

Abstract
Hydrophilic drugs are often poorly absorbed when administered orally. There has been considerable interest in the possibility of using absorption enhancers to promote absorption of polar molecules across membrane surfaces. The bile acids are one of the most widely investigated classes of absorption enhancers, but there is disagreement about what features of bile acid enhancers are responsible for their efficacy. We have designed a class of glycosylated bile acid derivatives to evaluate how increasing the hydrophilicity of the steroid nucleus affects the ability to transport polar molecules across membranes. Some of the glycosylated molecules are significantly more effective than taurocholate in promoting the intestinal absorption of a range of drugs, showing that hydrophobicity is not a critical parameter in transport efficacy, as previously suggested. Furthermore, the most effective glycosylated compound is also far less damaging to membranes than the best bile acid absorption promoters, presumably because it is more hydrophilic. The results reported here show that it is possible to decouple absorption-promoting activity from membrane damage, a finding that should spark interest in the design of new compounds to facilitate the delivery of polar drugs.
Keywords: drug delivery, absorption, calcitonin, gentamicin, bile acids
Biological membranes play critical roles in the homeostasis of all organisms. These natural barriers segregate important activities between and within cells and tissues. Small hydrophobic molecules can partition across biological membranes down a concentration gradient. Hydrophilic molecules generally require some sort of selective transport system to cross the lipid bilayer. Although the integrity of biological membranes is vital to the health of the organism, there are many instances in which it would be desirable to breach membrane barriers to allow the passage of otherwise impermeant molecules. Indeed, the ability to facilitate the absorption of hydrophilic compounds would revive interest in an enormous number of drug candidates that have failed because of delivery problems. In addition, effective strategies for breaching membrane barriers would permit the development of nontraditional drugs based on peptides, proteins, and nucleic acids (1). In this paper we report the characterization of a promising class of compounds designed to facilitate the transport of polar molecules across biological membranes.
These transport agents are based on a class of natural compounds, the bile acids, that have been shown to facilitate the passage of polar molecules across biological membranes (2–5). Carey and coworkers (2) have proposed that the bile acids partition into lipid bilayers as small aggregates in which the polar surfaces of the molecules are juxtaposed. They have suggested that these aggregates could alter membrane permeability by providing an ordered hydrophilic channel through which polar molecules pass or by disrupting bilayer packing so that there are water-filled defects in the membranes. Carey’s model emphasizes the importance of interactions between the polar surfaces of bile acid molecules. Some experiments, however, have shown that hydrophobic bile salts such as deoxycholate and chenodeoxycholate promote absorption more effectively than cholate, which has more polar surface area (6). It has, therefore, been suggested that transport activity is directly correlated with the hydrophobicity of the steroid nucleus (2). It is difficult to reconcile Carey’s conceptual model for how the bile acids might promote absorption with the apparent preference for a hydrophobic steroid nucleus.
Because it is often possible to learn details of structure and activity relationships through the design of unnatural analogs, we undertook the synthesis and evaluation of a new family of cholic acid-based transport agents. We increased the polarity of the hydrophilic surface of various cholic acid derivatives by attaching sugars to the C7 and C12 hydroxyls, and we evaluated the ability of the resulting compounds to promote absorption of polar drugs (7–9). Some of the glycosylated bile acid analogs were found to facilitate the transport of polar drugs across biological membranes more effectively than natural bile acid transport agents. This result shows that a hydrophobic steroid nucleus is not critical for efficacy in bile acid-based transport agents. In fact, through this work we have identified an effective but exceptionally hydrophilic absorption promoter that is much less disruptive to membranes than are standard bile acid transport agents.
MATERIALS AND METHODS
Materials.
Crystalline bovine insulin, gentamicin sulfate, sodium taurocholate, vancomycin hydrochloride, boric acid, o-phthalaldehyde, silicic acid, and sodium taurodeoxycholate were purchased from Sigma. Salmon calcitonin was purchased from Bachem. Sodium deoxycholate was purchased from Fluka. Orange OT (1-o-tolylazo-2-naphthol, 75%) was purchased from Aldrich and purified twice by precipitation from acetone with water, followed by recrystallization three times from ethanol. All other chemicals were also purchased from Aldrich. Defibrinated rabbit blood was purchased fresh from Remel Biological Products (Lenexa, KS).
Glycosylated steroids 5-7 (see Fig. 1) were synthesized as described previously (7, 8). For 9 (10), cholic acid was converted to methyl cholate by refluxing with BF3⋅Et2O in methanol and the C3 hydroxyl group was tosylated in 73% yield with tosyl chloride in pyridine in the presence of dimethylaminopyridine for 1 hr at 80°C. Treatment with sodium azide in dimethylformamide for 2 hr at 90°C afforded the C3 azide derivative. Glycosylation of the C3 azido cholate was performed by activation of perbenzylated glucose sulfoxide with triflic anhydride in the presence of 2,6-di-tert-butyl-4-methylpyridine in toluene at −78°C for 1 hr, and gradual warming to 25°C over 25 min to afford the bisglycosylated cholate derivative in 17% yield. Treatment with triphenyl phosphine in refluxing aqueous tetrahydrofuran (THF)/H2O (3:1, vol/vol) for 11 hr reduced the C3 azide to the C3 amine in 57% yield. The sugars were debenzylated with 20% palladium hydroxide on carbon in 10:1 C6H6/MeOH for 48 hr. Pure 9 was obtained by HPLC purification on a C18 Vydac column (250 × 10 mm, C18-coated 5 μm silica) with solvents 0.1% trifluoroacetic acid (TFA)/H2O (A) and 0.1% TFA/acetonitrile (B) with an isocratic gradient of 1% B/99% A for 8 min and then ramping to 99% B/1% A over 10 min (11). The compound was eluted at flow rate of 4.5 ml/min and detected by absorbance at 210 nm. Pure 9 (Mr 746) was eluted at 3.8–3.9 min and was lyophilized from water to yield a white powder. All compounds were fully characterized by 1H NMR, 13C NMR, TLC, and MS.
Figure 1.

Bile acids and glycosylated steroid amphiphiles.
Insulin Assay.
A series of samples was prepared by dissolving 0.5–5 mg of each appropriate transport agent in 0.5 ml of water containing 25 units of insulin (IU) yielding 0.1–1% (wt/vol) solutions of transport agent [at or near the critical micelle concentration (CMC)]. Solutions were adjusted to pH 7.8 and the samples were lyophilized for convenient storage. Immediately before use, the samples were redissolved in 0.5 ml of phosphate-buffered isotonic saline (pH 7.8), and appropriate volumes to deliver 8 IU/kg (≈0.06 ml ± 0.01 ml) were instilled into the nostrils of fasted female Sprague–Dawley rats (420–480 g) that had been partially anesthetized with sodium pentobarbital. The nostrils were sealed and blood glucose levels were determined from blood droplets expressed from the rats’ tails at 0, 15, 30, 60, 120, and 240 min after instillation, using a One Touch II glucose meter (Lifescan, Milpitas, CA). Two or three rats were tested for each concentration of each compound. Compounds that caused a greater drop in blood glucose levels than sodium deoxycholate were selected for further study.
Rat Intestinal Model.
Sprague–Dawley rats weighing 250–300 g were fasted overnight (18 hr), with free access to water. Anesthesia was induced by an intramuscular injection of ketamine and xylazine (60 mg/kg ketamine and 8 mg/kg xylazine) or sodium pentobarbital (50 mg/kg). Cannulas were implanted in the left or right jugular veins for blood sampling. A midline abdominal incision of 3–4 cm was made. The ileum was identified by locating the caecum and the dosing solution was injected 2–4 cm above the ileal–caecal junction by using a syringe with a 30 gauge needle. The rat ileal formulation was prepared by dissolving 5 mg of either gentamicin sulfate or vancomycin hydrochloride in 0.5 ml of distilled water. Alternatively, 0.2 mg of salmon calcitonin was dissolved in 0.5 ml of saline. Compound 9 or 6 was added as a powder to give a final concentration of 100 mM (37.3 mg per rat). The drug–steroid mixture was swirled briefly in a Vortex mixer and incubated at room temperature for 15 min prior to the administration. For the determination of i.v. plasma profiles, 1 mg of gentamicin or vancomycin or 4 μg of calcitonin was dissolved in 0.3 ml of distilled water and administered through the jugular vein.
Blood samples of 0.6 ml were withdrawn at 0, 1, 5, 10, 15, 30, and 45 min and 1, 1.5, 2, and 4 hr after ileal administrations into tubes containing EDTA. For comparison, blood samples were also obtained at 30 s, and 1.5, 3, 8, and 20 min after i.v. admistration. The blood was centrifuged (4,000 rpm, 5 min, in an Eppendorf microcentrifuge) and plasma samples were stored at −20°C. Glucose levels were measured with a Glucostix strip in an Ames Glucometer M blood glucose meter.
Gentamicin Assay.
Gentamicin concentration in plasma was determined by solid-phase extraction and derivatization with o-phthalaldehyde followed by HPLC according to the method of Maitra et al. (12) with the following exceptions. One-milliliter solid-phase extraction columns (Alltech Associates) and the o-phthalaldehyde derivatization reagent were prepared as described. Frozen plasma samples were thawed at room temperature and 0.25-ml aliquots were diluted with 1 ml of distilled water, mixed 30 s, and applied to the columns. Columns were placed on a vacuum manifold (Varian) under 5 mmHg (1 mmHg = 133 Pa) vacuum before derivatization, incubated with the o-phthalaldehyde reagent, and then, following the derivatization, dried under vacuum for 3 min and eluted with 100% methanol. Chromatographic separation was achieved with a Waters 600 E system controller and ultra WISP 715 sample processor on a Waters μBondapak C18 column (3.9 mm i.d. × 300 mm, 10 μm particle size). Samples (40 μl) were eluted with an aqueous mixture of 79% methanol and 1% triethylamine, adjusted to pH 6.2 with phosphoric acid. Gentamicin peaks in the sample were detected by a Waters 740 scanning fluorescence detector at excitation and emission wavelength of 360 and 418 nm, respectively.
Vancomycin Assay.
The vancomycin plasma assay was carried out exactly as described by Hosotsubo (13) except that 40 μl of the soluble fraction of the acetonitrile–plasma preparation was injected into a 15 × 0.46 cm Hypersil APS, 5 μm, column (Sigma-Aldrich). Sample components were separated as described (13).
Calcitonin Assay.
Plasma calcitonin levels were determined by a radioimmunoassay kit (DSL 1300 from DSL, Webster, TX), utilizing 125I-labeled salmon calcitonin and guinea pig anti-calcitonin serum. The radioactive material precipitated by PEG 6000 was quantitated and the concentration of calcitonin was calculated from a standard curve.
Data Analysis.
Ileal absorption is expressed as a percentage of the AUC observed after i.v. administration and is calculated as follows:
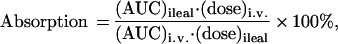 |
where AUC is the area under the curve from 0 and 240 min after dose administrations calculated from the trapezoidal rule. The relative absorption is the ratio of ileal drug absorption in individual animals in the presence of compound 9 to the mean ileal drug absorption in its absence, for the same drug dose.
Erythrocyte Lysis.
Erythrocyte lysis was determined by the method of Hirai et al. (4) with the following exceptions. One-milliliter samples of rabbit blood were washed three times with 1 ml of erythrocyte buffer (0.15 M NaCl/0.06 M K2HPO4, pH 7) and then diluted with 5–7 ml of erythrocyte buffer. Amphiphile solutions of various concentrations in erythrocyte buffer were mixed with 63 μl of dilute red blood cell suspension to a final volume of 500 μl. After incubation and centrifugation (as described), 400 μl of each supernatant was diluted with 700 μl of erythrocyte buffer. The absorbance of each solution (hemoglobin) was measured at 540 nm using a Cary Varian 1E spectrophotometer to determine the percentage of lysed cells. Trials were repeated for reproducibility; variance in data indicated ±4% error.
HPLC Assay.
A Hitachi L-6200A Intelligent Pump was used with a Protein and Peptide C18 Vydac column (250 × 10 mm, C18-coated 5-μm silica particles) with an isocratic aqueous mobile phase with 0.1% TFA/1% acetonitrile at a flow rate of 4.5 ml/min. Ten-milligram bile salt and 20-mg glycosylated steroid samples were dissolved in 1 ml 0.1% of TFA/water and filtered through Whatman poly(vinylidene difluoride) (PVDF) 0.45-μm filters; 50 μl of each solution was injected. The 210-nm absorbance was measured with a Waters 484 tunable absorbance detector, and peak retention times were recorded at an attenuation of 512 or 256 on a Waters 745 data module. The retention factors (k′) were calculated from solvent and peak retention times according to the method of Armstrong and Carey (14).
CMC Determination.
Dye solubilization. Glycosylated steroids and bile salts were dissolved in 1.0 ml of 0.15 M NaCl and filtered through Millex GV 0.22-μm Millipore filters into screw-cap vials. Fifty milligrams of solid organic dye orange OT was added to each solution and the vials were sealed and shaken on an Orbit shaker at 190 rpm for 48 hr. The solutions were refiltered to remove the solid dye and the UV absorbance of the solutions was measured at 492 nm with a Cary Varian 1E spectrophotometer. The first solution concentration that resulted in a nonzero absorbance was taken as the CMC. Surface tension measurements. Compounds were dissolved in 0.15 M NaCl at increasing concentrations. The solutions were delivered by means of a syringe pump at 1 drop/30 sec through a 5.0-mm brass tube. Five drops were collected per trial and the average mass per drop was calculated for each solution on the basis of two trials. The surface tension, γ, was calculated from: γ = mg/2πrf, using the correction factor, f, from the Harkins and Brown table (15). The surface tension was plotted as a function of the logarithm of the amphiphile concentration, giving the CMC at the break point of the graph.
RESULTS AND DISCUSSION
Design of the Compounds.
Although the bile acids are amphiphiles, they differ from conventional head-to-tail amphiphiles because the polar and nonpolar domains are separated along the longitudinal axis of the molecules. This gives rise to distinct polar and nonpolar faces. The facial amphiphilicity of the bile acids influences the way in which they organize in solution and presumably plays a role in their ability to promote the absorption of polar drugs across membranes. We attached glucose units to the C7 and C12 hydroxyl groups of cholic acid, 1 (Fig. 1), to evaluate how increasing the hydrophilicity of the polar surfaces would affect drug transport activity. The sugar hydroxyls create an extensive polar surface of hydrogen-bonding groups. We reasoned that if the hydrophobicity of the steroid nucleus is a major determinant of transport activity, these glycosylated bile acid derivatives should be poor absorption promoters. However, if interactions of the polar surfaces—either with themselves or with polar drugs—play an important role in the transfer of hydrophilic drugs across membranes, then perhaps these glycosylated molecules would actually work better than transport agents that have natural bile acid cores. To provide a more diverse set of compounds to evaluate, we also varied the substituents and stereochemistry at C3 and esterified the carboxylic acid on the steroid side chain.
Selection of Promising Transport Agents.
Because in vitro screens to evaluate absorption promoters do not adequately reflect their ability to promote transport across intact mucosa, we judged the potential efficacy of the different glycosylated compounds from their ability to increase the intranasal absorption of insulin in rats (4). Blood glucose levels were measured after intranasal administration of insulin (8 IU/kg) in the presence of a given transport agent (1.6 mg/kg). Decreased blood glucose levels were presumed to be due to the absorption of insulin into the circulation. Rats treated with insulin solutions ranging from 5 to 10 IU/kg in the absence of a transport agent showed no change in blood glucose level. Five glycosylated compounds, 5–9, were evaluated in the assay (Fig. 1). Sodium deoxycholate, an effective but irritating absorption promoter, was used as a standard for evaluating transport efficacy. In the presence of this compound, blood glucose levels dropped to 60% of initial levels after 30 min (results not shown). None of the carboxylate salts were effective compared with sodium deoxycholate. However, 6 and 9, which are both esterified on the side chain, caused blood glucose levels to drop to 55% and 80% of initial levels, respectively, 30 min after administration. Compounds 6 and 9 are identical except at C3 of the steroid nucleus. Compound 6 has a C3 α-hydroxyl group like the parent bile acid, while 9 has a C3 β-amino group. Both compounds were subjected to more intensive study to evaluate their efficacy for promoting absorption through the intestinal mucosa.
In Vivo Absorption of Polar Drugs.
Gentamicin, vancomycin, and calcitonin are polar drugs that have poor oral availability. Gentamicin is a broad-spectrum aminoglycoside antibiotic (Mr 550) and vancomycin is a glycopeptide antibiotic that has important utility in fighting antibiotic-resistant Gram-positive bacterial infections (Mr 1,400). Calcitonin is a peptide hormone that has been investigated for the treatment of osteoporosis (Mr 3,500). Gentamicin and vancomycin are currently administered by injection in a hospital setting. It might be desirable to have oral or nasal delivery routes for administering these compounds as well as calcitonin. This requires developing strategies to breach intestinal or nasal mucosa without seriously damaging the membranes. We evaluated the ability of compounds 6 and 9 to facilitate the ileal absorption of gentamicin in rats by using well established in vivo assays (Materials and Methods). The absorption of calcitonin and vancomycin was evaluated in this system only with compound 9, which was found to be significantly less damaging to membranes than compound 6 (see Hemolytic Potential below). Sodium taurocholate, 3, a well studied nonglycosylated bile acid enhancer, was used as a reference to evaluate the efficacy of the glycosylated bile acid derivatives for enhancing absorption.
All three drugs showed negligible absorption through the ileum when administered in either saline or water. Baseline absorptions were 4.1, 1.9, and 1.6% of the injected dose for gentamicin (5 mg), vancomycin (20 mg), and calcitonin (0.2 mg), respectively (Table 1). Both glycosylated steroids were significantly more effective than sodium taurocholate at enhancing the absorption of gentamicin (Table 1): only 30.4% of gentamicin reached the circulation when administered to the ileum in the presence of 100 mM sodium taurocholate, but at the same concentrations of either 6 or 9, 100% of the drug was absorbed. For compound 9 this corresponds to a 27-fold increase in gentamicin absorption relative to the baseline absorption level (Fig. 2). With vancomycin, only 2.3% of the 5-mg dose was absorbed in the presence of 100 mM sodium taurocholate 3, whereas 8.4% was absorbed with 100 mM 9, corresponding to a 13-fold increase over the vancomycin baseline absorption (Fig. 2). Calcitonin absorption also increased significantly, about 17-fold over baseline, when mixed with 100 mM 9. Ileal coadministration of each drug with 9 also resulted in increased peak concentrations of the drug in the circulation compared with the ileal controls. These concentrations were maintained for a significantly longer time than the peak concentrations achieved when the drug was administered i.v. as shown in Fig. 3. Therefore, the glycosylated steroid not only increased the amount of drug absorbed but also altered the pattern of absorption and clearance.
Table 1.
Absorption of gentamicin, vancomycin, and calcitonin through the rat ileum in the presence of sodium taurocholate, 3, glycosylated steroids 6 or 9, or no drug transport agent
| Drug | Absorption
|
|||
|---|---|---|---|---|
| Alone | With 3 | With 6 | With 9 | |
| Gentamicin | 4.1 ± 1.6 | 30.4 ± 6.8 | 104.7 ± 36.4 | 109.7 ± 15.2 |
| Vancomycin | 1.9 ± 0.5 | 2.3 ± 0.6 | ND | 8.4 ± 1.5 |
| Calcitonin | 1.6 ± 1.1 | ND | ND | 26.8 ± 8.7 |
Compound 3, 6, or 9, at a concentration of 100 mM, was combined with a 5-mg dose of either gentamicin or vancomycin, or 0.2 mg of calcitonin. Absorption was calculated as described in Materials and Methods. Results are mean ± SEM of three or four rats. ND, not determined.
Figure 2.

Relative absorption of 20 mg of vancomycin, 0.2 mg of calcitonin, or 5 mg of gentamicin in the presence of 100 mM compound 9. Each drug was mixed with 9 and administered to the rat ileum as described in the text. The relative absorption is the ratio of ileal drug absorption in individual animals in the presence of 9 to the mean ileal drug absorption in its absence, for the same drug dose. Relative absorption was calculated from the individual values of three or four animals and is shown as the mean ± SEM.
Figure 3.

Pharmacokinetics of vancomycin in the plasma of rats after ileal or i.v. administration. Vancomycin (20 mg) was mixed with either 100 mM compound 9 (▴) or water (•) and introduced into the ileum as described in the text. Alternatively, vancomycin (1 mg) was injected i.v. through the jugular vein (▪). The graphs represent the mean and SEM for three rats.
These in vivo results are not consistent with previous observations suggesting that the hydrophobicity of the steroid nucleus correlates with transport efficacy (2). Sodium taurocholate 3 contains a cholic acid steroid framework with three hydroxyl groups on the polar surface, whereas 6 and 9 contain a bis-glycosylated cholic acid framework with either eight or nine hydroxyls on the polar surface. Thus, 6 and 9 should present a much more hydrophilic steroid nucleus than sodium taurocholate. In addition, 9 contains a β-amino group at C3 that is charged at physiological pH. This charge would be expected to augment the hydrophilicity of the steroid nucleus of 9 considerably. The efficacy of the glycosylated compounds prompted us to investigate some of their physicochemical properties in an attempt to explain their apparently anomalous behavior.
Hydrophobic–Hydrophilic Balance.
Although it seemed obvious that the steroid nucleus of 6 and 9 would be more hydrophilic than that of sodium taurocholate 3, we wanted a quantitative comparison of the overall hydrophobic–hydrophilic balance of the molecules (2, 6). We also wanted to evaluate the effect of glycosylation on the hydrophobic–hydrophilic balance of the steroid framework. Various glycosylated bile acid derivatives were compared with nonglycosylated bile acid derivatives by using a reverse-phase HPLC assay which is commonly used to evaluate the hydrophilic–hydrophobic balance in sets of amphiphiles (14). In this assay, the retention factor of an amphiphilic molecule reflects the partitioning of the amphiphile between a hydrocarbon stationary phase and a polar mobile phase (14, 16). The more hydrophilic the molecule, the faster it is eluted from the column. Table 2 shows the relative ranking of 6, 9, the nonglycosylated diol 10, and the taurine conjugates of the bile salts. A comparison of 9 and 10 shows that glycosylation of the C7 and C12 hydroxyl groups substantially increases the hydrophilicity of the steroid molecule, as expected. We could not directly compare methyl cholate and 6 with this mobile phase (1% acetonitrile/water with 0.1% TFA) because of solubility problems with methyl cholate. However, the fact that 6 is highly water soluble whereas methyl cholate is not indicates that the sugars increase the hydrophilicity of the steroid nucleus significantly. As might be expected, esterifying the carboxylic acid substantially reduces the overall hydrophilicity of the molecule. Accordingly, bis-glycosylated compound 6 is more hydrophobic than either of the nonglycosylated taurine conjugates, which contain a negatively charged side chain. Because 6 is a more effective transport agent than the taurine conjugates, one might be tempted to conclude that even if the hydrophobicity of the steroid nucleus is not always important, the overall hydrophobicity of the molecule is critical for efficacy. However, compound 9 clearly shows that overall hydrophobicity does not correlate with transport efficacy. The equatorial C3 amine functionality of 9 increases its overall hydrophilicity so much that special HPLC conditions had to be developed to get any retention on a C18 reverse-phase column.
Table 2.
Hydrophilicity rankings and CMC determinations of transport agents
| Transport agent | Hydrophilicity*k′ | CMC, mM
|
|
|---|---|---|---|
| Orange OT† | Surface tension‡ | ||
| 3 | 0.55 | 7–8 | 8–9 |
| 4 | 1.92 | 1–2 | 1–2 |
| 6 | 3.38 | 1–2 | 1 |
| 9 | 0.43 | 11–12 | 11 |
| 10 | 1.85 | ND | ND |
ND, not determined.
Hydrophilic amphiphiles are represented by small retention factors, k′, signifying short retention times. Mobile phase: 0.1% TFA/1% CH3CN/water.
Solubilization of orange OT dye in 0.15 M NaCl, pH 7.0.
Surface tension measurements in 0.15 M NaCl.
CMCs of the Glycosylated Steroids.
The ability to self-associate is a property that may play an important role in the transport efficacy of absorption promoters (2). Carey’s model for how the bile acids function emphasizes the formation of aggregates in which the polar surfaces are self-associated (2). The ability to self-associate can be evaluated from the concentrations at which amphiphiles form micelles in water. Accordingly, the CMCs of the glycosylated steroids were measured using both dye-solubilization (10, 17) and surface-tension methods (18).
The dye-solubilization assay is a simple technique in which the CMC is determined from the minimum concentration of amphiphile that solubilizes a detectable amount of a hydrophobic dye. The dye is not solubilized in water until an aggregate with a hydrophobic interior sufficiently large to encapsulate the dye is formed. The CMCs of the absorption promoters and bile salts are shown in Table 2. The measured values for the natural bile salts are in excellent agreement with earlier reports (19), giving us some confidence in the CMCs obtained for the glycosylated molecules. However, the addition of the hydrophobic dye could affect the results by providing a hydrophobic surface to promote aggregation of the steroids or by requiring an unnaturally large amphiphile aggregate to accommodate the dye molecule. Therefore, we employed the noninvasive surface-tension technique to verify the results. With this method, the CMC is derived from the change in the surface tension of a solution as amphiphile concentrations are increased (15). The CMC values obtained from surface-tension data are in excellent agreement with the dye-solubilization data (20), and they show that the CMC of 6 is 1–2 mM whereas the CMC of 9 is 11 mM. Therefore, although the glycosylated compounds must be administered at concentrations above their CMCs to be effective, there is no direct correlation between transport efficacy and CMC as there reportedly is with other bile acid-based transport agents (2, 5).
Although CMC values do not predict the transport efficacy of the glycosylated steroids, they can provide some insight into the molecular interactions that may be involved in the absorption process. The CMC values for both 6 and 9 are surprisingly low, given the number of additional hydroxyl groups incorporated into the molecules upon glycosylation. Compound 9 has a CMC only slightly higher than sodium cholate (8 mM) even though it has six additional hydroxyls and contains a full positive charge at C3 (9, 19). In a previous study on the aggregation of various bile acid derivatives in water, we found that bis-glycosylated bile acids consistently have lower CMCs than their nonglycosylated counterparts in which every other feature of the molecules is identical (9). Thus, the glycosylated compounds self-associate more effectively in water than their nonglycosylated counterparts. Because only the hydrophilic faces of the glycosylated and nonglycosylated molecules are different, we have suggested that the polar surfaces of the glycosylated molecules interact more favorably than those of the nonglycosylated bile acids (9). This leads to the rather surprising conclusion that the formation of multiple hydrogen bonds facilitates self-association in water. Crystal structures of glycosylated steroids support this idea, revealing that the solid-state structures are ordered by the formation of multiple hydrogen bonds between sugars on opposing molecules (8). Thus, hydrogen bonding complementarity may explain the relatively low CMC values of these apparently polar molecules. As proposed by Carey, the ability of the polar surfaces to self-associate may play an important role in transport efficacy (2). However it cannot be the only important factor because 9 is at least as good a transport agent as 6 even though its CMC is severalfold higher.
Hemolytic Potential.
The requirement that the glycosylated transport agents be administered at concentrations above their CMCs raises questions about the effect of the amphiphiles on membranes (2, 5). Although it may be acceptable to cause transient and rapidly reversible increases in membrane permeability by a variety of mechanisms, it is not acceptable to solubilize membranes substantially. Many compounds that enhance absorption seem to do so by extracting membrane lipids into mixed micelles with the absorption promoters (4, 6, 21). These compounds irritate intact mucosa and raise concerns about the long-term effects of repeat administration. Therefore, we compared the membrane-damaging effects of 6 and 9 to those of various bile acid transport agents.
We used a well established assay involving the lysis of red blood cells to evaluate the membranolytic potential of 6 and 9 (4). In this experiment red blood cells are incubated with glycosylated transport agents or bile acid transport agents at various concentrations. Solutions that disrupt cell membranes substantially cause hemoglobin to be released from the cells into the surrounding buffer. Intact cells can be separated from the buffer, and the released hemoglobin can be detected by monitoring the absorbance at 540 nm. The percentage of total cellular hemoglobin released can be used as a measure of cell damage at the given amphiphile concentration. As shown in Fig. 4, 6 damages blood cells at concentrations comparable to hydrophobic bile salts such as sodium deoxycholate 2, which is an effective absorption promoter but is irritating to membranes. Compound 6 also has a CMC very similar to the CMCs of the hydrophobic bile salts. The behavior of 6 in the hemolysis assay suggests that its ability to promote absorption could be related to its ability to disrupt cell membranes significantly. Compound 9, however, behaves very differently from 6. It does not fully lyse red blood cells even at concentrations as high as 100 mM—more than twice the concentration at which sodium taurocholate 3 completely obliterates all blood cells. Others have noted an inverse correlation between membrane-damaging effects and the polarity of absorption enhancers (5, 6). It is generally the case that increasing the polarity of an enhancer decreases its damaging effects on cell membranes, but with the unfortunate consequence of dramatically reducing its efficacy. Compound 9 is far more polar than 6 or even sodium taurocholate, so the hemolysis results are not surprising; however, it is surprising that 9 promotes absorption as well or better than many compounds that have lower CMCs and that substantially damage membranes. Clearly, transport efficacy does not have to correlate with membranolytic effects.
Figure 4.

Percentage of erythrocytes lysed as a function of amphiphile concentration in 0.15 M NaCl/0.06 M K2HPO4, pH 7.0. Associated error ±4%.
CONCLUSION
More work is necessary to understand the complex relationship between the physicochemical properties of the glycosylated steroids and their efficacy as transport agents. For example, it will also be important to establish whether transport is transcellular or paracellular for both 6 and 9. Nevertheless, from the work reported above we have drawn an important conclusion that has implications for the study and design of absorption promoters. Neither the hydrophobicity of the steroid nucleus nor the overall hydrophobicity of the molecule is predictive of the transport efficacy of bile acid-based enhancers. The example of 9 shows that it is possible to find compounds that are hydrophilic and yet are highly effective in transporting polar drugs across membranes barriers. Furthermore, although the ability to self-associate may be an important property of effective absorption promoters, the concentration at which micelles form does not correlate directly with efficacy. Therefore, it is possible to design absorption promoters that do not disrupt cell membranes significantly or cause substantial cell damage.
A comparison of 6 and 9 suggests one factor that may be involved in the success of 9. Compounds 6 and 9 differ only in the substituent and stereochemistry at the C3 position. The amine at C3 clearly increases the hydrophilicity of 9 relative to 6 and is presumably responsible for the corresponding decrease in cell membrane damage. At the same time, the presence of the amine improves the transport efficacy, although it is not clear how. The positive charge sets 9 apart from the taurine conjugates and other negatively charged bile salts, and suggests that it is worth investigating whether positively charged groups are useful features to include in the design of other absorption promoters. Cationic lipids and other cationic polymers have been shown to increase the uptake of plasmid DNA and other negatively charged oligonucleotides in various cell types (22–28). The efficacy of cationic polymers in DNA delivery has been partly attributed to their attraction for negatively charged cell membranes. Ionic pairing of oppositely charged lipids with proteins and nucleic acids has also been shown to promote transport through hydrophobic solvents and biomimetic membranes (29, 30). However, the incorporation of positive charges into small-molecule delivery agents has not been investigated. The behavior and properties of compound 9 as an absorption promoter raise new possibilities for the delivery of hydrophilic drugs across membrane barriers.
Further study of hydrophilic positively charged amphiphiles may lead to a better understanding of the structural features critical for effective transport and the development of additional compounds with superior activity. Nevertheless, our findings on the behavior and properties of compound 9 may revive expectations for absorption promoters as a way to increase the bioavailability of polar drugs and to make possible the use of nonparenteral delivery routes.
Acknowledgments
We thank Rebecca Goodwin for technical support. We gratefully acknowledge Prof. Alfred Stracher and Prof. Leo Kesner at the Health Science Center, State University of New York, Brooklyn, NY, who carried out the insulin absorption studies. We thank the Office of Naval Research for partial support of this work.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: TFA, trifluoroacetic acid; IU, insulin unit; CMC, critical micelle concentration.
References
- 1.Aungst B J. J Pharm Sci. 1993;82:979–987. [PubMed] [Google Scholar]
- 2.Gordon G S, Moses A C, Silver R D, Flier J S, Carey M C. Proc Natl Acad Sci USA. 1985;82:7419–7423. doi: 10.1073/pnas.82.21.7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hirai S, Yashiki T, Mima H. Int J Pharm. 1981;9:165–172. [Google Scholar]
- 4.Hirai S, Yashiki T, Mima H. Int J Pharm. 1981;9:173–184. [Google Scholar]
- 5.Swenson E S, Curatolo W J. Adv Drug Delivery Rev. 1992;8:39–92. [Google Scholar]
- 6.Murakami T, Sasaki Y, Yamajo R, Yata N. Chem Pharm Bull. 1984;32:1948–1955. doi: 10.1248/cpb.32.1948. [DOI] [PubMed] [Google Scholar]
- 7.Kahne D, Walker S, Cheng Y, Van Engen D. J Am Chem Soc. 1989;111:6881–6882. [Google Scholar]
- 8.Cheng Y, Ho D M, Gottlieb C R, Kahne D, Bruck M A. J Am Chem Soc. 1992;114:7319–7320. [Google Scholar]
- 9.Venkatesan P, Cheng Y, Kahne D. J Am Chem Soc. 1994;116:6955–6956. [Google Scholar]
- 10.Venkatesan P. Ph.D. thesis. Princeton, NJ: Princeton Univ.; 1995. [Google Scholar]
- 11.Bowe C L. Ph.D. thesis. Princeton, NJ: Princeton Univ.; 1996. [Google Scholar]
- 12.Maitra S K, Yoshikawa T T, Hansen J L, Nilsson-Ehle I, Palin W J, Schotz M C, Guze L. Clin Chem. 1977;23:2275–2278. [PubMed] [Google Scholar]
- 13.Hosotsubo H. J Chromatogr. 1989;487:421–427. doi: 10.1016/s0378-4347(00)83049-2. [DOI] [PubMed] [Google Scholar]
- 14.Armstrong M J, Carey M C. J Lipid Res. 1982;23:70–80. [PubMed] [Google Scholar]
- 15.Wilkinson M C. J Colloid Interface Sci. 1972;40:14–26. [Google Scholar]
- 16.Shaw R, Rivetna M, Elliott W H. J Chromatogr. 1980;202:347–361. [Google Scholar]
- 17.Schott H. J Phys Chem. 1966;70:2966–2973. [Google Scholar]
- 18.Adamson A W. Physical Chemistry of Surfaces. New York: Wiley; 1990. pp. 21–23. [Google Scholar]
- 19.Roda A, Hofmann A F, Mysels K J. J Biol Chem. 1983;258:6362–6370. [PubMed] [Google Scholar]
- 20.Mokhtarzadeh, L. (1996) B.A. thesis (Princeton Univ., Princeton, NJ).
- 21.Longenecker J P. In: Delivery Systems for Peptide Drugs. Davis S S, Illum L, Tomlinson E, editors. New York: Plenum; 1987. pp. 211–220. [Google Scholar]
- 22.Felgner P L, Gadek T R, Holm M, Roman R, Chan H W, Wenz M, Northrop J P, Ringold G M, Danielsen M. Proc Natl Acad Sci USA. 1987;84:7413–7417. doi: 10.1073/pnas.84.21.7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Behr J-P, Demeneix B, Loeffler J-P, Perez-Mutul J. Proc Natl Acad Sci. 1989;86:6982–6986. doi: 10.1073/pnas.86.18.6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leventis R, Silvius J R. Biochim Biophys Acta. 1990;1023:124–132. doi: 10.1016/0005-2736(90)90017-i. [DOI] [PubMed] [Google Scholar]
- 25.Zhou X, Huang L. Biochim Biophys Acta. 1994;1189:195–203. doi: 10.1016/0005-2736(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 26.Remy H H, Sirlin C, Vierling P, Behr J-P. Bioconjugate Chem. 1994;5:647–654. doi: 10.1021/bc00030a021. [DOI] [PubMed] [Google Scholar]
- 27.Felgner J H, Kumar R, Sridhar C N, Wheeler C J, Tsai Y J, Border R, Ramsey P, Martin M, Felgner P L. J Biol Chem. 1994;269:2550–2561. [PubMed] [Google Scholar]
- 28.Walker S, Sofia M J, Kakarla R, Kogan N A, Wierichs L, Longley C B, Bruker K, Axelrod H R, Midha S, Babu S, Kahne D. Proc Natl Acad Sci USA. 1996;93:1585–1590. doi: 10.1073/pnas.93.4.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bromberg L E, Klibanov A M. Proc Natl Acad Sci USA. 1994;91:143–147. doi: 10.1073/pnas.91.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bromberg L E, Klibanov A M. Proc Natl Acad Sci USA. 1995;92:1262–1266. doi: 10.1073/pnas.92.5.1262. [DOI] [PMC free article] [PubMed] [Google Scholar]