

Abstract
HIV-1 envelope (Env) glycoprotein gp120 induces release of pro-inflammatory cytokines including IL-1β from macrophages, independent of infection, which are implicated in the pathogenesis of HIV-associated dementia (HAD). However, the signal transduction pathways involved have not been fully defined. Previously, our lab reported that soluble gp120 activates multiple protein kinases in primary human monocyte-derived macrophages (MDMs) including the Src family kinase Lyn, PI3K and focal adhesion-related kinase Pyk2. Here we showed that gp120 induces IL-1β release from macrophages in a time- and concentration-dependent manner through binding to chemokine receptor CCR5 and coupling to Giα protein. Utilizing pharmacological inhibitors and siRNA gene knockdown, we demonstrated that concomitant activation of Lyn, Pyk2 and class IA PI3K are required for gp120-induced IL-1β production. By co-immunoprecipitation and immunofluorescence confocal microscopy, we showed that CCR5 activation by gp120 triggered the assembly of a signaling complex involving endogenous Lyn, PI3K and Pyk2, associated with PI3K and Pyk2 translocation from the cytoplasm to the membrane where it co-localized with Lyn. Finally, we demonstrated that virion-associated gp120 induced similar response, as structurally intact whole virions also triggered IL-1β release and re-localization of PI3K and Pyk2. This study identifies a novel signaling mechanism for HIV-1-induced IL-1β production by primary human macrophages that may be involved in the neuropathogensis of HAD.
Keywords: Human, Monocytes/Macrophages, Immunodeficiency Diseases, Cytokines, Signal Transduction
Introduction
HIV-1-associated dementia (HAD) is a common neurological complication associated with HIV and is estimated to develop in ∼20% of infected patients (1). Unlike many viral encephalopathies, neurons are not productively infected by HIV. HAD is characterized by the infiltration of blood-derived macrophages at perivascular sites in the CNS, formation of multinucleated giant cells and neuronal death, as well as widespread activation of brain macrophages (2). Although infection of macrophages/microglia in the CNS is a prerequisite for the development of HAD, macrophage activation appears to play a critical role, as the number of activated macrophages in the CNS is the best correlate of HAD (3-5) and it is believed that release of pro-inflammatory cytokines from activated macrophages/microglia is a principal mechanism of neuronal injury (6). Both infected and uninfected (bystander) macrophages are activated in HAD (7), but the mechanisms responsible are incompletely defined.
IL-1β and TNF-α are highly expressed in the CNS and in patients with HAD, correlate with neuronal injury, and have been implicated as important pro-inflammatory cytokines involved in HAD pathogenesis (8, 9). IL-1β is up-regulated in vivo in both cerebral spinal fluid (CSF) and brain of HAD patients (10-12), and macrophages/microglia are the primary cell type in CNS responsible for its production (7, 13). Exposure of macrophages to whole HIV-1 virions or envelope (Env) glycoprotein gp120 has been shown in many in vitro systems to induce IL-1β release independent from productive infection (14, 15). In animal models, intracerebral injection of gp120 can also induce neuronal apoptosis (16), which is mediated through IL-1β release from macrophages/microglia since it can be prevented by pretreatment with the blocker for IL-1β receptor or interleukin-1beta-converting enzyme (ICE) (17). Despite the important role of IL-1β in neuronal injury and HAD pathogenesis, how gp120 induces IL-1β release from macrophages is not well understood.
HIV-1 entry is initiated by gp120 binding first to cellular CD4 followed by structural changes that enable interaction with the chemokine receptors CCR5 (R5 strains), CXCR4 (X4 strains) or both (R5X4 strains). Most HIV-1 isolates from the CNS or associated with HAD use CCR5 (R5 strains). Our laboratory has previously reported that HIV-1 R5 gp120 can activate CCR5-mediated signaling pathways in monocyte-derived macrophages (MDMs) including the Src family kinase (SFK) Lyn, PI3K, focal adhesion-related proline-rich tyrosine kinase (Pyk2) and several ionic currents, and that Lyn and PI3K are involved in gp120-induced TNF-α production by macrophages (18-21). However, the signaling pathways regulating IL-1β release by macrophages in response to gp120, and the mechanisms of interaction among signaling molecules activated by gp120, have not been fully defined. In this study, we set out to determine the role of Lyn, PI3K and Pyk2 in gp120-induced IL-1β release by macrophages, and determine whether these kinases physically associate to form a signaling complex in response to activation of CCR5 by HIV-1 gp120. We also investigated the effect of virion-associated gp120, since macrophages in vivo may be exposed to both monomeric (shed) and virion-associated envelope glycoproteins. We found that HIV-1 gp120 induces IL-1β release by primary macrophages through binding to CCR5 and coupling of Giα protein, and requires concomitant activation of Lyn, PI3K and Pyk2, and their subcellular redistribution and physical association to form a multimeric signaling complex. We further demonstrated that, like monomeric gp120, virion-associated gp120 also triggers IL-1β release, as well as translocation of PI3K and Pyk2 from the cytoplasm to the plasma membrane where they co-localize with Lyn in a CCR5-dependent manner.
Materials and Methods
Reagents and cells
Recombinant gp120 (CM235) was obtained from the NIH AIDS Reagent Repository (Bethesda, MD) and was tested negative for endotoxin contamination (limit of detection = 1.5 pg/ml). CCL4 (MIP-1β) was from PeproTech (Rocky Hill, NJ). The CCR5 antagonist M657 was kindly provided by M. Miller of Merck & Co. (Whitehorse Station, NJ) (22). Pertussis toxin (PTX) was purchased from Sigma (St. Louis, MO). Isoform-specific PI3K inhibitors were from Echelon (Salt Lake City, UT). All other kinase inhibitors, their inactive analogs and Pansorbin beads were from EMD Chemicals (San Diego, CA). The Lyn-specific peptide inhibitor KRX-123.302 and control peptide KRX-107.110 were synthesized by Bachem (King of Prussia, PA) according to previously published peptide sequences (23).
Affinity-purified anti-gp120 polyclonal IgG derived from pooled AIDS patient sera and anti-gp120 rabbit polyclonal antibody were kindly provided by the late K. Steimer of Chiron Corp. (Emeryville, CA). Rabbit polyclonal antibodies to Lyn, Hck, PI3Kp85, PI3Kp101 and anti-phosphotyrosine antibodies were from Millipore Corp. (Billerica, MA). Mouse monoclonal Pyk2 antibody was from BD Bioscience (San Jose, CA). Rabbit polyclonal β-actin antibody, HRP-conjugated anti-rabbit and anti-mouse IgG were from Cell Signaling Technology Inc. (Danvers, MA). Goat polyclonal PI3Kp85 antibody and control mouse, rabbit, goat IgG were from Santa Cruz Biotechnologies (Santa Cruz, CA). Alexa Fluor 555, 488, 633-conjugated IgG were purchased from Molecular Probes (Eugene, OR).
Monocytes were isolated by elutriation (24) or by selective adherence (25) from healthy donors who were screened for the CCR5Δ32 mutation by PCR (26). Only donors homozygous for the wild-type allele were used, unless otherwise specified. Monocytes were cultured in 100 mm petri dishes for 7 days to differentiate into macrophages in conditions described previously (18), harvested by gentle scraping, resuspended and replated for IL-1β ELISA, kinase activation, co-immunoprecipitation or confocal immunofluorescence microscopy studies.
Quantitation of IL-β release
MDMs plated in 24-well plates (5 × 105 cells/well) were incubated in serum-free media overnight, exposed to 20 nM gp120 for 16 h (unless stated otherwise), and IL-1β levels in cell culture media were determined by ELISA (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions. The limit of detection for IL-1β ELISA is less than 1 pg/ml. For blocking studies, antagonists, inhibitors or control vehicle (0.1% DMSO) were added 15 min to 2 h prior to and maintain throughout the period of stimulation. To ensure that inhibitory effects of the pharmacological agents on IL-1β production were not due to cytotoxicity, whenever inhibitors were used, cell viability assay was performed using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI) in parallel. 105 MDMs in 100 μl of serum-free media were added to the 96-well plates and incubated with the inhibitors for the same duration as the ELISA. Cell viability was measured spectrophotometrically according to the manufacturer's instructions.
siRNA gene knockdown
Pre-designed siRNA specific for Lyn, PI3Kp85, Pyk2 and non-targeting control siRNA were purchased from Dharmacon (Chicago, IL). Transfection of siRNAs in primary MDMs was performed using the Human Macrophage Nucleofector Kit (Amaxa, Gaithersburg, MD) according to the manufacturer's instructions. Seven-day-old MDMs were resuspended (5 × 106 cells/ml) in 100 μl of nucleofection solution with 1 to 3 μg of siRNA and electroporated using program Y-10. After transfection, MDMs were cultured for 2 days (chosen for maximal protein knockdown) before harvesting to assay in parallel both functional response by ELISA and protein expression by immunoblot.
Immunoprecipitation and immunoblot analysis
MDMs plated in 12-well plates (106 cells/well) were incubated in serum-free media overnight, exposed to stimuli for indicated durations, and lysed in RIPA buffer supplemented with protease inhibitor cocktail as previously described (18). Protein concentrations of whole cell lysates (WCL) were determined by BCA protein assay (Pierce, Rockford, IL). For immunoprecipitation, equal amounts (400 μg) of cell lysates incubated at 4°C with 1 μg of Lyn or Pyk2 antibody for 2 h followed by 50 μl of Pansorbin beads for 2 h. Immune complexes were washed twice with lysis buffer, eluted by boiling in sample buffer for 5 min and subjected to immunoblot analysis.
Immunoblot analysis was carried out on immunoprecipitates generated as described above or directly on cell lysates containing 20 μg protein. Samples were denatured by boiling in Laemmli buffer for 5 min, resolved by 8% SDS-PAGE and transferred to nitrocellulose membranes. The conditions for blocking, washing and antibody dilution were based on instructions provided by the manufacturer of the antibodies. Immunoreactive proteins were visualized by ECL detection reagents (GE Healthcare, Waukesha, WI) on autoradiographic films. Multiple exposures of the films were used to ensure that quantitations were conducted within the linear range of the images. The films were digitally scanned and the relative intensities of the protein bands were quantitated by Image J (NIH) software. In some experiments, membranes were stripped with Restore Western Blot Stripping Buffer (Pierce, Rockford, IL) and reprobed with a second antibody.
Immunofluorescence laser scanning confocal microscopy
MDMs were plated onto glass coverslips and cultured overnight in serum-free media, exposed to stimuli, then fixed in 4% paraformaldehyde for 20 min and permeabilized with 0.2% Triton X-100 for 10 min. Fixed cells were blocked with donkey serum for 20 min prior to incubation for 1 hr with the following primary antibodies (all 1:100 dilution): rabbit polyclonal anti-Lyn; goat polyclonal anti-PI3Kp85 and mouse monoclonal anti-Pyk2. Controls consisted of replacing the primary antibodies with non-specific IgG from the corresponding species or omitting primary antibodies. MDMs were then incubated for 30 min with the corresponding secondary antibodies (all 1:100 dilution) conjugated to the fluorophores. Between all incubation steps, cells were washed three times for 5 min with PBS. All incubations were carried out at room temperature. Coverslips were mounted onto slides with VectorShield mounting medium (Vector Laboratories, Burlingame, CA). Confocal images were captured with a Zeiss LSM 510 Meta laser scanning confocal device attached to an Axioplan 2 microscope using a 63 × Plan-Apochromat oil objective. To avoid the bleed-through effects, each fluorophore was scanned independently using the multitracking function of the LSM 510 device. Images were electronically merged using the LSM Image Browser software and saved as TIFF files.
HIV-1 virions
Virions were generated using an Env trans-complementation method. HEK293T cells were co-transfected with the envelope-deleted pNL-Luc backbone (27) and a plasmid expressing the envelope gene of HIV-1 R5 strain BaL. To make envelope-negative (bald) virions as a control, the pNL-Luc backbone was co-transfected with an empty plasmid vector. Cell culture supernatants were harvested 72 h post-transfection and concentrated by ultracentrifugation through 20% sucrose (150,000 × g, 2 h). The pelleted virions were resuspended and quantitated using a p24gag ELISA assay (Perkin Elmer, Waltham, MA). Incorporation of Env into pseudotype but not control virions was confirmed by Western blot. Equal amounts of each virion preparation based on p24gag antigen content were subjected to SDS-PAGE alongside known amounts of recombinant gp120 as standards, and subjected to immunoblot analysis with rabbit anti-gp120 and mouse anti-p24 antibodies.
Statistical analysis
Quantitative data are presented as mean ± S.E. of three independent experiments using cells from different blood donors. Multiple group comparisons were analyzed by one-way analysis of variance (ANOVA), followed by the Bonferroni for comparison of means. For all tests, P value of less than 0.05 was considered statistically significant. Immunoblot and immunofluorescent staining images shown are representative of experiments done using cells from at least three different donors.
Results
HIV-1 envelope glycoprotein gp120 induces IL-1β release by primary human macrophages
To characterize the effect of HIV-1 gp120 stimulation of IL-1β in primary human macrophages, we first performed time course and concentration-response experiments. As shown in Fig. 1A, MDM stimulation with gp120 from a R5 strain HIV-1 triggered an increase in IL-1β release in a time-dependent manner with maximal response at 16 h, a 3.5-fold increase over the basal level. IL-1β production by MDMs in response to gp120 was also concentration-dependent (Fig. 1B), with maximal stimulation observed at 20 nM gp120 and no further increase with higher concentrations. As shown in Fig 1C, IL-1β release was completely abrogated if gp120 was heat-inactivated (ΔHI; 100°C for 30 min), confirming that it was not due to contamination with a heat-stable component such as endotoxin. IL-1β release was also eliminated if gp120 was preincubated with affinity-purified anti-gp120 polyclonal IgG derived from pooled AIDS patient sera, while incubation with control IgG exhibited no blocking effect. These results confirmed that IL-1β release by primary human macrophages is elicited by HIV-1 gp120, which is consistent with longstanding data from previous studies (14, 15).
FIGURE 1.

HIV-1 gp120 induces IL-1β release by primary human macrophages. A, MDMs were treated without (gray bars) or with (black bars) gp120 (20 nM) for indicated times. B, MDMs were stimulated for 16 h with indicated concentrations of gp120. C, gp120 was subjected to heat inactivation (ΔHI) for 30 min or incubation with anti-gp120 IgG or control human IgG for 1 h prior to exposure to MDMs. D, MDMs were pretreated without or with the CCR5 antagonist (M657, 1 μM) for 1 h, or inhibitors for Gi (PTX, 100 ng/ml) or Gs (NF449, 200 nM) for 2 h, prior to stimulate with gp120. E, MDMs from donors lacking CCR5 (CCR5Δ32) or wild type (WT) were stimulated with gp120, LPS (0.1 ng/ml) or CCL4 (MIP-1β, 10 nM). Cell culture supernatants were collected and IL-1β levels were quantitated by ELISA. (*, P<0.05; **, P<0.01; ***, P<0.001).
HIV-1 gp120 induces IL-1β release by MDMs through binding to CCR5 and coupling of Giα protein
We employed both pharmacological and genetic approaches to define the role of chemokine receptor CCR5 in gp120-induced IL-1β release in MDMs. As shown in Fig. 1D, pretreatment of macrophages with the CCR5 antagonist M657 completely abolished the IL-1β response to gp120. In addition, CCR5-deficient MDMs from donors homozygous for the Δ32 deletion allele failed to release IL-1β in response to gp120, although they responded normally to lipopolysaccharide (LPS) stimulation (Fig. 1E). Macrophages lacking CCR5 also failed to produce IL-1β in response to CCL4 (MIP-1β), the most specific chemokine agonist of CCR5, whereas MDMs from wild type donors responded to CCL4 with IL-1β production at levels similar to that seen with R5 gp120.
Although Giα is the primary G protein coupled to CCR5 (28), other G proteins have been implicated in CCR5 signaling (29, 30). Therefore, we determined whether CCR5-mediated IL-1β release by MDMs in response to gp120 requires coupling of Giα. Pretreatment with Gi/o inhibitor PTX markedly impeded the gp120-induced IL-1β production, while the Gs-specific inhibitor NF449 (31) showed no effect (Fig. 1D). Together, these results indicate that gp120 induces IL-1β release by MDMs through binding to CCR5 and coupling of Giα protein.
gp120-induced IL-1β release by MDMs requires activation of Src family kinase Lyn
We previously reported that gp120-triggered CCR5-mediated TNF-α production in MDMs was mediated through SFK member Lyn, based on pharmacological inhibition (18). To determine whether Lyn is involved in gp120-induced IL-1β release, MDMs were pretreated with the broad spectrum SFK inhibitor PP2. As shown in Fig. 2A, PP2 completely abrogated gp120-induced IL-1β production whereas its inactive analog PP3 showed no blocking effect. For greater specificity, we also tested a pseudosubstrate peptide inhibitor specific to Lyn, KRX-123.302 (23), as well as a negative control peptide KRX-107.110. IL-1β release was also markedly attenuated by KRX-123.302 while no inhibition was observed with the negative control peptide KRX-107.110. Parallel experiments to evaluate the viability of MDMs in the presence of pharmacological inhibitors ruled out inhibitor-induced cytotoxicity as a cause of the decreased IL-1β response (data not shown).
FIGURE 2.

gp120-induced IL-1β production in macrophages requires activation of Src family kinase Lyn. A, MDMs were pretreated for 1 to 2 h with the broad-spectrum SFK inhibitor PP2 or its inactive analog PP3 (10 μM), a Lyn-specific peptide inhibitor KRX-123.302 or its negative control peptide KRX-107.110 (10 μM), or control vehicle alone, then stimulated with gp120. B, MDMs were transfected with non-targeting control or Lyn-specific siRNA prior to stimulation without (gray bars) or with (black bars) gp120. Inset, immunoblots (IB) showing Pyk2, PI3Kp85, Lyn, Hck and β-actin protein levels in siRNA-transfected MDMs from representative parallel transfection. (***, P<0.001).
To further substantiate the role of Lyn in gp120-induced IL-1β release, we then employed siRNA-mediated gene silencing. As shown in Fig. 2B, siRNA knockdown suppressed Lyn protein expression in primary MDMs by an average of 91% based on quantitation of immunoblots, whereas other proteins such as Pyk2, PI3K, the SFK Hck and β-actin were unaffected. Down-regulation of Lyn significantly impaired the ability of MDMs to produce IL-1β in response to gp120 (Fig. 2B). Suppression of cytokine production was not the result of decreased cell viability, as the viability of both control and Lyn siRNA transfected MDMs were equivalent (data not shown). We also confirmed that Lyn is activated by gp120 based on tyrosine phosphorylation (data not shown), which is consistent with results previously observed by kinase assay (18).
Activation of PI3K is necessary for gp120-triggered IL-1β induction in MDMs
PI3K has been implicated as a mediator of CCR5-elicited signals leading to both survival and TNF-α production in macrophages (19, 32). Therefore, we tested if PI3K is important for gp120-induced IL-1β production. As shown in Fig. 3A, LY294002 and wortmannin, two potent PI3K inhibitors with different mechanisms of action, significantly attenuated the macrophage IL-1β response to gp120. We verified that gp120 activated PI3K in MDMs based on phosphorylation of Akt, a major downstream target of PI3K (data not shown), which is consistent with our prior published results (19).
FIGURE 3.

IL-1β release by gp120-stimulated macrophages requires PI3K activation. A, MDMs were pretreated for 1 to 2 h with the broad PI3K inhibitor LY294002 (10 μM), wortmannin (100 nM) or control vehicle alone prior to exposure to gp120. B, MDMs were treated with pharmacologic inhibitors specific for class IA PI3K (PI-103; 40 nM) or class IB (AS605240; 10 nM), or both, or control vehicle only, prior to stimulation with gp120. C, MDMs were transfected with non-targeting control or PI3Kp85-specific siRNA prior to stimulation without (gray bars) or with (black bars) gp120. Inset, immunoblots (IB) showing Pyk2, PI3Kp85, PI3Kp101, Lyn, Hck and β-actin protein levels in siRNA-transfected MDMs from representative parallel transfection. (*, P<0.05; **, P<0.01; ***, P<0.001).
Classically, class IA PI3K isoforms (p85-p110α,β,δ) are activated by receptor tyrosine kinases whereas class IB PI3Ks (p101-p110γ) are activated by G protein-coupled receptors (GPCRs) including chemokine receptors (33, 34). However, some reports suggested that class IA PI3K might also be activated by GPCRs and may play a role in chemokine-receptor mediated IL-1β induction in primary human monocytes (35-38). To define the role of specific PI3K isoforms on gp120-stimulated IL-1β release, we used class IA PI3K specific inhibitor PI-103 (39) and class IB PI3K specific inhibitor AS605240 (40, 41). As shown in Fig. 3B, inhibition of class IA PI3K markedly attenuated the IL-1β response to gp120, while the IB inhibitor had no effect.
We then used siRNA directed against p85 subunit of PI3K, which achieved ∼85% knockdown of PI3K protein expression as shown in Fig. 3C, while the expression of class IB PI3Kp101 and other proteins including Pyk2, Lyn and Hck were unaffected. IL-1β induction by gp120 was markedly attenuated in PI3Kp85-depleted MDMs relative to MDMs transfected with control siRNA (Fig. 3C), confirming a role for PI3K in gp120-stimulated IL-1β release. Together, these results suggest that class IA PI3K, rather than the more typically GPCR-associated class IB isoforms, are the primary isoform responsible for IL-1β production in response to gp120.
IL-1β induction by gp120 involves focal adhesion-related kinase Pyk2
Previous data from our lab and others have reported that CCR5 activates tyrosine kinase Pyk2 (20, 42, 43), but the role of Pyk2 in gp120-triggered IL-1β release in primary macrophages has not been examined. Since specific inhibitors for Pyk2 are currently not available, we used two pharmacological agents that target upstream activators of Pyk2 through different mechanisms. Dantrolene inhibits Pyk2 by blocking ryanodine-receptor-mediated intracellular calcium release, which is required for Pyk2 activation (44-46). Another agent often used as a Pyk2 inhibitor is AG17, which blocks calcium release-activated calcium (CRAC) channel-mediated intracellular calcium (47-49). As shown in Fig 4A, both dantrolene and AG17 significantly inhibited IL-1β response, whereas inactive analog AG43 exhibited no effect.
FIGURE 4.

Macrophage IL-1β release triggered by gp120 requires the proline-rich tyrosine kinase Pyk2. A, MDMs were pretreated for 15 to 60 min with the upstream Pyk2 inhibitor dantrolene (10 μM), AG17 or its inactive analog AG43 (both 20 μM), or control vehicle alone prior to stimulation with gp120. B, MDMs were pretreated for 1 h with the CaMKII inhibitor KN62 (1 μM), GSK3 inhibitor BIO (10 nM), or control vehicle alone prior to stimulation with gp120. C, MDMs were transfected with non-targeting control or Pyk2-specific siRNA prior to stimulation without (gray bars) or with (black bars) gp120. To monitor protein expression, immunoblots (IB) were performed in parallel for Pyk2, PI3Kp85, Lyn, Hck and β-actin on the same batch of transfected MDMs used or IL-1β ELISA. (***, P<0.001).
Owing to the calcium-dependence of gp120-induced Pyk2 activation suggested by these results and to the ability of gp120 to open ion channels to increase intracellular calcium levels in MDMs demonstrated in previous studies (20, 21), we then asked whether gp120-induced IL-1β release involves calcium/calmodulin-dependent protein kinase II (CaMKII), which has been implicated in ATP-induced IL-1β release by MDMs (50) and Pyk2 activation in some cell models (51, 52). We also examined the role of glycogen synthase kinase 3 (GSK3), a downstream target of PI3K that modulates cytokine production in several model systems (53-55). Neither the CaMKII inhibitor KN62 nor the GSK3 inhibitor BIO affected gp120-induced IL-1β induction in MDMs (Fig. 4B). In addition to excluding a role for CaMKII and GSK3 in gp120-induced IL-1β production, these results also serve to illustrate, in part, the specific requirement for Lyn, PI3K and Pyk2 activation in IL-1β induction by gp120.
Since the available pharmacological inhibitors of Pyk2 are not completely specific, we next confirmed the role of Pyk2 in gp120-induced IL-1β production by transfecting primary MDMs with Pyk2-specific siRNA. This knockdown suppressed Pyk2 protein expression by ∼72% and was gene-specific, as the expression of other proteins were unaffected (Fig. 4C). Pyk2 knockdown markedly attenuated gp120-induced IL-1β induction as shown in Fig. 4C. Immunoprecipitation/immunoblot analysis confirmed tyrosine phosphorylation of Pyk2 in a time-dependent manner concordant with activation in response to gp120 exposure to MDMs (data not shown) as reported previously (20).
Lyn, PI3K and Pyk2 physically interact in response to HIV-1 gp120 stimulation in primary MDMs
Our data demonstrated that Lyn, PI3K and Pyk2 are each important for gp120-induced IL-1β production in MDMs. All three kinases contain structural motifs that facilitate protein-protein interactions including SH2 and SH3 domains of Lyn and PI3K, as well as the proline-rich motifs of PI3K and Pyk2. Therefore, we determined whether gp120 triggered a physical association between the endogenous Lyn, PI3K and Pyk2 in macrophages (Fig. 5).
FIGURE 5.
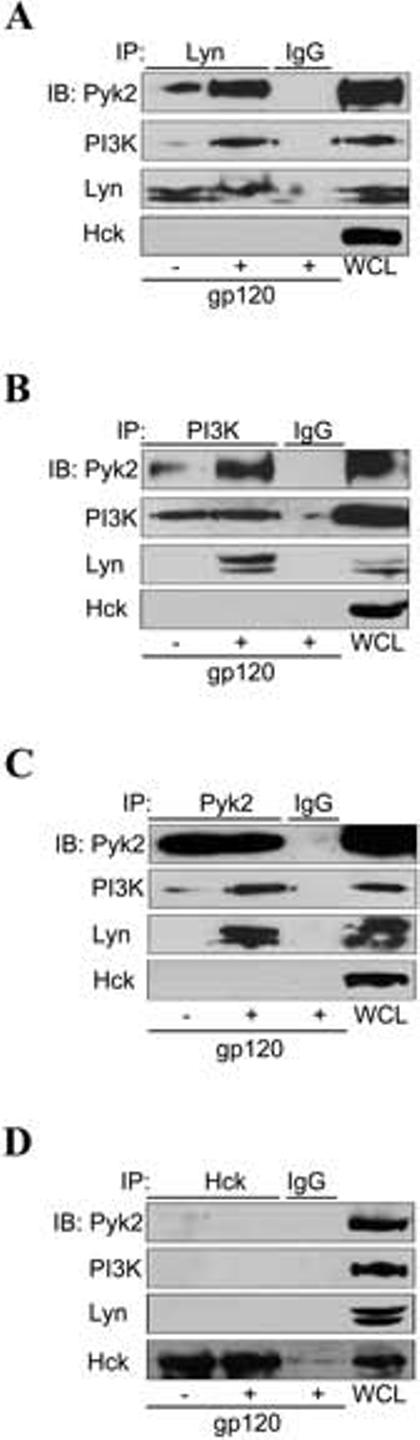
Lyn, PI3K and Pyk2 physically associated to form a multi-kinase signaling complex in response to HIV-1 gp120 stimulation. MDMs were treated without or with gp120 (20 nM) for 10 min prior to cell lysis. Cell lysates were immunoprecipitated (IP) with antibody specific for (A) Lyn, (B) PI3Kp85, (C) Pyk2, (D) Hck, or with control IgG. Immune complexes were washed, resolved on SDS-PAGE and subjected to immunoblot (IB) with antibodies specific for Pyk2, PI3Kp85, Lyn and Hck. Whole cell lysates (WCL) of unstimulated MDMs served as a positive control for immunoblotting.
When endogenous Lyn protein was immunoprecipitated with Lyn antibody in unstimulated MDMs (Fig. 5A), low levels of PI3K and Pyk2 could be detected in the immune complex (first lane), suggesting some constitutive association among these proteins. Exposure of MDMs to gp120 resulted in a marked increase in co-precipitation (Fig. 5A, second lane), indicating that gp120 stimulation up-regulated the formation of this complex.
We next used both PI3K and Pyk2 antibody for immunoprecipitation to perform the reciprocal experiments (Fig. 5B and 5C). Immunoprecipitation with anti-PI3K antibody demonstrated gp120-enhanced Pyk2 and Lyn co-precipitation (Fig. 5B), and similar results were observed with Pyk2 co-immunoprecipitation studies (Fig. 5C). In contrast, immunoprecipitation with antibody specific to Hck, another SFK member that is abundantly expressed in MDMs, showed no constitutive or stimulation-induced association with PI3K or Pyk2 protein (Fig. 5D), nor did probing the Lyn, PI3K or Pyk2 immunoprecipitation blots with anti-Hck antibody yield any signal (bottom panels of Figs. 5A, B, C). Of note, the rapidity of complex formation (10 minutes) indicates an increase in the complex-associated fraction for each kinase following gp120 exposure rather than upregulated total protein levels, a mechanism that is concordant with our prior results showing an increase in activated but not total Lyn and Pyk2 following gp120 exposure (18, 20). Thus, Lyn, PI3K and Pyk2 specifically associate to form a signaling complex in response to gp120 stimulation.
HIV-1 gp120 induces co-localization of Lyn, PI3K and Pyk2 in primary macrophages
Since a physical association among Lyn, PI3K and Pyk2 was suggested by our co-immunoprecipitation results, we next performed triple-labeling immunocytochemistry to determine the subcellular distribution of these kinases and the putative signaling complex (Fig. 6). In unstimulated MDMs (top panels, A-D), Lyn was primarily localized at the plasma membrane (Fig. 6A), PI3Kp85 was distributed both at the plasma membrane and in the cytoplasm (Fig. 6B), while Pyk2 was mainly found in the cytoplasm (Fig. 6C). Co-localization of the three kinases, which should appear as white, was not observed in the merge image in unstimulated MDMs (Fig. 6D). Control experiments in which the primary antibodies were omitted or substituted with isotype control IgG of the appropriate species showed no fluorescent staining (data not shown).
FIGURE 6.

HIV-1 gp120 triggers PI3K and Pyk2 redistribution and co-localization with Lyn in primary MDMs. MDMs were pretreated without or with the CCR5 antagonist M657 (1 μM) for 1 h prior to stimulation with gp120 for 10 min. MDMs were fixed, permeabilized and triple-labeled with Lyn, PI3Kp85 and Pyk2 antibodies before examination by confocal microscopy. Subcellular distribution of Lyn (green; A, E, I, M), PI3K (blue; B, F, J, N) and Pyk2 (red; C, G, K, O) are shown in the single channel images. Co-localization of Lyn, PI3K and Pyk2 are indicated by arrowheads shown in the merge images (white; D, H, L, P).
When MDMs were stimulated with gp120, most of the PI3K and Pyk2 was re-localized at the plasma membrane (Fig. 6F-G), while Lyn remained in its previous membrane distribution (Fig. 6E). This gp120-induced PI3K and Pyk2 translocation resulted in co-localization of the three kinases at the membrane, as highlighted by arrowheads seen in the merge image (white, Fig. 6H). Re-localization was blocked if MDMs were incubated with the CCR5 antagonist M657 prior to stimulation with gp120 (Fig. 6I-L), confirming that gp120-induced translocation of these kinases and complex formation were mediated through CCR5.
Virion-associated gp120 also triggers redistribution of PI3K and Pyk2, co-localization with Lyn and IL-1β release in primary macrophages
These results demonstrated that soluble monomeric gp120 triggers co-localization of Lyn, PI3K and Pyk2 in MDMs. In addition to shed monomeric gp120, macrophages in vivo may also be exposed to gp120 on the surface of HIV-1 virions, where it exists as a trimer (56). Therefore, to extend our findings to this physiologically relevant stimulus, we then asked if virion-associated gp120 would also alter PI3K and Pyk2 subcellular distribution and co-localization with Lyn. Pseudotype virions were generated either carrying Env from the HIV-1 R5 strain BaL, or lacking Env (bald virus) to serve as a control. MDMs were exposed to equal amounts of Env-containing and bald virus, based on p24 gag antigen content, and subjected to triple-labeling immunofluorescent staining and confocal microscopy (Fig. 7A). Compared with unstimulated macrophages (micrograph a-d), MDMs exposed to gp120-containing virions showed marked increase in PI3K and Pyk2 distribution at the membrane, resulting in co-localization of Lyn, PI3K and Pyk2 as shown in the merge image (micrograph e to h). This pattern was identical to that seen following MDM exposure to monomeric gp120 (Fig. 6). In contrast, virion particles lacking Env did not induce co-localization (micrograph i-l), confirming gp120 dependence of the stimulus. Of note, bald virions also serves as an important control for potential virion-incorporated host-membrane proteins (57). Treatment of macrophages with the CCR5 antagonist M657 completely blocked virion-induced co-localization (micrograph m-p), confirming further that it is mediated through CCR5.
FIGURE 7.
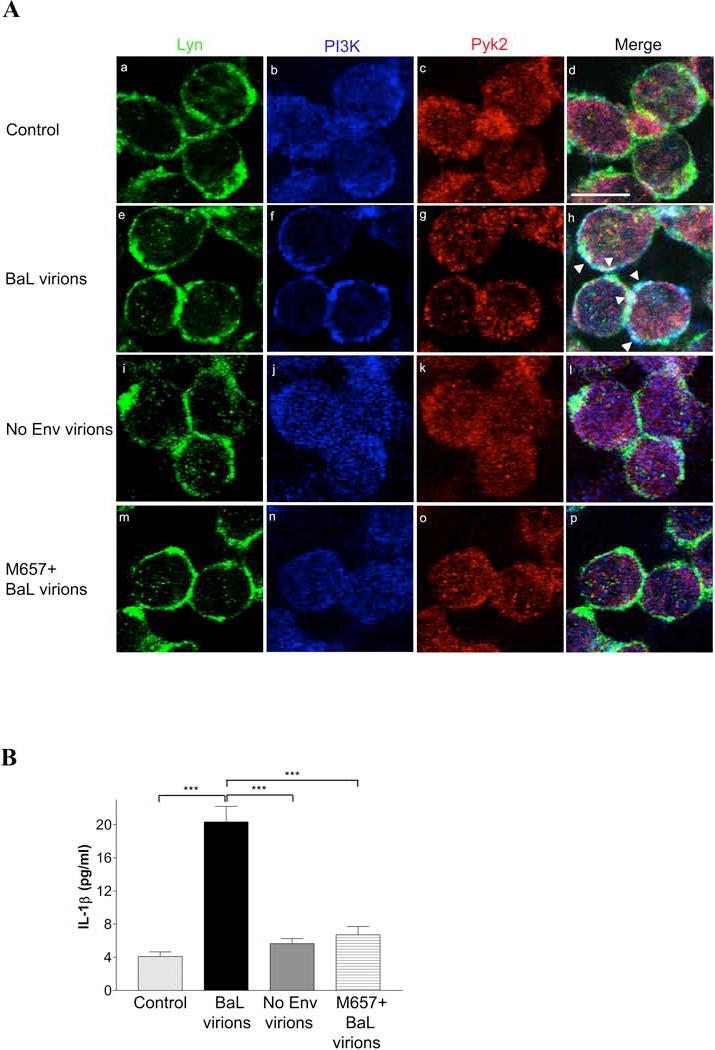
Virion-associated gp120 induces CCR5-mediated redistribution of PI3K and Pyk2, co-localization with Lyn and IL-1β release in primary MDMs. MDMs were stimulated without or with HIV-1 pseudotype virions carrying envelope from the R5 strain BaL, or lacking envelope (bald virus), using 10 ng of p24gag antigen content per virus. In parallel, MDMs were treated with the CCR5 antagonist M657 (1 μM) prior to exposure to BaL pseudotype virion. A, After 10 min of virion exposure, MDMs were fixed, permeabilized and triple-labeled with Pyk2, PI3Kp85 and Lyn antibodies before examination by confocal microscopy. B, Cell culture supernatants were collected after 16 h of virion stimulation and IL-1β levels were quantitated by ELISA. (***, P<0.001).
We also demonstrated that virion-associated gp120 induces IL-1β release in macrophages (Fig. 7B). MDM exposure to gp120-containing virions resulted in IL-1β production at levels comparable to that observed with soluble gp120. This IL-1β induction was both gp120- and CCR5-dependent, as bald virions failed to trigger cytokine release and IL-1β release in response to Env-containing virions was abolished by pretreatment of macrophages with the CCR5 antagonist M657.
Discussion
IL-1β is one of the important inflammatory and neurotoxic products secreted by immune activated macrophages/microglia that has been implicated in the pathogenesis of HIV-1-associated dementia (HAD) (6, 58). In this study we demonstrated that HIV-1 gp120-induced IL-1β release by macrophages is mediated through CCR5 and coupling of Giα protein; requires concomitant activation of Lyn, PI3K and Pyk2; that these kinases act coordinately through formation of a multi-kinase signaling complex; and that both monomeric and virion-associated gp120 activate these pathways (Fig. 8). IL-1β release by HIV-1 gp120- or virion-stimulated macrophages is a long-standing observation believed to be important in pathogenesis (9, 14, 15) and this is the first study to define the signal transduction mechanism responsible.
FIGURE 8.

Model for signaling mechanism mediating gp120-induced IL-1β release in primary human macrophages. Binding of monomeric or virion-associated HIV-1 gp120 to macrophage CCR5 triggers Gi-mediated PI3Kp85 and Pyk2 re-localization to the membrane and formation of a signaling complex with Lyn. Activation of this complex then leads to IL-1β production, likely through the action of downstream MAP kinases and nuclear transcription factors (NTFs).
In the brain, IL-1β is expressed at low level under physiological conditions and is induced in response to both acute and chronic inflammation (59). This is in agreement with immunohistochemical study of autopsy brain tissues from AIDS patients showing intense IL-1β staining in macrophages/microglia only from those with pathological evidence of HIV encephalitis, but not in those without neurological complications or in healthy individuals (10, 11). Similarly, CSF IL-1β levels are also increased in HIV-infected individuals with clinical HAD but undetectable in those without (12). Results from our primary human macrophage cell model are consistent with these observations showing the release of low level IL-1β in unstimulated MDMs, which is increased by stimulation by gp120 and gp120-coating virions. Hence, primary MDMs provide a physiologically relevant primary human cell model to examine the signaling pathways mediating gp120-induced IL-1β release.
Src family kinase members and PI3K have each individually been implicated in modulating IL-1β production in both primary and monocyte/macrophage cell lines in response to LPS, which acts through TLR4 (60-63). Our results extended that data to chemokine receptor-elicited IL-1β response, and further identify Pyk2 as a component of the pathway.
Multiple kinases often organize to form a signaling complex to modulate both efficiency and specificity of signaling response. Here we identify gp120/CCR5 interaction promote physical association between Lyn, PI3K and Pyk2 to assemble a new signaling complex. Lyn, PI3K and Pyk2 all possess structural motifs known to facilitate protein-protein interactions. Lyn and PI3K each contain both SH2 domains that may interact with phosphorylated tyrosine residues, and SH3 domains that can bind to proline-rich motifs (64, 65), while PI3K and Pyk2 possess proline-rich motifs that could interact with SH3 domains (66, 67). Furthermore, all three kinases contain tyrosine residues that undergo phosphorylation upon activation (68-70). Hence, it is possible that these three kinases could directly interact with each other to form a complex in response to CCR5 activation in macrophages. Alternatively, Lyn, PI3K and Pyk2 can be associated indirectly through binding to a common docking protein. We recently found that arrestin scaffolding proteins are involved in CCL4-induced Lyn/PI3K/Pyk2 complex formation (manuscript submitted) and thus it is likely that gp120-triggered complex assembly is also arrestin-dependent, although a role for the direct interaction between these three kinases cannot be excluded.
How activation of PI3K, Lyn and Pyk2 regulate IL-1β release in MDMs is not clear. One likely mechanism is that activation of these kinases may lead to phosphorylation of transcription factors that interact with the IL-1β promoter region to induce transcriptional activation. NF-kB and AP-1 are two main cis-acting elements identified in the IL-1β promoter region (71, 72) and both Lyn and PI3K have been shown to activate NF-kB whereas Pyk2 is involved in AP-1 activation (73-75). Further defining the downstream pathways by which each of these kinases regulate IL-1β in macrophages requires further investigation.
In summary, we have identified specific signaling pathways and evidence for interactions of multiple kinases that are activated by HIV-1 envelope glycoprotein both in soluble and native virion-associated forms, which lead to release of inflammatory and neurotoxic IL-1β by primary human macrophages. These findings provide a mechanism that may be involved in the development of the HIV-elicited bystander macrophage activation and neuronal injury that contribute to the pathogenesis of HAD.
Acknowledgments
We thank Dr. M. Malik for advice and discussions; M. Kiezle and J. Issacman-Beck for technical assistance; and the Virus/Molecular and Immunology Cores of the Penn Center for AIDS Research for technical support.
This work was supported by National Institute of Health grants MH061139, NS027405 and AI 35502 to R.G.C. and CA108552 to A.P. R.C. is the recipient of the Canadian Institutes of Health Research postdoctoral fellowship award.
Abbreviations
- MDMs
monocyte-derived macrophages
- HAD
HIV-associated dementia
- CSF
cerebral spinal fluid
- Env
envelope
- GPCRs
G protein-coupled receptors
- PTX
pertussis toxin
- LPS
lipopolysaccharide
- SFK
Src family kinase
- Pyk2
proline-rich tyrosine kinase 2
- CaMKII
calcium/calmodulin-dependent protein kinase II
- GSK3
glycogen synthase kinase 3
- siRNA
small interfering RNA
- SH2
Src homology 2
- ICE
interleukin-1beta-converting enzyme
Footnotes
Disclosures The authors have no financial conflict of interest.
References
- 1.Wiley CA, Masliah E, Morey M, Lemere C, DeTeresa R, Grafe M, Hansen L, Terry R. Neocortical damage during HIV infection. Ann Neurol. 1991;29:651–657. doi: 10.1002/ana.410290613. [DOI] [PubMed] [Google Scholar]
- 2.Gray F, Adle-Biassette H, Chretien F, Lorin de la Grandmaison G, Force G, Keohane C. Neuropathology and neurodegeneration in human immunodeficiency virus infection. Pathogenesis of HIV-induced lesions of the brain, correlations with HIV-associated disorders and modifications according to treatments. Clin Neuropathol. 2001;20:146–155. [PubMed] [Google Scholar]
- 3.Glass JD, Fedor H, Wesselingh SL, McArthur JC. Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol. 1995;38:755–762. doi: 10.1002/ana.410380510. [DOI] [PubMed] [Google Scholar]
- 4.Achim CL, Wiley CA. Inflammation in AIDS and the role of the macrophage in brain pathology. Curr Opin Neurol. 1996;9:221–225. doi: 10.1097/00019052-199606000-00013. [DOI] [PubMed] [Google Scholar]
- 5.Adle-Biassette H, Chretien F, Wingertsmann L, Hery C, Ereau T, Scaravilli F, Tardieu M, Gray F. Neuronal apoptosis does not correlate with dementia in HIV infection but is related to microglial activation and axonal damage. Neuropathol Appl Neurobiol. 1999;25:123–133. doi: 10.1046/j.1365-2990.1999.00167.x. [DOI] [PubMed] [Google Scholar]
- 6.Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- 7.Nuovo GJ, Alfieri ML. AIDS dementia is associated with massive, activated HIV-1 infection and concomitant expression of several cytokines. Mol Med. 1996;2:358–366. [PMC free article] [PubMed] [Google Scholar]
- 8.Epstein LG, Gendelman HE. Human immunodeficiency virus type 1 infection of the nervous system: pathogenetic mechanisms. Ann Neurol. 1993;33:429–436. doi: 10.1002/ana.410330502. [DOI] [PubMed] [Google Scholar]
- 9.Brabers NA, Nottet HS. Role of the pro-inflammatory cytokines TNF-alpha and IL-1beta in HIV-associated dementia. Eur J Clin Invest. 2006;36:447–458. doi: 10.1111/j.1365-2362.2006.01657.x. [DOI] [PubMed] [Google Scholar]
- 10.Tyor WR, Glass JD, Griffin JW, Becker PS, McArthur JC, Bezman L, Griffin DE. Cytokine expression in the brain during the acquired immunodeficiency syndrome. Ann Neurol. 1992;31:349–360. doi: 10.1002/ana.410310402. [DOI] [PubMed] [Google Scholar]
- 11.Sippy BD, Hofman FM, Wallach D, Hinton DR. Increased expression of tumor necrosis factor-alpha receptors in the brains of patients with AIDS. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;10:511–521. [PubMed] [Google Scholar]
- 12.Gallo P, Frei K, Rordorf C, Lazdins J, Tavolato B, Fontana A. Human immunodeficiency virus type 1 (HIV-1) infection of the central nervous system: an evaluation of cytokines in cerebrospinal fluid. J Neuroimmunol. 1989;23:109–116. doi: 10.1016/0165-5728(89)90029-5. [DOI] [PubMed] [Google Scholar]
- 13.Wesselingh SL, Takahashi K, Glass JD, McArthur JC, Griffin JW, Griffin DE. Cellular localization of tumor necrosis factor mRNA in neurological tissue from HIV-infected patients by combined reverse transcriptase/polymerase chain reaction in situ hybridization and immunohistochemistry. J Neuroimmunol. 1997;74:1–8. doi: 10.1016/s0165-5728(96)00160-9. [DOI] [PubMed] [Google Scholar]
- 14.Herbein G, Keshav S, Collin M, Montaner LJ, Gordon S. HIV-1 induces tumour necrosis factor and IL-1 gene expression in primary human macrophages independent of productive infection. Clin Exp Immunol. 1994;95:442–449. doi: 10.1111/j.1365-2249.1994.tb07016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Merrill JE, Koyanagi Y, Chen IS. Interleukin-1 and tumor necrosis factor alpha can be induced from mononuclear phagocytes by human immunodeficiency virus type 1 binding to the CD4 receptor. J Virol. 1989;63:4404–4408. doi: 10.1128/jvi.63.10.4404-4408.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bagetta G, Corasaniti MT, Berliocchi L, Navarra M, Finazzi-Agro A, Nistico G. HIV-1 gp120 produces DNA fragmentation in the cerebral cortex of rat. Biochem Biophys Res Commun. 1995;211:130–136. doi: 10.1006/bbrc.1995.1787. [DOI] [PubMed] [Google Scholar]
- 17.Bagetta G, Corasaniti MT, Berliocchi L, Nistico R, Giammarioli AM, Malorni W, Aloe L, Finazzi-Agro A. Involvement of interleukin-1beta in the mechanism of human immunodeficiency virus type 1 (HIV-1) recombinant protein gp120-induced apoptosis in the neocortex of rat. Neuroscience. 1999;89:1051–1066. doi: 10.1016/s0306-4522(98)00363-7. [DOI] [PubMed] [Google Scholar]
- 18.Tomkowicz B, Lee C, Ravyn V, Cheung R, Ptasznik A, Collman RG. The Src kinase Lyn is required for CCR5 signaling in response to MIP-1beta and R5 HIV-1 gp120 in human macrophages. Blood. 2006;108:1145–1150. doi: 10.1182/blood-2005-12-012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee C, Tomkowicz B, Freedman BD, Collman RG. HIV-1 gp120-induced TNF-{alpha} production by primary human macrophages is mediated by phosphatidylinositol-3 (PI-3) kinase and mitogen-activated protein (MAP) kinase pathways. J Leukoc Biol. 2005;78:1016–1023. doi: 10.1189/jlb.0105056. [DOI] [PubMed] [Google Scholar]
- 20.Del Corno M, Liu QH, Schols D, de Clercq E, Gessani S, Freedman BD, Collman RG. HIV-1 gp120 and chemokine activation of Pyk2 and mitogen-activated protein kinases in primary macrophages mediated by calcium-dependent, pertussis toxin-insensitive chemokine receptor signaling. Blood. 2001;98:2909–2916. doi: 10.1182/blood.v98.10.2909. [DOI] [PubMed] [Google Scholar]
- 21.Liu QH, Williams DA, McManus C, Baribaud F, Doms RW, Schols D, De Clercq E, Kotlikoff MI, Collman RG, Freedman BD. HIV-1 gp120 and chemokines activate ion channels in primary macrophages through CCR5 and CXCR4 stimulation. Proc Natl Acad Sci U S A. 2000;97:4832–4837. doi: 10.1073/pnas.090521697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finke PE, Oates B, Mills SG, MacCoss M, Malkowitz L, Springer MS, Gould SL, DeMartino JA, Carella A, Carver G, Holmes K, Danzeisen R, Hazuda D, Kessler J, Lineberger J, Miller M, Schleif WA, Emini EA. Antagonists of the human CCR5 receptor as anti-HIV-1 agents. Part 4: synthesis and structure-activity relationships for 1-[N-(methyl)-N-(phenylsulfonyl)amino]-2-(phenyl)-4-(4-(N-(alkyl)-N-(benzyloxycarbonyl)amino)piperidin-1-yl)butanes. Bioorg Med Chem Lett. 2001;11:2475–2479. doi: 10.1016/s0960-894x(01)00492-9. [DOI] [PubMed] [Google Scholar]
- 23.Goldenberg-Furmanov M, Stein I, Pikarsky E, Rubin H, Kasem S, Wygoda M, Weinstein I, Reuveni H, Ben-Sasson SA. Lyn is a target gene for prostate cancer: sequence-based inhibition induces regression of human tumor xenografts. Cancer Res. 2004;64:1058–1066. doi: 10.1158/0008-5472.can-03-2420. [DOI] [PubMed] [Google Scholar]
- 24.Weiner RS, Shah VO. Purification of human monocytes: isolation and collection of large numbers of peripheral blood monocytes. J Immunol Methods. 1980;36:89–97. doi: 10.1016/0022-1759(80)90034-4. [DOI] [PubMed] [Google Scholar]
- 25.Collman R, Hassan NF, Walker R, Godfrey B, Cutilli J, Hastings JC, Friedman H, Douglas SD, Nathanson N. Infection of monocyte-derived macrophages with human immunodeficiency virus type 1 (HIV-1). Monocyte-tropic and lymphocyte-tropic strains of HIV-1 show distinctive patterns of replication in a panel of cell types. J Exp Med. 1989;170:1149–1163. doi: 10.1084/jem.170.4.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, Muyldermans G, Verhofstede C, Burtonboy G, Georges M, Imai T, Rana S, Yi Y, Smyth RJ, Collman RG, Doms RW, Vassart G, Parmentier M. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 27.Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206:935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- 28.Zhao J, Ma L, Wu YL, Wang P, Hu W, Pei G. Chemokine receptor CCR5 functionally couples to inhibitory G proteins and undergoes desensitization. J Cell Biochem. 1998;71:36–45. doi: 10.1002/(sici)1097-4644(19981001)71:1<36::aid-jcb4>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 29.Gosling J, Monteclaro FS, Atchison RE, Arai H, Tsou CL, Goldsmith MA, Charo IF. Molecular uncoupling of C-C chemokine receptor 5-induced chemotaxis and signal transduction from HIV-1 coreceptor activity. Proc Natl Acad Sci U S A. 1997;94:5061–5066. doi: 10.1073/pnas.94.10.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mueller A, Strange PG. CCL3, acting via the chemokine receptor CCR5, leads to independent activation of Janus kinase 2 (JAK2) and Gi proteins. FEBS Lett. 2004;570:126–132. doi: 10.1016/j.febslet.2004.04.100. [DOI] [PubMed] [Google Scholar]
- 31.Hohenegger M, Waldhoer M, Beindl W, Boing B, Kreimeyer A, Nickel P, Nanoff C, Freissmuth M. Gsalpha-selective G protein antagonists. Proc Natl Acad Sci U S A. 1998;95:346–351. doi: 10.1073/pnas.95.1.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tyner JW, Uchida O, Kajiwara N, Kim EY, Patel AC, O'Sullivan MP, Walter MJ, Schwendener RA, Cook DN, Danoff TM, Holtzman MJ. CCL5-CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat Med. 2005;11:1180–1187. doi: 10.1038/nm1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D. Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science. 2000;287:1046–1049. doi: 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- 34.Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- 35.Turner L, Ward SG, Westwick J. RANTES-activated human T lymphocytes. A role for phosphoinositide 3-kinase. J Immunol. 1995;155:2437–2444. [PubMed] [Google Scholar]
- 36.Turner SJ, Domin J, Waterfield MD, Ward SG, Westwick J. The CC chemokine monocyte chemotactic peptide-1 activates both the class I p85/p110 phosphatidylinositol 3-kinase and the class II PI3K-C2alpha. J Biol Chem. 1998;273:25987–25995. doi: 10.1074/jbc.273.40.25987. [DOI] [PubMed] [Google Scholar]
- 37.Yano N, Ianus V, Zhao TC, Tseng A, Padbury JF, Tseng YT. A Novel Signaling Pathway for {beta}-Adrenergic Receptor-Mediated Activation of Phosphoinositide 3-Kinase in H9c2 Cardiomyocytes. Am J Physiol Heart Circ Physiol. 2007;293:H385–H387. doi: 10.1152/ajpheart.01318.2006. [DOI] [PubMed] [Google Scholar]
- 38.Pan ZK, Chen LY, Cochrane CG, Zuraw BL. fMet-Leu-Phe stimulates proinflammatory cytokine gene expression in human peripheral blood monocytes: the role of phosphatidylinositol 3-kinase. J Immunol. 2000;164:404–411. doi: 10.4049/jimmunol.164.1.404. [DOI] [PubMed] [Google Scholar]
- 39.Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barber DF, Bartolome A, Hernandez C, Flores JM, Redondo C, Fernandez-Arias C, Camps M, Ruckle T, Schwarz MK, Rodriguez S, Martinez AC, Balomenos D, Rommel C, Carrera AC. PI3Kgamma inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat Med. 2005;11:933–935. doi: 10.1038/nm1291. [DOI] [PubMed] [Google Scholar]
- 41.Camps M, Ruckle T, Ji H, Ardissone V, Rintelen F, Shaw J, Ferrandi C, Chabert C, Gillieron C, Francon B, Martin T, Gretener D, Perrin D, Leroy D, Vitte PA, Hirsch E, Wymann MP, Cirillo R, Schwarz MK, Rommel C. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med. 2005;11:936–943. doi: 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- 42.Davis CB, Dikic I, Unutmaz D, Hill CM, Arthos J, Siani MA, Thompson DA, Schlessinger J, Littman DR. Signal transduction due to HIV-1 envelope interactions with chemokine receptors CXCR4 or CCR5. J Exp Med. 1997;186:1793–1798. doi: 10.1084/jem.186.10.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ganju RK, Brubaker SA, Chernock RD, Avraham S, Groopman JE. Beta-chemokine receptor CCR5 signals through SHP1, SHP2, and Syk. J Biol Chem. 2000;275:17263–17268. doi: 10.1074/jbc.M000689200. [DOI] [PubMed] [Google Scholar]
- 44.Bandyopadhyay G, Sajan MP, Kanoh Y, Standaert ML, Quon MJ, Reed BC, Dikic I, Farese RV. Glucose activates protein kinase C-zeta/lambda through proline-rich tyrosine kinase-2, extracellular signal-regulated kinase, and phospholipase D: a novel mechanism for activating glucose transporter translocation. J Biol Chem. 2001;276:35537–35545. doi: 10.1074/jbc.M106042200. [DOI] [PubMed] [Google Scholar]
- 45.Sajan MP, Bandyopadhyay G, Kanoh Y, Standaert ML, Quon MJ, Reed BC, Dikic I, Farese RV. Sorbitol activates atypical protein kinase C and GLUT4 glucose transporter translocation/glucose transport through proline-rich tyrosine kinase-2, the extracellular signal-regulated kinase pathway and phospholipase D. Biochem J. 2002;362:665–674. doi: 10.1042/0264-6021:3620665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao F, Li P, Chen SR, Louis CF, Fruen BR. Dantrolene inhibition of ryanodine receptor Ca2+ release channels. Molecular mechanism and isoform selectivity. J Biol Chem. 2001;276:13810–13816. doi: 10.1074/jbc.M006104200. [DOI] [PubMed] [Google Scholar]
- 47.Evangelista V, Pamuklar Z, Piccoli A, Manarini S, Dell'elba G, Pecce R, Martelli N, Federico L, Rojas M, Berton G, Lowell CA, Totani L, Smyth SS. Src family kinases mediate neutrophil adhesion to adherent platelets. Blood. 2007;109:2461–2469. doi: 10.1182/blood-2006-06-029082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shin SY, Kim CG, Ko J, Min DS, Chang JS, Ohba M, Kuroki T, Choi YB, Kim YH, Na DS, Kim JW, Lee YH. Transcriptional and post-transcriptional regulation of the PKC delta gene by etoposide in L1210 murine leukemia cells: implication of PKC delta autoregulation. J Mol Biol. 2004;340:681–693. doi: 10.1016/j.jmb.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 49.Marhaba R, Mary F, Pelassy C, Stanescu AT, Aussel C, Breittmayer JP. Tyrphostin A9 inhibits calcium release-dependent phosphorylations and calcium entry via calcium release-activated channel in Jurkat T cells. J Immunol. 1996;157:1468–1473. [PubMed] [Google Scholar]
- 50.Baraldi PG, del Carmen Nunez M, Morelli A, Falzoni S, Di Virgilio F, Romagnoli R. Synthesis and biological activity of N-arylpiperazine-modified analogues of KN-62, a potent antagonist of the purinergic P2X7 receptor. J Med Chem. 2003;46:1318–1329. doi: 10.1021/jm021049d. [DOI] [PubMed] [Google Scholar]
- 51.Ginnan R, Singer HA. CaM kinase II-dependent activation of tyrosine kinases and ERK1/2 in vascular smooth muscle. Am J Physiol Cell Physiol. 2002;282:C754–761. doi: 10.1152/ajpcell.00335.2001. [DOI] [PubMed] [Google Scholar]
- 52.Guo J, Meng F, Fu X, Song B, Yan X, Zhang G. N-methyl-D-aspartate receptor and L-type voltage-gated Ca2+ channel activation mediate proline-rich tyrosine kinase 2 phosphorylation during cerebral ischemia in rats. Neurosci Lett. 2004;355:177–180. doi: 10.1016/j.neulet.2003.10.076. [DOI] [PubMed] [Google Scholar]
- 53.Rodionova E, Conzelmann M, Maraskovsky E, Hess M, Kirsch M, Giese T, Ho AD, Zoller M, Dreger P, Luft T. GSK-3 mediates differentiation and activation of proinflammatory dendritic cells. Blood. 2007;109:1584–1592. doi: 10.1182/blood-2006-06-028951. [DOI] [PubMed] [Google Scholar]
- 54.Cuzzocrea S, Genovese T, Mazzon E, Esposito E, Muia C, Abdelrahman M, Di Paola R, Bramanti P, Thiemermann C. Glycogen synthase kinase-3beta inhibition attenuates the development of bleomycin-induced lung injury. Int J Immunopathol Pharmacol. 2007;20:619–630. doi: 10.1177/039463200702000320. [DOI] [PubMed] [Google Scholar]
- 55.Cuzzocrea S, Mazzon E, Esposito E, Muia C, Abdelrahman M, Di Paola R, Crisafulli C, Bramanti P, Thiemermann C. Glycogen synthase kinase-3beta inhibition attenuates the development of ischaemia/reperfusion injury of the gut. Intensive Care Med. 2007;33:880–893. doi: 10.1007/s00134-007-0595-1. [DOI] [PubMed] [Google Scholar]
- 56.Wyatt R, Sodroski J. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science. 1998;280:1884–1888. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- 57.Arthur LO, Bess JW, Jr., Sowder RC, 2nd, Benveniste RE, Mann DL, Chermann JC, Henderson LE. Cellular proteins bound to immunodeficiency viruses: implications for pathogenesis and vaccines. Science. 1992;258:1935–1938. doi: 10.1126/science.1470916. [DOI] [PubMed] [Google Scholar]
- 58.Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- 59.Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol. 2005;5:629–640. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- 60.Shapira L, Takashiba S, Champagne C, Amar S, Van Dyke TE. Involvement of protein kinase C and protein tyrosine kinase in lipopolysaccharide-induced TNF-alpha and IL-1 beta production by human monocytes. J Immunol. 1994;153:1818–1824. [PubMed] [Google Scholar]
- 61.Stefanova I, Corcoran ML, Horak EM, Wahl LM, Bolen JB, Horak ID. Lipopolysaccharide induces activation of CD14-associated protein tyrosine kinase p53/56lyn. J Biol Chem. 1993;268:20725–20728. [PubMed] [Google Scholar]
- 62.Su SC, Hua KF, Lee H, Chao LK, Tan SK, Lee H, Yang SF, Hsu HY. LTA and LPS mediated activation of protein kinases in the regulation of inflammatory cytokines expression in macrophages. Clin Chim Acta. 2006;374:106–115. doi: 10.1016/j.cca.2006.05.045. [DOI] [PubMed] [Google Scholar]
- 63.Molnarfi N, Gruaz L, Dayer JM, Burger D. Opposite regulation of IL-1beta and secreted IL-1 receptor antagonist production by phosphatidylinositide-3 kinases in human monocytes activated by lipopolysaccharides or contact with T cells. J Immunol. 2007;178:446–454. doi: 10.4049/jimmunol.178.1.446. [DOI] [PubMed] [Google Scholar]
- 64.Hibbs ML, Dunn AR. Lyn, a src-like tyrosine kinase. Int J Biochem Cell Biol. 1997;29:397–400. doi: 10.1016/s1357-2725(96)00104-5. [DOI] [PubMed] [Google Scholar]
- 65.Funaki M, Katagiri H, Inukai K, Kikuchi M, Asano T. Structure and function of phosphatidylinositol-3,4 kinase. Cell Signal. 2000;12:135–142. doi: 10.1016/s0898-6568(99)00086-8. [DOI] [PubMed] [Google Scholar]
- 66.Pleiman CM, Hertz WM, Cambier JC. Activation of phosphatidylinositol-3' kinase by Src-family kinase SH3 binding to the p85 subunit. Science. 1994;263:1609–1612. doi: 10.1126/science.8128248. [DOI] [PubMed] [Google Scholar]
- 67.Seabold GK, Burette A, Lim IA, Weinberg RJ, Hell JW. Interaction of the tyrosine kinase Pyk2 with the N-methyl-D-aspartate receptor complex via the Src homology 3 domains of PSD-95 and SAP102. J Biol Chem. 2003;278:15040–15048. doi: 10.1074/jbc.M212825200. [DOI] [PubMed] [Google Scholar]
- 68.Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B, Schlessinger J. Protein tyrosine kinase PYK2 involved in Ca(2+)-induced regulation of ion channel and MAP kinase functions. Nature. 1995;376:737–745. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- 69.Dikic I, Tokiwa G, Lev S, Courtneidge SA, Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature. 1996;383:547–550. doi: 10.1038/383547a0. [DOI] [PubMed] [Google Scholar]
- 70.Felsch JS, Cachero TG, Peralta EG. Activation of protein tyrosine kinase PYK2 by the m1 muscarinic acetylcholine receptor. Proc Natl Acad Sci U S A. 1998;95:5051–5056. doi: 10.1073/pnas.95.9.5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cogswell JP, Godlevski MM, Wisely GB, Clay WC, Leesnitzer LM, Ways JP, Gray JG. NF-kappa B regulates IL-1 beta transcription through a consensus NF-kappa B binding site and a nonconsensus CRE-like site. J Immunol. 1994;153:712–723. [PubMed] [Google Scholar]
- 72.Bensi G, Mora M, Raugei G, Buonamassa DT, Rossini M, Melli M. An inducible enhancer controls the expression of the human interleukin 1 beta gene. Cell Growth Differ. 1990;1:491–497. [PubMed] [Google Scholar]
- 73.Saijo K, Schmedt C, Su IH, Karasuyama H, Lowell CA, Reth M, Adachi T, Patke A, Santana A, Tarakhovsky A. Essential role of Src-family protein tyrosine kinases in NF-kappaB activation during B cell development. Nat Immunol. 2003;4:274–279. doi: 10.1038/ni893. [DOI] [PubMed] [Google Scholar]
- 74.Reddy SA, Huang JH, Liao WS. Phosphatidylinositol 3-kinase as a mediator of TNF-induced NF-kappa B activation. J Immunol. 2000;164:1355–1363. doi: 10.4049/jimmunol.164.3.1355. [DOI] [PubMed] [Google Scholar]
- 75.Cuschieri J, Gourlay D, Garcia I, Jelacic S, Maier RV. Slow channel calcium inhibition blocks proinflammatory gene signaling and reduces macrophage responsiveness. J Trauma. 2002;52:434–442. doi: 10.1097/00005373-200203000-00004. [DOI] [PubMed] [Google Scholar]