24 ± 1.0 |
Values are means ± s.e.m. EH, episodic hypoxia; CH, continuous hypoxia; Control, all measured in bursts per minute.
P < 0.05, significantly different compared to baseline.
There were no long-term changes in either inspiratory amplitude or neural minute ventilation following exposure to either moderate or mild levels (both moderate and mild, two-way RM ANOVA, P > 0.05; data not shown). However, both moderate and mild hypoxia induced a persistent augmentation of respiratory frequency (moderate: n = 6, P < 0.05; mild: n = 10, P < 0.05; Fig. 7). Exposure to moderate hypoxia increased respiratory frequency by 40 ± 2.1% above baseline conditions at 60 min (P = 0.005) while exposure to mild levels increased respiratory frequency by 25 ± 2.4% above baseline conditions (P < 0.001 at 60 min) (Table 2).
Figure 7. Effects of severe, moderate and mild hypoxia on the magnitude of respiratory LTF.

Mean group data demonstrating the effects of severe (10% O2; n = 8), moderate (20% O2; n = 6) or mild (40% O2; n = 10) episodic hypoxia on the magnitude of frequency LTF. Severe and moderate hypoxia significantly increased respiratory frequency (Rf) above baseline values by 40 ± 3.3% and 40 ± 2.1% (at 60 min post-hypoxia), respectively. In the severe hypoxic group, Rf significantly increased above baseline at 15, 30, 45 and 60 min post-hypoxia; however, in the moderate group, it did not increase until minutes 45 and 60 post-hypoxia. Mild episodic hypoxia induced the smallest degree of LTF, increasing Rf by 25 ± 2.4% above baseline levels at 60 min post-hypoxia. As in the moderate hypoxic group, mild hypoxia did not induce a statistically significant increase in Rf until 45 and 60 min post-hypoxia. Data are expressed as mean percentage changes from baseline (± s.e.m.). *, †, ‡ denote a significant increase (P < 0.05) above baseline values for the severe, moderate and mild hypoxic groups, respectively.
Table 2.
Measurements of respiratory frequency before, and for 60 min after episodic hypoxia at various intensities
| Protocol | Baseline | 15 min | 30 min | 45 min | 60 min |
|---|---|---|---|---|---|
| Severe (n = 8) | 24 ± 2.3 | 28 ± 3.6* | 30 ± 3.6* | 32 ± 3.2* | 34 ± 3.3* |
| Moderate (n = 6) | 25 ± 3.4 | 26 ± 2.4 | 29 ± 2.5 | 31 ± 2.7* | 34 ± 3.1* |
| Mild (n = 10) | 24 ± 2.0 | 25 ± 2.4 | 27 ± 2.6 | 29 ± 2.7* | 30 ± 2.4* |
Values are means ± s.e.m. Severe, mild and moderate hypoxia measured in bursts per minute.
P < 0.05, significantly different compared to baseline.
Figure 7 demonstrates that episodic hypoxia-induced LTF behaves in a dose-dependent manner, with severe hypoxia inducing the greatest degree of respiratory augmentation (40 ± 3.3% above baseline at 60 min post-hypoxia; P < 0.001) and mild hypoxia producing the least (25 ± 2.4% above baseline at 60 min post-hypoxia; P = 0.001). Hypoxic intensity also affects the time course of LTF, with severe hypoxia inducing a faster rate of facilitation (i.e. LTF) than either moderate or mild hypoxia (Fig. 7). In the severe hypoxia group, respiratory frequency significantly increased above baseline levels at minutes 15, 30, 45 and 60 post-hypoxia (P < 0.003 at all four time points); however, in the moderate and mild groups, respiratory frequency did not increase significantly until 45 min post-hypoxia (Fig. 7 and Table 2). In the moderate hypoxia group, respiratory frequency increased to 32 ± 2.8% (P = 0.03) and 40 ± 3.1% (P = 0.005) above baseline at 45 and 60 min post-hypoxia, respectively; and in the mild hypoxia group it increased to 19 ± 2.7% (P = 0.015) and 25 ± 2.4% (P = 0.001) above baseline at 45 and 60 min post-hypoxia, respectively.
Continuous hypoxia
Respiratory motor output is suppressed by continuous hypoxia
There was no observable difference between the response patterns to severe, moderate or mild levels of hypoxia and therefore only the results for severe hypoxia are presented. Continuous hypoxia suppressed respiratory motor output during the 15 min hypoxic exposure (Fig. 1B), during which time both respiratory frequency and inspiratory amplitude declined and then after 9 ± 1.6 min stopped (n = 8, one-way RM ANOVA, P = 0.001 for both frequency and inspiratory amplitude). When baseline conditions were reinstated, both variables increased toward pre-hypoxic values (Fig. 1B); however, unlike respiratory frequency, which returned to baseline conditions (P = 0.10 at 15 min post-hypoxia), inspiratory amplitude remained suppressed for the remaining 60 min (26 ± 1.5% below baseline at 15 min post-hypoxia; P < 0.001; Figs 5 and 6B).
Continuous hypoxia does not induce long-term facilitation
Unlike episodic hypoxia, which evoked a long-lasting augmentation of respiratory frequency, continuous hypoxia had no such effect (n = 8, two-way ANOVA, P > 0.05; Figs 5 and 6A, and Table 1). However, inspiratory amplitude remained significantly below baseline levels post-hypoxia (38 ± 1.7% at 60 min; P < 0.001; Fig. 6B) as did neural minute ventilation (38 ± 9.2% at 60 min; P < 0.001; Fig. 6C).
Time control
Stability of respiratory motor output
Both respiratory frequency and inspiratory amplitude remained constant over the 150 min period (n = 9, two-way RM ANOVA, P > 0.05; Figs 5 and 8C and Table 1), thereby illustrating the stability of the in situ preparation.
Figure 8. Both inspiratory activity and the respiratory response to 10% CO2 remain stable across the 150 min recording period.

A, a typical example showing that 10% CO2 increases inspiratory phrenic nerve activity (integrated and in arbitrary units, a.u.) above baseline levels (5% CO2), and that the response is of a similar magnitude at the beginning (0 min) and end (150 min) of experiments. B, mean group data (n = 5) demonstrating that the respiratory response to 10% CO2 is the same at 0 min and 150 min. Although a 5 min exposure to 10% CO2 induced a significant increase in inspiratory amplitude (Iamp; with no change in respiratory frequency (Rf), data not shown), it did not change across the experimental recording period (i.e. 0 min versus 150 min). C, mean group data (n = 9) showing that neither Rf nor Iamp changed across the experimental recording period. *Statistically significant (P < 0.05) change from baseline values.
Stability of the CO2 response
While exposure to 10% CO2 significantly increased inspiratory amplitude above baseline values (n = 5; paired t test, P = 0.013; Fig. 8A and B), it had no statistically significant effect on respiratory frequency (P = 0.06; group data not shown). Exposure to 10% CO2 at the beginning of experiments (i.e. 0 min) increased inspiratory amplitude by 120 ± 3.3% (P = 0.018) above baseline levels and increased it by 116 ± 3.0% (P = 0.03) above baseline values at the end of experiments (i.e. 150 min). The magnitude of the responses at 0 min and 150 min were not significantly different from one another (paired t test, P = 0.259; Fig. 8B).
Discussion
This study is the first to dissociate the relative roles played by hypoxia and respiratory activation in hypoxia-induced LTF. Here, we demonstrate that in postnatal rats, episodic hypoxia evokes respiratory LTF despite the fact that it potently inhibits inspiratory activity during individual hypoxic exposures. Specifically, we show that episodic hypoxia induces a persistent increase in respiratory drive, which is manifested by increased respiratory frequency, with no change in inspiratory amplitude. LTF could only be induced by episodic hypoxia since a continuous hypoxic exposure had no lasting effect on respiratory frequency. We also demonstrate that the magnitude of LTF is determined by the intensity of the hypoxic stimulus. LTF magnitude as well as its time course depend on the hypoxic severity, with more potent hypoxia inducing a more rapid and pronounced increase in respiratory frequency. These data therefore demonstrate that LTF can be induced despite potent respiratory suppression during hypoxia, and we therefore suggest that respiratory activation is not required for LTF. Accordingly, we conclude that hypoxia per se is the physiological stimulus for eliciting LTF in the in situ model system.
LTF expression
Many studies, most in anaesthetized, vagotomized and mechanically ventilated adult rats, report hypoxia-induced LTF as an increase in either respiratory frequency or inspiratory amplitude or increases in both (Baker & Mitchell, 2000; Olson et al. 2001; McKay et al. 2004). Some studies report amplitude increases in either phrenic or hypoglossal (or genioglossus muscle EMG) (McKay et al. 2004; Harris et al. 2006) nerves or in both (Fuller, 2005). However, we found that LTF was manifested as an increase in respiratory frequency with no change in inspiratory amplitude. The lack of amplitude LTF is not the result of an inability to elevate amplitude since we demonstrate that exposure to 10% CO2 at the end of experiments induced a robust increase in inspiratory amplitude – the time when LTF was maximal. Further, the lack of amplitude LTF is not due to time-dependent reductions in tissue  because alveolar lung ventilation was absent in this model system and therefore
because alveolar lung ventilation was absent in this model system and therefore  was clamped at a constant level via the perfusion medium (i.e. 5% CO2).
was clamped at a constant level via the perfusion medium (i.e. 5% CO2).
However, the lack of amplitude LTF may be related to the absence of anaesthesia since LTF in unanaesthetized and behaving mammals (e.g. rats, mice, goats and dogs) is manifested as a persistent increase in respiratory frequency (frequency LTF) with minimal or no change in either tidal volume or inspiratory amplitude (Cao et al. 1992; Turner & Mitchell, 1997; Olson et al. 2001; Kline et al. 2002; McGuire et al. 2002). Anaesthesia not only alters the manner in which the CNS responds to hypoxia, but it also has marked affects on serotonin neurotransmission (Whittington & Virag, 2006). This issue is relevant to LTF because serotonin receptor activation is required for the expression of hypoxia-induced LTF (Fuller et al. 2001).
Developmental considerations
Our findings echo those of two previous studies in immature rats. McGuire & Ling (2005) show that in 2-month-old awake rats, hypoxia-induced LTF is manifested primarily as an increase in respiratory frequency, with minor changes in inspiratory amplitude. Similarly, McKay et al. (2004) found that episodic hypoxia induces LTF in anaesthetized neonatal rats. However, unlike our findings, they report that LTF is attributable to increased inspiratory genioglossus activity (amplitude LTF), with no appreciable change in respiratory frequency. This difference could be related to developmental age or arousal state since our observations were made in unanaesthetized, decerebrate 15- to 25-day-old rats, whereas theirs were in anaesthetized 2- to 3-day-olds. A similarity between studies is that neither observed changes in either tidal volume or phrenic inspiratory amplitude (surrogate of tidal volume) following episodic hypoxia.
Another consideration is the influence that postnatal development may have on hypoxia-induced LTF. Respiratory regulation, and particularly the hypoxic ventilatory response, is powerfully affected by developmental age. For example, in adult rats, hypoxia augments breathing, whereas in postnatal rats (< 30 days old) it has a biphasic effect, initially increasing breathing (for 1–2 min) and then decreasing it. Indeed, this type of respiratory response was the primary impetus for studying postnatal rats because it provided a natural model for determining whether LTF could be induced by a stimulus that normally depresses respiratory motor output.
In this study we found that discrete hypoxic episodes inhibited respiratory motor output by suppressing both respiratory frequency and inspiratory amplitude. This observation confirms previous work from both intact (e.g. anaesthetized) and reduced (e.g. en bloc and in situ models) rat preparations, which demonstrate that hypoxia acts to depress respiratory activity during the postnatal period (0- to 30-day-old rats) (Eden & Hanson, 1987; Fung et al. 1996; St-John & Paton, 2000; Blitz & Ramirez, 2002; Liu et al. 2006). Although inspiratory activity was powerfully suppressed during discrete hypoxic episodes, recurrent exposures nonetheless lead to a potent and persistent augmentation of respiratory motor output that resembles LTF in adult rodents. The episodic nature of the hypoxic stimulus is pivotal in eliciting this neuroplastic response because a single continuous hypoxic exposure did not augment respiratory activity. We therefore conclude that although the neurocircuitry which mediates the hypoxic ventilatory response is affected by developmental status, respiratory LTF is not. Accordingly, we suggest that LTF is an ontogenetically preserved behaviour that could protect against repetitive hypoxia such as in sudden infant death syndrome.
Mechanisms of LTF
A variety of different episodic stimuli, including intermittent hypoxia, carotid sinus nerve stimulation or serotonergic/noradrenergic receptor activation (Bach & Mitchell, 1996; Ling et al. 1997; Neverova et al. 2007), triggers respiratory LTF either in vivo or in vitro. Since all such stimuli cause episodic respiratory enhancement, it raises the possibility that LTF may be the result of non-specific activity-dependent inspiratory activation. In adult rats, it is impossible to uncouple the relative roles played by inspiratory activation and episodic hypoxia in eliciting LTF because hypoxia potently augments inspiratory activity. The present study overcame this limitation by studying postnatal rats, which exhibit hypoxia-induced ventilatory depression. We demonstrate that episodic hypoxia evokes respiratory LTF despite the fact that it also inhibits inspiratory activity during individual hypoxic exposures.
Other studies support our findings that respiratory activation alone is not sufficient to produce LTF. For example, Baker et al. (2001) demonstrate that in adult rats in vivo LTF is induced by repetitive hypoxic, but not by repetitive hypercapnic exposures suggesting that hypoxia triggers a defined brain region (e.g. medullary raphe) to initiate LTF (Baker et al. 2001). Recent evidence from the in vitro isolated respiratory network demonstrates that LTF of hypoglossal motor output cannot be elicited by repetitive non-specific activation of the respiratory network (via elevated K+ concentrations); rather it requires direct activation of either noradrenergic (e.g. α1) or serotonergic (e.g. 5-HT2) receptors on hypoglossal motoneurons (McGuire et al. 2004; Neverova et al. 2007).
Although we argue that the observed LTF is elicited by episodic hypoxia and not hypoxic-induced respiratory modulation, it is conceivable that intermittent respiratory suppression could initiate LTF because Zhang et al. (2003) showed that in ventilated anaesthetized adult rats in vivo, episodic vagus nerve stimulation, which simultaneously caused inspiratory inhibition (but without hypoxia), also led to LTF (Zhang et al. 2003). We do not believe that episodic inspiratory suppression causes hypoxia-induced LTF because we found that the magnitude of LTF was associated with hypoxic severity not with the degree of ventilatory suppression during hypoxia (see Fig. 4).
LTF and hypoxic severity
We show for the first time that the magnitude and the time course of LTF are dependent on the severity of episodic hypoxia. In particular, we demonstrate that more severe hypoxic levels induce greater LTF, with more intense hypoxia inducing a faster rate of facilitation as well as a larger augmentation of respiratory frequency. By contrast, exposure to continuous hypoxia had no facilitatory effect on respiratory motor output. It therefore appears that the respiratory control system is sensitive to and adjusts breathing in response to varying levels of episodic, but not continuous, hypoxia.
While it is unknown how different hypoxic intensities evoke varying degrees of LTF, we hypothesize that more intense hypoxia leads to greater and more rapid activation of raphe and/or locus coeruleus neurons and hence serotonin/noradrenaline release onto the respiratory network. Hypoxia-induced LTF is probably mediated by a central mechanism in the in situ model system because it was recently demonstrated that episodic hypoxic activation of carotid bodies does not elicit LTF, suggesting that hypoxia evokes LTF by direct activation of serotonergic and/or noradrenergic neurons (Day & Wilson, 2005).
The dose dependence of LTF is in agreement with observations that the magnitude of phrenic nerve LTF is positively correlated with levels of BDNF (a protein necessary for amplitude LTF) in the phrenic motor nucleus (Baker-Herman et al. 2004). Together, these data indicate that the magnitude of LTF can be manipulated by either hypoxic intensity or BDNF levels. However, our data do not support the hypothesis that the magnitude of LTF is correlated with the magnitude of the hypoxic ventilatory response (Fuller et al. 2000) because we observed that severe, moderate and mild hypoxia induced the same degree of ventilatory suppression during hypoxic episodes despite evoking different levels and rates of LTF.
Summary
The current results are important because they dissect the individual roles played by hypoxia and respiratory activation in hypoxia-induced LTF. They show that in the in situ model system, episodic hypoxia evokes respiratory LTF despite the fact that it inhibits inspiratory activity during individual hypoxic exposures. In particular, they demonstrate that episodic hypoxia induces a persistent augmentation of respiratory frequency. Importantly, they also show that the magnitude and time course of LTF depend on hypoxic severity, with more intense hypoxia inducing stronger LTF.
These observations are important because they demonstrate that although hypoxia's immediate effects are depressive, this same stimulus, when presented episodically, can paradoxically elicit long-term enhancement of respiratory motor output. This finding indicates that fundamentally different mechanisms mediate the hypoxic ventilatory response and hypoxia-induced LTF during early postnatal development. We suggest that LTF may serve a vital role in preserving respiratory drive during periods of recurrent hypoxia such as in obstructive sleep apnoea or in sudden infant death syndrome.
Acknowledgments
This research was supported by grants from the National Science and Engineering Research Council of Canada (NSERC), Canadian Institutes of Health Research (CIHR), Canadian Foundation for Innovation (CFI), Ontario Thoracic Society (OTS) and the Parker B. Francis Foundation as well as the University of Toronto.
References
- Babcock M, Shkoukani M, Aboubakr SE, Badr MS. Determinants of long-term facilitation in humans during NREM sleep. J Appl Physiol. 2003;94:53–59. doi: 10.1152/japplphysiol.00476.2002. [DOI] [PubMed] [Google Scholar]
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol. 1996;104:251–260. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- Baker TL, Fuller DD, Zabka AG, Mitchell GS. Respiratory plasticity: differential actions of continuous and episodic hypoxia and hypercapnia. Respir Physiol. 2001;129:25–35. doi: 10.1016/s0034-5687(01)00280-8. [DOI] [PubMed] [Google Scholar]
- Baker TL, Mitchell GS. Episodic but not continuous hypoxia elicits long-term facilitation of phrenic motor output in rats. J Physiol. 2000;529:215–219. doi: 10.1111/j.1469-7793.2000.00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J Neurosci. 2002;22:6239–6246. doi: 10.1523/JNEUROSCI.22-14-06239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavis RW, Mitchell GS. Intermittent hypoxia induces phrenic long-term facilitation in carotid-denervated rats. J Appl Physiol. 2003;94:399–409. doi: 10.1152/japplphysiol.00374.2002. [DOI] [PubMed] [Google Scholar]
- Beecroft J, Duffin J, Pierratos A, Chan CT, McFarlane P, Hanly PJ. Enhanced chemo-responsiveness in patients with sleep apnoea and end-stage renal disease. Eur Respir J. 2006;28:151–158. doi: 10.1183/09031936.06.00075405. [DOI] [PubMed] [Google Scholar]
- Blitz DM, Ramirez JM. Long-term modulation of respiratory network activity following anoxia in vitro. J Neurophysiol. 2002;87:2964–2971. doi: 10.1152/jn.2002.87.6.2964. [DOI] [PubMed] [Google Scholar]
- Bocchiaro CM, Feldman JL. Synaptic activity-independent persistent plasticity in endogenously active mammalian motoneurons. Proc Natl Acad Sci U S A. 2004;101:4292–4295. doi: 10.1073/pnas.0305712101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao KY, Zwillich CW, Berthon-Jones M, Sullivan CE. Increased normoxic ventilation induced by repetitive hypoxia in conscious dogs. J Appl Physiol. 1992;73:2083–2088. doi: 10.1152/jappl.1992.73.5.2083. [DOI] [PubMed] [Google Scholar]
- Day TA, Wilson RJ. Specific carotid body chemostimulation is sufficient to elicit phrenic poststimulus frequency decline in a novel in situ dual-perfused rat preparation. Am J Physiol Regul Integr Comp Physiol. 2005;289:R532–R544. doi: 10.1152/ajpregu.00812.2004. [DOI] [PubMed] [Google Scholar]
-
Day TA, Wilson RJA. Brainstem
 modulates phrenic responses to specific carotid body hypoxia in an in situ dual perfused rat preparation. J Physiol. 2006;578:843–857. doi: 10.1113/jphysiol.2006.119594. [DOI] [PMC free article] [PubMed] [Google Scholar]
modulates phrenic responses to specific carotid body hypoxia in an in situ dual perfused rat preparation. J Physiol. 2006;578:843–857. doi: 10.1113/jphysiol.2006.119594. [DOI] [PMC free article] [PubMed] [Google Scholar] - Eden GJ, Hanson MA. Maturation of the respiratory response to acute hypoxia in the newborn rat. J Physiol. 1987;392:1–9. doi: 10.1113/jphysiol.1987.sp016765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci. 2003;26:239–266. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Neverova NV, Saywell SA. Modulation of hypoglossal motoneuron excitability by intracellular signal transduction cascades. Respir Physiol Neurobiol. 2005;147:131–143. doi: 10.1016/j.resp.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Fuller DD. Episodic hypoxia induces long-term facilitation of neural drive to tongue protrudor and retractor muscles. J Appl Physiol. 2005;98:1761–1767. doi: 10.1152/japplphysiol.01142.2004. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Bach KB, Baker TL, Kinkead R, Mitchell GS. Long term facilitation of phrenic motor output. Respir Physiol. 2000;121:135–146. doi: 10.1016/s0034-5687(00)00124-9. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Johnson SM, Mitchell GS. Respiratory long-term facilitation is associated with enhanced spinally evoked phrenic potentials. Abstr Soc Neurosci. 2002 363.1. [Google Scholar]
- Fuller DD, Zabka AG, Baker TL, Mitchell GS. Phrenic long-term facilitation requires 5-HT receptor activation during but not following episodic hypoxia. J Appl Physiol. 2001;90:2001–2006. doi: 10.1152/jappl.2001.90.5.2001. discussion 2000. [DOI] [PubMed] [Google Scholar]
- Fung ML, Wang W, Darnall RA, St John WM. Characterization of ventilatory responses to hypoxia in neonatal rats. Respir Physiol. 1996;103:57–66. doi: 10.1016/0034-5687(95)00077-1. [DOI] [PubMed] [Google Scholar]
- Harris DP, Balasubramaniam A, Badr MS, Mateika JH. Long-term facilitation of ventilation and genioglossus muscle activity is evident in the presence of elevated levels of carbon dioxide in awake humans. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1111–R1119. doi: 10.1152/ajpregu.00896.2005. [DOI] [PubMed] [Google Scholar]
- Hilaire G, Duron B. Maturation of the mammalian respiratory system. Physiol Rev. 1999;79:325–360. doi: 10.1152/physrev.1999.79.2.325. [DOI] [PubMed] [Google Scholar]
- Janssen PL, Fregosi RF. No evidence for long-term facilitation after episodic hypoxia in spontaneously breathing, anesthetized rats. J Appl Physiol. 2000;89:1345–1351. doi: 10.1152/jappl.2000.89.4.1345. [DOI] [PubMed] [Google Scholar]
- Jordan AS, Catcheside PG, O'Donoghue FJ, McEvoy RD. Long-term facilitation of ventilation is not present during wakefulness in healthy men or women. J Appl Physiol. 2002;93:2129–2136. doi: 10.1152/japplphysiol.00135.2002. [DOI] [PubMed] [Google Scholar]
- Kasparov S, Paton JF. Changes in baroreceptor vagal reflex performance in the developing rat. Pflugers Arch. 1997;434:438–444. doi: 10.1007/s004240050418. [DOI] [PubMed] [Google Scholar]
- Kline DD, Overholt JL, Prabhakar NR. Mutant mice deficient in NOS-1 exhibit attenuated long-term facilitation and short-term potentiation in breathing. J Physiol. 2002;539:309–315. doi: 10.1113/jphysiol.2001.014571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Olson EB, Jr, Vidruk EH, Mitchell GS. Integrated phrenic responses to carotid afferent stimulation in adult rats following perinatal hyperoxia. J Physiol. 1997;500:787–796. doi: 10.1113/jphysiol.1997.sp022058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Lowry TF, Wong-Riley MT. Postnatal changes in ventilation during normoxia and acute hypoxia in the rat: implication for a sensitive period. J Physiol. 2006;577:957–970. doi: 10.1113/jphysiol.2006.121970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire M, Ling L. Ventilatory long-term facilitation is greater in 1- vs. 2-mo-old awake rats. J Appl Physiol. 2005;98:1195–1201. doi: 10.1152/japplphysiol.00996.2004. [DOI] [PubMed] [Google Scholar]
- McGuire M, Zhang Y, White DP, Ling L. Effect of hypoxic episode number and severity on ventilatory long-term facilitation in awake rats. J Appl Physiol. 2002;93:2155–2161. doi: 10.1152/japplphysiol.00405.2002. [DOI] [PubMed] [Google Scholar]
- McGuire M, Zhang Y, White DP, Ling L. Chronic intermittent hypoxia enhances ventilatory long-term facilitation in awake rats. J Appl Physiol. 2003;95:1499–1508. doi: 10.1152/japplphysiol.00044.2003. [DOI] [PubMed] [Google Scholar]
- McGuire M, Zhang Y, White DP, Ling L. Serotonin receptor subtypes required for ventilatory long-term facilitation and its enhancement after chronic intermittent hypoxia in awake rats. Am J Physiol Regul Integr Comp Physiol. 2004;286:R334–R341. doi: 10.1152/ajpregu.00463.2003. [DOI] [PubMed] [Google Scholar]
- McKay LC, Janczewski WA, Feldman JL. Episodic hypoxia evokes long-term facilitation of genioglossus muscle activity in neonatal rats. J Physiol. 2004;557:13–18. doi: 10.1113/jphysiol.2004.064006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahamed S, Hanly PJ, Gabor J, Beecroft J, Duffin J. Overnight changes of chemoreflex control in obstructive sleep apnoea patients. Respir Physiol Neurobiol. 2005;146:279–290. doi: 10.1016/j.resp.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Millhorn DE, Eldridge FL, Waldrop TG. Prolonged stimulation of respiration by a new central neural mechanism. Respir Physiol. 1980;41:87–103. doi: 10.1016/0034-5687(80)90025-0. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB., Jr Invited review: Intermittent hypoxia and respiratory plasticity. J Appl Physiol. 2001a;90:2466–2475. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Powell FL, Hopkins SR, Milsom WK. Time domains of the hypoxic ventilatory response in awake ducks: episodic and continuous hypoxia. Respir Physiol. 2001b;124:117–128. doi: 10.1016/s0034-5687(00)00197-3. [DOI] [PubMed] [Google Scholar]
- Mortola JP. How newborn mammals cope with hypoxia. Respir Physiol. 1999;116:95–103. doi: 10.1016/s0034-5687(99)00038-9. [DOI] [PubMed] [Google Scholar]
- Neverova NV, Saywell SA, Nashold LJ, Mitchell GS, Feldman JL. Episodic stimulation of α1-adrenoreceptors induces protein kinase C-dependent persistent changes in motoneuronal excitability. J Neurosci. 2007;27:4435–4442. doi: 10.1523/JNEUROSCI.2803-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson EB, Jr, Bohne CJ, Dwinell MR, Podolsky A, Vidruk EH, Fuller DD, Powell FL, Mitchel GS. Ventilatory long-term facilitation in unanesthetized rats. J Appl Physiol. 2001;91:709–716. doi: 10.1152/jappl.2001.91.2.709. [DOI] [PubMed] [Google Scholar]
- Paton JF. A working heart-brainstem preparation of the mouse. J Neurosci Meth. 1996;65:63–68. doi: 10.1016/0165-0270(95)00147-6. [DOI] [PubMed] [Google Scholar]
- Paton JF, Abdala AP, Koizumi H, Smith JC, St-John WM. Respiratory rhythm generation during gasping depends on persistent sodium current. Nat Neurosci. 2006;9:311–313. doi: 10.1038/nn1650. [DOI] [PubMed] [Google Scholar]
- Potts JT, Rybak IA, Paton JF. Respiratory rhythm entrainment by somatic afferent stimulation. J Neurosci. 2005;25:1965–1978. doi: 10.1523/JNEUROSCI.3881-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell FL, Milsom WK, Mitchell GS. Time domains of the hypoxic ventilatory response. Respir Physiol. 1998;112:123–134. doi: 10.1016/s0034-5687(98)00026-7. [DOI] [PubMed] [Google Scholar]
- St-John WM, Paton JF. Characterizations of eupnea, apneusis and gasping in a perfused rat preparation. Respir Physiol. 2000;123:201–213. doi: 10.1016/s0034-5687(00)00177-8. [DOI] [PubMed] [Google Scholar]
- Turner DL, Mitchell GS. Long-term facilitation of ventilation following repeated hypoxic episodes in awake goats. J Physiol. 1997;499:543–550. doi: 10.1113/jphysiol.1997.sp021947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington RA, Virag L. Isoflurane decreases extracellular serotonin in the mouse hippocampus. Anesth Analg. 2006;103:92–98. doi: 10.1213/01.ane.0000221488.48352.61. [DOI] [PubMed] [Google Scholar]
-
Wilson RJ, Remmers JE, Paton JF. Brain stem
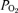 and pH of the working heart-brain stem preparation during vascular perfusion with aqueous medium. Am J Physiol Regul Integr Comp Physiol. 2001;281:R528–R538. doi: 10.1152/ajpregu.2001.281.2.R528. [DOI] [PubMed] [Google Scholar]
and pH of the working heart-brain stem preparation during vascular perfusion with aqueous medium. Am J Physiol Regul Integr Comp Physiol. 2001;281:R528–R538. doi: 10.1152/ajpregu.2001.281.2.R528. [DOI] [PubMed] [Google Scholar] - Zhang Y, McGuire M, White DP, Ling L. Episodic phrenic-inhibitory vagus nerve stimulation paradoxically induces phrenic long-term facilitation in rats. J Physiol. 2003;551:981–991. doi: 10.1113/jphysiol.2003.048157. [DOI] [PMC free article] [PubMed] [Google Scholar]