

Abstract
We previously reported that hypoxia-mediated reductions in α-adrenoceptor sensitivity do not explain the augmented vasodilatation during hypoxic exercise, suggesting an enhanced vasodilator signal. We hypothesized that β-adrenoceptor activation contributes to augmented hypoxic exercise vasodilatation. Fourteen subjects (age: 29 ± 2 years) breathed hypoxic gas to titrate arterial O2 saturation (pulse oximetry) to 80%, while remaining normocapnic via a rebreath system. Brachial artery and antecubital vein catheters were placed in the exercising arm. Under normoxic and hypoxic conditions, baseline and incremental forearm exercise (10% and 20% of maximum) was performed during control (saline), α-adrenoceptor inhibition (phentolamine), and combined α- and β-adrenoceptor inhibition (phentolomine/propranolol). Forearm blood flow (FBF), heart rate, blood pressure, minute ventilation, and end-tidal CO2 were determined. Hypoxia increased heart rate (P < 0.05) and minute ventilation (P < 0.05) at rest and exercise under all drug infusions, whereas mean arterial pressure was unchanged. Arterial adrenaline (P < 0.05) and venous noradrenaline (P < 0.05) were higher with hypoxia during all drug infusions. The change (Δ) in FBF during 10% hypoxic exercise was greater with phentolamine (Δ306 ± 43 ml min−1) vs. saline (Δ169 ± 30 ml min−1) or combined phentolamine/propranolol (Δ213 ± 25 ml min−1; P < 0.05 for both). During 20% hypoxic exercise, ΔFBF was greater with phentalomine (Δ466 ± 57 ml min−1; P < 0.05) vs. saline (Δ346 ± 40 ml min−1) but was similar to combined phentolamine/propranolol (Δ450 ± 43 ml min−1). Thus, in the absence of overlying vasoconstriction, the contribution of β-adrenergic mechanisms to the augmented hypoxic vasodilatation is dependent on exercise intensity.
In healthy human subjects, the net effect of exposure to acute hypoxia is a peripheral vasodilatation in several vascular beds, including skeletal muscle (Rowell et al. 1986, 1989; Blauw et al. 1995; Weisbrod et al. 2001; Dinenno et al. 2003). This augmented muscle blood flow occurs in the face of an enhanced sympathetic vasoconstrictor signal (Saito et al. 1988; Somers et al. 1989; Leuenberger et al. 1991) and is not due to a hypoxia-linked blunting of α-adrenoceptor-mediated vasoconstriction (Dinenno et al. 2003; Wilkins et al. 2006). Additionally, pharmacological inhibition of α-adrenergic receptors during hypoxia reveals an even more marked skeletal muscle vasodilatation (Weisbrod et al. 2001), suggesting the enhanced sympathetic outflow acts to limit hypoxic vasodilatation. These observations imply that enhanced release of vasodilator substances, such as adenosine (MacLean et al. 1998; Leuenberger et al. 1999) and adrenaline (Blauw et al. 1995; Weisbrod et al. 2001), probably contribute to hypoxic vasodilatation in resting human skeletal muscle.
During hypoxic exercise, muscle blood flow is augmented relative to the same level of exercise under normoxic conditions (Hartley et al. 1973; Rowell et al. 1986; Mazzeo et al. 1995; Calbet et al. 2003; Wilkins et al. 2006). This enhanced hypoxic exercise hyperaemia is proportional to the hypoxia-induced fall in arterial oxygen content, enabling maintenance of muscle oxygen delivery and consumption during hypoxic exercise. Similar to resting conditions, we recently reported that the augmented muscle blood flow during hypoxic exercise was not explained by blunted α-adrenoceptor vasoconstriction (Wilkins et al. 2006), suggesting a role for an enhanced vasodilator signal. Along these lines, we observed an exercise intensity-dependent rise in arterial adrenaline, suggesting that β-adrenergic receptor-mediated vasodilatation might contribute to the augmented hypoxic exercise hyperaemia.
With this information as background, we designed an experiment to test the hypothesis that there can be marked β-adrenoceptor-mediated vasodilatation during hypoxic forearm exercise. Because the enhanced sympathetic outflow with systemic hypoxia may mask vasodilator responses, we removed this vasoconstrictor influence via non-specific α-adrenoceptor blockade. This approach allowed for the isolation of β-adrenoceptor-mediated vasodilatation in the absence of any competing vasoconstrictor tone.
Methods
Institutional review board approval was obtained and subjects gave informed, written consent prior to participation. All studies were performed according to the Declaration of Helsinki.
Subjects
Three female subjects (mean age 26 ± 4 years) and 11 male subjects (mean age 30 ± 2 years) volunteered to participate in this study. Subjects underwent a standard health screening and were healthy, non-obese, non-smokers, and were not taking any medications (except for oral contraceptives in some women). Subjects arrived in the laboratory at least 4 h postprandial after refraining from exercise or caffeine for at least 24 h. Female subjects were studied during the early follicular phase of the menstrual cycle or the placebo phase of oral contraceptives (Minson et al. 2000a,b).
Arterial and venous catheterization
For administration of study drugs and to obtain arterial blood samples, a 20 gauge, 5 cm catheter was placed in the brachial artery of the exercising arm under aseptic conditions after local anaesthesia (2% lidocaine (lignocaine)). The catheter was connected to a three-port connector in series, as previously described in detail (Dietz et al. 1994). One port was linked to a pressure transducer to allow measurement of arterial pressure and was continuously flushed (3 ml h−1) with heparinized saline with a stop-cock system to enable arterial blood sampling. The remaining two ports allowed arterial drug administration. Deep venous blood was sampled via an 18 gauge, 3 cm catheter inserted retrograde in an antecubital vein (Joyner et al. 1992).
Forearm blood flow
Brachial artery mean blood velocity and brachial artery diameter were determined with a 12 MHz linear-array Doppler probe (12 MHz linear array, Model M12L, Vivid 7, General Electric, Milwaukee, WI, USA). Brachial artery blood velocity was measured throughout each condition with a probe insonation angle previously calibrated to 60 deg. Brachial artery diameter measurements were obtained at end-diastole and between contractions during steady-state conditions (rest or exercise). Diameter measurement typically results in the loss of the pulse wave signal for 15–20 s. Forearm blood flow (FBF) was calculated by multiplying mean blood velocity (cm s−1) by brachial artery cross-sectional area (cm2) then multiplied by 60 to present values as millilitres per minute.
Rhythmic forearm exercise
Forearm exercise was performed with a hand grip device by the non-dominant arm at 10% and 20% of each subject/s maximal voluntary contraction (MVC, mean 45 ± 4 kg, range 25–64 kg), determined at the beginning of each experiment. The weight was lifted 4–5 cm over a pulley at a duty cycle of 1 s contraction/2 s relaxation (20 contractions per minute) using a metronome to ensure correct timing. The average weight used for forearm exercise was 4.5 ± 0.3 and 9.1 ± 0.6 kg for 10% and 20% MVC, respectively.
Systemic hypoxia
To isolate the effects of systemic normocapnic hypoxia in modulating skeletal muscle blood flow we adapted a self-regulating partial rebreath system developed by Banzett et al. (2000). This method maintains constant alveolar fresh air ventilation independent of changes in breathing frequency or tidal volume and allowed us to clamp end-tidal CO2 despite large changes in minute ventilation in response to hypoxia (Banzett et al. 2000; Weisbrod et al. 2001; Wilkins et al. 2006). The amount of oxygen provided in the inspiratory gas was controlled by mixing N2 with medical air via an anaesthesia gas blender. During the hypoxic condition, the level of inspired O2 was titrated to achieve an arterial O2 saturation (assessed via pulse oximetry) of 80%. Subjects breathed on a scuba mouthpiece with a nose clip to prevent nasal breathing. Carbon dioxide concentrations were monitored by an anaesthesia monitor (Cardiocap/5, Datex-Ohmeda, Louisville, CO, USA) and ventilation was assessed via a pneumotach (model VMM-2a, Interface Associates, Laguna Nigel, CA, USA).
Pharmacological infusions
Phentolamine, a non-selective α-adrenoceptor antagonist, was administered to the exercising forearm via the brachial artery catheter as a loading dose (10 μg (dl forearm volume)−1 min−1 for 5 min) followed by a continuous maintenance dose (25 μg min−1). This dose of phentolamine has been shown to effectively inhibit α-receptor vasoconstriction (Eklund & Kaijser, 1976). Propranolol, a non-selective β-adrenergic receptor antagonist, was administered to the forearm via the brachial catheter as a loading dose (20 μg (dl forearm volume)−1 min−1 for 5 min) followed by a continuous maintenance dose (25 μg min−1). This dose of propranolol has been shown to block forearm vasodilatation to the β-adrenoceptor agonist isoproterenol (isoprenaline) (Johnsson, 1967; Eklund & Kaijser, 1976).
Blood gas and catecholamine analysis
Brachial artery and deep venous blood samples were analysed with a clinical blood gas analyser (Bayer 855 Automatic Blood Gas System, Boston, MA, USA) for partial pressures of O2 and CO2 (PO2 and PCO2), O2 content, pH, and O2 saturation (SO2). Arterial and venous plasma catecholamine (adrenaline (epinephrine) and noradrenaline (norepinephrine)) levels were determined by HPLC with electrochemical detection.
Experimental protocol
A schematic diagram of the general experimental design is depicted in Fig. 1. Each subject completed a resting baseline condition (5 min) followed by rhythmic forearm exercise at 10% (5 min) which was immediately increased to 20% (5 min) during normoxia and normocapnic hypoxia. Exposure to normoxia or hypoxia was alternated and randomized. Resting baseline and each exercise intensity (normoxia and hypoxia) was performed during a control (saline) infusion, followed by phentolamine infusion, followed by infusion of combined phentolamine and propranolol. Due to the long half-lives, study drugs were always administered in the same order. A rest period of at least 20 min was allowed between conditions under each drug infusion. During each infusion (saline, phentolamine, phentolamine/propranolol) and each condition (normoxia and hypoxia) arterial and venous blood was sampled at rest and at steady-state exercise for blood gas analysis and plasma catecholamine determination (Fig. 1).
Figure 1. Schematic diagram of experimental protocol.

Measurements were obtained at baseline and incremental exercise (10% and 20% of maximum) under normoxic and hypoxic conditions. Protocol was performed during control (saline), phentolamine alone, and combined phentolamine/propranolol infusions.
Data analysis and statistics
Data were collected and stored on a computer at 200 Hz and analysed off-line using signal-processing software (Widaq, DATAQ Instruments, Akron, OH, USA). Mean arterial pressure was determined from the brachial artery pressure waveform and heart rate was determined from the electrocardiogram. Values for minute ventilation, end-tidal CO2, and O2 saturation (pulse oximetry) were determined by averaging minutes 4 and 5 at rest and each exercise bout. Forearm blood flow and mean arterial pressure were determined by averaging values from the 4th minute at rest and each exercise bout. Forearm vascular conductance (FVC) was calculated by dividing FBF by mean arterial pressure and expressed as ml min−1 (100 mmHg)−1. Due to the altered baseline blood flow with drug infusion, the change (Δ) in FBF and FVC due to hypoxia at rest and due to hypoxic exercise (10% and 20%) was calculated by subtracting resting FBF and FVC during normoxia at each drug infusion (saline, phentolamine, or combined phentolamine/propranolol) from FBF and FVC values obtained during hypoxia (at rest and during exercise) within each drug infusion. Blood gas and catecholamine values were determined from blood samples obtained during normoxia and hypoxia with each drug infusion. Arteriovenous oxygen difference during forearm exercise was calculated by the difference between arterial and venous O2 content.
All values are expressed as means ±s.e.m. To determine the effect of hypoxia with each pharmacological treatment, differences in absolute FBF and FVC at rest (normoxia and hypoxia) and differences in ΔFBF and ΔFVC at rest and during each exercise intensity (normoxia and hypoxia) were determined via repeated measures analysis of variance (ANOVA). Haemodynamic, respiratory, blood gases and catecholamine variables were compared via repeated measures ANOVA to detect differences between responses during hypoxia at rest and during exercise across pharmacological infusions. Appropriate post hoc analysis was used to determine where statistical differences occurred. Statistical difference was set a priori at P < 0.05.
Results
Twelve of the 14 subjects completed the study protocol. Those subjects who did not complete the protocol had symptoms of vasovagal syncope (precipitous fall in blood pressure and heart rate) at some point during a hypoxic condition. Occurrence of symptoms did not coincide with any specific drug infusion (phentolamine or propranolol) and data collected from these subjects were not included in the group analysis. Those subjects completing the protocol were 29 ± 2 years of age, 178 ± 3 cm in height, and weighed 78 ± 3 kg (BMI: 25 ± 1 kg m−2).
Systemic haemodynamic and respiratory responses (Table 1)
Table 1.
Systemic haemodynamic and respiratory responses at rest and increasing exercise intensity during normoxia and hypoxia with each drug infusion
| Normoxia | Hypoxia | |||||
|---|---|---|---|---|---|---|
| Rest | 10% | 20% | Rest | 10% | 20% | |
| Control (no drug) | ||||||
| Mean arterial pressure (mmHg) | 86 ± 2 | 88 ± 2 | 90 ± 2 | 86 ± 2 | 88 ± 2 | 90 ± 2 |
| Heart rate (beats min−1) | 62 ± 3 | 67 ± 3 | 69 ± 3 | 77 ± 4* | 77 ± 4* | 80 ± 5* |
| Minute ventilation (l min−1; BTPS) | 7.1 ± 0.3 | 7.9 ± 0.4 | 8.4 ± 0.5† | 10.5 ± 1.1* | 12.1 ± 1.4* | 13.6 ± 2.2*† |
| End-tidal CO2 (%) | 5.4 ± 0.1 | 5.4 ± 0.1 | 5.5 ± 0.1 | 5.4 ± 0.1 | 5.4 ± 0.1 | 5.4 ± 0.1 |
| O2 saturation (pulse ox.) (%) | 99 ± 0 | 99 ± 0 | 99 ± 0 | 79 ± 1* | 79 ± 0* | 80 ± 1* |
| O2 consumption (ml min−1) | 3.0 ± 0.4 | 19.1 ± 2.8† | 37.7 ± 5.2†‡ | 4.4 ± 0.6 | 20.0 ± 2.9† | 34.6 ± 4.6†‡ |
| Phentolamine | ||||||
| Mean arterial pressure (mmHg) | 85 ± 2 | 87 ± 2 | 88 ± 2 | 85 ± 2 | 86 ± 2 | 87 ± 2 |
| Heart rate (beats min−1) | 62 ± 3 | 66 ± 3 | 69 ± 3 | 81 ± 3* | 81 ± 4* | 82 ± 5* |
| Minute ventilation (l min−1; BTPS) | 7.9 ± 0.6 | 8.9 ± 0.6 | 9.5 ± 0.7† | 14.8 ± 1.4* | 15.7 ± 1.1* | 17.4 ± 1.6*† |
| End-tidal CO2 (%) | 5.4 ± 0.1 | 5.4 ± 0.1 | 5.4 ± 0.1 | 5.3 ± 0.1 | 5.4 ± 0.1 | 5.3 ± 0.1 |
| O2 saturation (pulse ox.) (%) | 99 ± 0 | 99 ± 0 | 99 ± 0 | 80 ± 1* | 80 ± 0* | 81 ± 0* |
| O2 consumption (ml min−1) | 4.5 ± 0.9 | 20.9 ± 3.0† | 43.1 ± 5.8†‡ | 4.3 ± 0.0.8 | 20.0 ± 4.4† | 37.0 ± 6.3†‡ |
| Phentolamine/propranolol | ||||||
| Mean arterial pressure (mmHg) | 86 ± 2 | 87 ± 3 | 91 ± 2 | 87 ± 3 | 88 ± 3 | 90 ± 3 |
| Heart rate (beats min−1) | 59 ± 3 | 63 ± 3 | 65 ± 3 | 74 ± 3* | 74 ± 3* | 75 ± 4* |
| Minute ventilation (l min−1; BTPS) | 8.5 ± 0.9 | 9.5 ± 1.0 | 10.9 ± 1.1† | 15.0 ± 1.9* | 13.8 ± 1.5* | 15.0 ± 2.0* |
| End-tidal CO2 (%) | 5.4 ± 0.1 | 5.4 ± 0.1 | 5.4 ± 0.1 | 5.4 ± 0.1 | 5.3 ± 0.1 | 5.4 ± 0.1 |
| O2 saturation (pulse ox.) (%) | 98 ± 0 | 98 ± 0 | 99 ± 0 | 80 ± 0* | 80 ± 0* | 80 ± 0* |
| O2 consumption (ml min−1) | 5.0 ± 1.2 | 33.6 ± 6.9† | 49.9 ± 7.3†‡ | 7.7 ± 1.4 | 29.9 ± 6.1† | 53.1 ± 5.3†‡ |
Values are means ±s.e.m.
Main effect of hypoxia, P < 0.05 vs. normoxia
P < 0.05 vs. rest
P < 0.05 vs. 10% exercise.
During control (saline), phentolamine alone and combined phentolamine/propranolol infusions, systemic normocapnic hypoxia increased heart rate at rest and with each exercise intensity (P < 0.01). Infusion of combined phentolamine/propranolol did not statistically decrease heart rate during normoxia and hypoxia relative to saline and phentolamine infusions (P = 0.16) and the rise in heart rate due to systemic hypoxia was similar (Table 1). There was no difference in mean arterial pressure during normoxic and hypoxic conditions. In addition, drug infusions during forearm exercise did not differentially affect the mean arterial pressure response (Table 1).
We accomplished our goal of maintaining O2 saturation at ∼80%, monitored via pulse oximetry, at rest and with incremental exercise. During control (saline), phentolamine, and combined phentolamine/propranolol infusions, minute ventilation was higher with systemic hypoxia at rest and with increasing exercise intensity (P < 0.01). Despite the large increases in minute ventilation, we were able to maintain end-tidal CO2 at rest and with increasing exercise intensity under normoxic and hypoxic conditions (P = 0.46, Table 1).
Although no statistical main effect was observed (P = 0.14), oxygen consumption was higher at rest and during hypoxic exercise with combined α- and β-receptor blockade (Table 1). This is unlikely to be due to an elevated workload relative to the other conditions, and is more likely to be due to restricted venous sampling. That is, sampling from one deep vein during a pharmacologically induced over-perfusion may not reflect the mean arteriovenous (a–v) O2 difference of the whole exercising forearm.
Blood gases (Table 2)
Table 2.
Arterial and venous blood gas responses at rest and with incremental exercise during normoxia and hypoxia under each drug condition
| Normoxia | Hypoxia | |||||
|---|---|---|---|---|---|---|
| Rest | 10% | 20% | Rest | 10% | 20% | |
| Control (no drug) | ||||||
| [Hb]a (g dl−1) | 13.9 ± 0.4 | 14.2 ± 0.3 | 14.2 ± 0.4 | 14.1 ± 0.4 | 14.1 ± 0.4 | 14.3 ± 0.5 |
| SaO2 (%) | 97 ± 0 | 97 ± 0 | 97 ± 0 | 80 ± 1* | 82 ± 1* | 81 ± 1* |
| PaO2 (Torr) | 101 +3 | 107 ± 7 | 105 ± 3 | 45 ± 1* | 48 ± 1* | 49 ± 2* |
| PaO2 (Torr) | 38 ± 3 | 25 ± 1† | 25 ± 1† | 27 ± 2* | 24 ± 2* | 23 ± 1*† |
| Arterial O2 content (ml l−1) | 190 ± 5 | 194 ± 5 | 195 ± 5 | 158 ± 4* | 163 ± 5* | 164 ± 4* |
| Venous O2 content (ml l−1) | 125 ± 11 | 77 ± 7† | 73 ± 4† | 86 ± 9* | 63 ± 7*† | 64 ± 5*† |
| a–v O2 (ml l−1) | 65 ± 10 | 117 ± 6† | 121 ± 5† | 72 ± 9 | 99 ± 8*† | 102 ± 5*† |
| PaCO2 (Torr) | 40 ± 1 | 40 ± 1 | 41 ± 1 | 38 ± 1 | 39 ± 1 | 40 ± 1 |
| PvCO2 (Torr) | 48 ± 1 | 55 ± 1† | 61 ± 1‡ | 48 ± 2 | 53 ± 1*† | 58 ± 2*‡ |
| pHa | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 |
| pHv | 7.4 ± 0.0 | 7.3 ± 0.0 | 7.3 ± 0.0 | 7.4 ± 0.0 | 7.3 ± 0.0 | 7.3 ± 0.0 |
| Phentolamine | ||||||
| [Hb]a (g dl−1) | 14.2 ± 0.5 | 14.2 ± 0.5 | 14.3 ± 0.5 | 14.2 ± 0.3 | 14.2 ± 0.3 | 14.4 ± 0.3 |
| SaO2 (%) | 97 ± 0 | 97 ± 0 | 97 ± 0 | 81 ± 1* | 82 ± 0* | 83 ± 1* |
| PaO2 (Torr) | 102 ± 3 | 104 ± 3 | 103 ± 2 | 45 ± 1* | 47 ± 0* | 49 ± 1* |
| PvO2 (Torr)§ | 54 ± 2 | 36 ± 1† | 32 ± 1‡ | 40 ± 1* | 30 ± 1*† | 28 ± 1*† |
| Arterial O2 content (ml l−1) | 194 ± 6 | 194 ± 6 | 196 ± 6 | 161 ± 4* | 163 ± 4* | 167 ± 4* |
| Venous O2 content (ml l−1)§ | 173 ± 6 | 127 ± 5† | 100 ± 6† | 140 ± 8* | 102 ± 7*† | 87 ± 6*† |
| a-v O2 (ml l−1)§ | 21 ± 2 | 67 ± 5† | 96 ± 7† | 13 ± 2* | 61 ± 7*† | 80 ± 7*† |
| PaCO2 (Torr) | 40 ± 1 | 41 ± 1 | 40 ± 0 | 39 ± 1 | 39 ± 0 | 40 ± 1 |
| PvCO2 (Torr)§ | 43 ± 1 | 49 ± 1† | 53 ± 1‡ | 42 ± 1 | 46 ± 1*† | 50 ± 1*‡ |
| pHa | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 |
| pHv§ | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.3 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 |
| Phentolamine/propranolol | ||||||
| [Hb]a (g dl−1) | 13.8 ± 0.5 | 14.0 ± 0.4 | 14.1 ± 0.4 | 14.2 ± 0.4 | 14.2 ± 0.4 | 14.3 ± 0.3 |
| SaO2 (%) | 97 ± 0 | 97 ± 0 | 97 ± 0 | 82 ± 1* | 81 ± 1* | 82 ± 1* |
| PaO2 (Torr) | 97 ± 3 | 102 ± 2 | 100 ± 2 | 47 ± 1* | 47 ± 1* | 49 ± 1* |
| PvO2 (Torr)§ | 55 ± 3 | 35 ± 3† | 31 ± 2† | 38 ± 1* | 28 ± 2*† | 27 ± 2*† |
| Arterial O2 content (ml l−1) | 186 ± 7 | 190 ± 6 | 192 ± 6 | 162 ± 5* | 160 ± 4* | 166 ± 4* |
| Venous O2 content (ml l−1)§ | 167 ± 8 | 107 ± 8† | 101 ± 7† | 138 ± 6* | 94 ± 9*† | 90 ± 8*† |
| a–v O2 (ml l−1)§ | 21 ± 3 | 83 ± 7† | 91 ± 7† | 24 ± 3 | 66 ± 8*† | 76 ± 8*† |
| PaCO2 (Torr) | 39 ± 1 | 40 ± 0 | 40 ± 1 | 39 ± 0 | 39 ± 0 | 40 ± 0 |
| PvCO2 (Torr)§ | 42 ± 1 | 48 ± 1† | 54 ± 1‡ | 41 ± 1 | 46 ± 1*† | 52 ± 2*‡ |
| pHa | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 |
| pHv§ | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.3 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.3 ± 0.0 |
Values are means ±s.e.m.
Main effect of hypoxia, P < 0.05 vs. normoxia
P <0.05 vs. rest
P < 0.05 vs. 10% exercise; § main effect of drug, P < 0.05 vs. control.
As expected, systemic hypoxia reduced arterial PO2 (main effect, P < 0.01) and arterial O2 content (main effect, P < 0.01) at rest and with forearm exercise during all drug infusions. Acute hypoxia also decreased venous O2 content (P < 0.05) during each drug infusion, suggesting greater extraction of O2 with exercise under hypoxic conditions. Due to the lower arterial and venous O2 content during hypoxic exercise, the a–v O2 difference was substantially reduced (P < 0.01) during each drug infusion, compared to normoxic exercise. Because blood flow was higher during phentolamine and combined phentolamine/propranolol infusions, values (resting and exercise) for venous O2 content were higher (P < 0.05) and values for a–v O2 difference were lower (P < 0.05). Arterial PCO2 (P = 0.63) and arterial pH (P = 0.85) were similar at rest and with incremental exercise under all drug infusions (Table 2).
Catecholamines (Table 3)
Table 3.
Venous noradrenaline and arterial adrenaline at rest and increasing exercise intensity during normoxia and hypoxia with each drug infusion
| Normoxia | Hypoxia | |||||
|---|---|---|---|---|---|---|
| Rest | 10% | 20% | Rest | 10% | 20% | |
| Control (saline) | ||||||
| Arterial noradrenaline (pg ml−1) | 129 ± 14 | 116 ± 10 | 136 ± 13 | 141 ± 15* | 147 ± 17* | 149 ± 13* |
| Venous noradrenaline (pg ml−1) | 176 ± 6 | 168 ± 4 | 180 ± 5 | 251 ± 9* | 203 ± 8*† | 194 ± 6*† |
| v–a noradrenaline difference (pg ml−1) | 38 ± 12 | 35 ± 5 | 33 ± 7 | 99 ± 20* | 54 ± 12*† | 35 ± 9*† |
| Arterial adrenaline (pg ml−1) | 43 ± 7 | 51 ± 5 | 49 ± 5 | 64 ± 9* | 73 ± 10* | 86 ± 18* |
| Venous adrenaline (pg ml−1) | 16 ± 1 | 27 ± 1 | 38 ± 2 | 23 ± 1* | 52 ± 3*† | 71 ± 5*† |
| Phentolamine | ||||||
| Arterial noradrenaline (pg ml−1) | 171 ± 16 | 167 ± 20 | 175 ± 21 | 179 ± 30 | 200 ± 26 | 214 ± 23 |
| Venous noradrenaline (pg ml−1)§ | 282 ± 5 | 261 ± 11 | 248 ± 8 | 297 ± 13 | 270 ± 9 | 262 ± 10 |
| v–a noradrenaline difference (pg ml−1) | 93 ± 12 | 79 ± 21 | 58 ± 11† | 85 ± 17 | 56 ± 8† | 57 ± 8† |
| Arterial adrenaline (pg ml−1) | 42 ± 5 | 54 ± 6 | 60 ± 6 | 74 ± 10* | 76 ± 5* | 95 ± 15* |
| Venous adrenaline (pg ml−1) | 27 ± 1 | 35 ± 1 | 45 ± 2 | 47 ± 3* | 53 ± 2*† | 71 ± 4*† |
| Phentolamine/propranolol | ||||||
| Arterial noradrenaline (pg ml−1) | 176 ± 21 | 180 ± 21 | 192 ± 17 | 166 ± 19 | 167 ± 18 | 175 ± 20 |
| Venous noradrenaline (pg ml−1)§ | 261 ± 9 | 277 ± 9 | 229 ± 7 | 258 ± 9 | 242 ± 9 | 217 ± 7 |
| v–a noradrenaline difference (pg ml−1) | 78 ± 16 | 76 ± 10 | 42 ± 9† | 83 ± 14 | 68 ± 14 | 34 ± 18† |
| Arterial adrenaline (pg ml−1)§‡ | 67 ± 8 | 80 ± 9 | 98 ± 11† | 136 ± 20* | 163 ± 19*† | 167 ± 26*† |
| Venous adrenaline (pg ml−1)§‡ | 47 ± 2 | 64 ± 4 | 76 ± 4† | 88 ± 5* | 123 ± 4*† | 137 ± 8*† |
Values are means ±s.e.m.
Main effect of hypoxia, P < 0.05 vs. normoxia
P < 0.05 vs. rest
main effect of drug, P < 0.05 vs. control
main effect of drug, P < 0.05 vs. phentolamine.
Systemic hypoxia increased venous noradrenaline levels (P < 0.01). Venous noradrenaline decreased with hypoxic exercise intensity (P < 0.05), but remained higher than the same intensity of normoxic exercise. Despite higher venous noradrenaline with phentolamine and combined phentolamine/propranolol infusions (main effect; P < 0.01), acute hypoxia did not further increase noradrenaline spillover (P = 0.48). During the control (saline) infusion, systemic hypoxia increased arterial adrenaline at rest and during exercise (P < 0.01). Arterial adrenaline increased with hypoxic exercise intensity, but this increase was not statistically significant (P = 0.10). Hypoxia also increased arterial adrenaline concentration (P < 0.01) with phentolamine infusion. Combined phentolamine/propranolol infusion increased arterial adrenaline at rest and during incremental exercise under normoxic conditions (P < 0.05). Despite the higher normoxic adrenaline levels, hypoxia increased circulating adrenaline during combined phentolamine/propranolol infusion (Table 3, P < 0.05).
Forearm vasodilatation
Presented in Table 4 are mean ±s.e.m. forearm haemodynamics at rest and increasing exercise intensity during normoxia and hypoxia with each drug infusion. Systemic isocapnic hypoxia increased resting FBF and FVC during all drug infusions (main effect, P < 0.01; Table 4). Figure 2 shows the change (Δ) in FBF (Fig. 2A) and FVC (Fig. 2B) due to hypoxia at rest. During the control saline infusion, resting FBF under normoxic conditions was 62 ± 11 ml min−1 and FVC was 73 ± 12 ml min−1 (100 mmHg)−1. Hypoxia increased resting FBF to 76 ± 13 ml min−1 (Δ13 ± 6 ml min−1) and increased FVC to 89 ± 16 ml min−1 (100 mmHg)−1 (Δ16 ± 8 ml min−1 (100 mmHg)−1). Infusion of phentolamine increased resting FBF under the normoxic condition to 224 ± 20 ml min−1 (P < 0.01 vs. saline) and FVC increased to 265 ± 24 ml min−1 (100 mmHg)−1 (P < 0.01 vs. saline). Hypoxia further increased resting FBF to 378 ± 39 ml min−1 (Δ154 ± 27 ml min−1) and FVC to 448 ± 46 ml min−1 (100 mmHg)−1 (Δ183 ± 31 ml min−1 (100 mmHg)−1). The change in resting FBF (Fig. 2A) and FVC (Fig. 2B) due to systemic hypoxia during phentolamine infusion was significantly greater than during the control (saline) infusion (P < 0.01).
Table 4.
Forearm haemodynamics at rest and increasing exercise intensity during normoxia and hypoxia with each drug condition. Δ from normoxia rest: absolute change from normoxia rest within each drug infusion
| Δ from normoxia rest | |||||
|---|---|---|---|---|---|
| Rest | 10% | 20% | 10% | 20% | |
| Forearm blood flow (ml min−1) | |||||
| Control | |||||
| Normoxia† | 62 ± 11 | 189 ± 23 | 350 ± 40 | 126 ± 16 | 287 ± 35 |
| Hypoxia*† | 76 ± 13 | 231 ± 39 | 409 ± 46 | 169 ± 30 | 346 ± 40 |
| Phentolamine | |||||
| Normoxia† | 224 ± 20‡ | 356 ± 31‡ | 502 ± 44‡ | 131 ± 21 | 286 ± 41 |
| Hypoxia*† | 378 ± 39‡ | 509 ± 46‡ | 669 ± 33‡ | 306 ± 43‡ | 466 ± 57‡ |
| Phentolamine/propranolol | |||||
| Normoxia† | 280 ± 40‡ | 420 ± 51‡ | 593 ± 61‡ | 139 ± 25 | 312 ± 45 |
| Hypoxia*† | 382 ± 46‡ | 494 ± 50‡ | 751 ± 42‡§ | 213 ± 25§ | 450 ± 43‡ |
| Forearm vascular conductance (ml min−1 (100 mmHg)−1) | |||||
| Control | |||||
| Normoxia† | 73 ± 12 | 213 ± 26 | 387 ± 43 | 139 ± 18 | 314 ± 38 |
| Hypoxia*† | 89 ± 16 | 263 ± 43 | 450 ± 47 | 190 ± 32 | 377 ± 40 |
| Phentolamine | |||||
| Normoxia† | 265 ± 24‡ | 411 ± 36‡ | 570 ± 53‡ | 147 ± 25 | 315 ± 50 |
| Hypoxia*† | 448 ± 46‡ | 591 ± 55‡ | 759 ± 69‡ | 354 ± 53‡ | 521 ± 63‡ |
| Phentolamine/propranolol | |||||
| Normoxia† | 325 ± 47‡ | 477 ± 58‡ | 652 ± 65‡ | 152 ± 29 | 326 ± 51 |
| Hypoxia*† | 443 ± 55‡ | 565 ± 54‡ | 825 ± 52‡ | 240 ± 27§ | 479 ± 53‡ |
Values are mean ±s.e.m.
Main effect of hypoxia, P < 0.05
main effect of exercise, P < 0.05
P < 0.05 vs. control
P < 0.05 vs. phentolamine.
Figure 2. Change (Δ) in forearm blood flow (FBF; A) and forearm vascular conductance (FVC; B) due to hypoxia at rest.

Blockade of α-adrenergic receptors (phentolamine) revealed a substantial resting hypoxic vasodilatation. Combined α- and β-adrenergic receptor blockade (phentolamine/propranolol) reduced hypoxic vasodilatation compared to α-adrenergic blockade alone. *P < 0.01 vs. control (saline); †P < 0.05 vs. phentolamine.
During the combined phentolamine/propranolol infusion, resting FBF under the normoxic condition was 280 ± 40 ml min−1 (P < 0.01 vs. saline and P = 0.17 vs. phentolamine) and FVC was 325 ± 47 ml min−1 (100 mmHg)−1 (P < 0.01 vs. saline and P = 0.14 vs. phentolamine). Hypoxia increased resting FBF to 382 ± 46 ml min−1 (Δ102 ± 24 ml min−1 (100 mmHg)−1) and increased FVC to 443 ± 55 ml min−1 (100 mmHg)−1 (Δ118 ± 30 ml min−1 (100 mmHg)−1). The combination of phentolamine/propranolol reduced the hypoxia-induced change in resting FBF (Fig. 2A) and FVC (Fig. 2B) compared to phentolamine alone (P < 0.05). However, the hypoxia-induced changes in resting FBF (Fig. 2A) and FVC (Fig. 2B) remained greater than the control (saline) infusion (P < 0.01).
Figure 3A–D shows the group data for the changes (Δ) in FBF and FVC (relative to normoxic baseline values during each respective drug infusion) with exercise at 10% and 20% MVC, under normoxic and hypoxic conditions, during each infusion. The ΔFBF (Fig. 3A) and ΔFVC (Fig. 3B) was greater during hypoxic exercise at 10% MVC compared to the normoxic exercise of the same intensity (main effect of hypoxia, P < 0.01). Phentolamine infusion did not affect ΔFBF and ΔFVC during normoxic exercise at 10%. However, during phentolamine infusion, ΔFBF and ΔFVC was significantly greater than that observed during 10% hypoxic exercise with the saline infusion (P < 0.01, Fig. 3A and B). Combined phentolamine/propranolol infusion did not affect ΔFBF or ΔFVC during normoxic exercise at 10% MVC (Fig. 3A and B). However, the addition of propranolol (combined phentolamine/propranolol) decreased ΔFBF and ΔFVC with hypoxic exercise at 10% MVC compared to phentolamine alone (P < 0.05, Fig. 3A and B).
Figure 3. Change (Δ) in forearm blood flow (FBF) and forearm vascular conductance (FVC) during exercise at 10% (A and B) and 20% (C and D).

At 10% forearm exercise, blockade of α-adrenergic receptors (phentolamine) revealed a substantial hypoxic exercise vasodilatation (A and B) which was reduced with combined α- and β-adrenergic receptor blockade (phentolamine/propranolol). At 20% forearm exercise, blockade of α-adrenergic receptors revealed a substantial hypoxic exercise vasodilatation. However, combined α- and β-adrenergic receptor blockade did not reduced forearm blood flow (C) or vascular conductance (D) compared to phentolamine alone. Neither phentolamine nor combined phentolamine/propranolol had an affect on vasodilatation during normoxic exercise. * Main effect of hypoxia, P < 0.01 vs. normoxia; †P < 0.05 vs. control (saline); ‡P < 0.05 vs. phentolamine.
Neither phentolamine alone nor combined phentolamine/propranolol affected ΔFBF or ΔFVC during normoxic exercise at 20% MVC (Fig. 3C and D). Similar to exercise at 10% MVC, ΔFBF (Fig. 3C) and ΔFVC (Fig. 3D) due to hypoxic exercise at 20% MVC was greater than the same normoxic exercise intensity (main effect of hypoxia, P < 0.01). During phentolamine infusion, ΔFBF and ΔFVC was greater than hypoxic exercise at 20% during saline infusion (P < 0.01). In contrast to hypoxic exercise at 10% MVC, combined phentolamine/propranolol infusion did not statistically decrease ΔFBF or ΔFVC during hypoxic exercise at 20% MVC compared to values observed with phentolamine infusion alone (P = 0.44 for ΔFBF; P = 0.39 for ΔFVC; Fig. 3C and D).
Discussion
The primary novel findings from this study were: (i) the augmented hypoxic exercise hyperaemia is mediated by β-adrenergic mechanisms during mild forearm exercise, (ii) the β-adrenergic component of the augmented hypoxic vasodilatation decreases with increased exercise intensity, and (iii) similar to findings under resting conditions (Weisbrod et al. 2001), α-adrenergic receptor blockade reveals a marked vasodilatation during hypoxic forearm exercise. Thus, in the absence of overlying sympathetic vasoconstriction, the β-adrenergic component of the augmented hypoxic vasodilatation observed at rest remained evident during hypoxic exercise at 10% MVC, but was absent during hypoxic exercise at 20% MVC (Fig. 3).
Results from the current study and previous reports (Weisbrod et al. 2001) demonstrate the important contribution of enhanced systemic adrenaline release as a signal for vasodilatation under hypoxic conditions at rest and during hypoxic exercise. Despite the graded release of adrenaline with incremental hypoxic forearm exercise (Wilkins et al. 2006; Table 3), the direct contribution for adrenaline to the augmented hypoxic exercise hyperaemia decreases with exercise intensity (Fig. 3). This suggests that the contribution of a local vasodilator signal (i.e. adenosine, NO, prostaglandins, or ATP) increases with exercise intensity to ensure appropriate matching of oxygen delivery to the demand. Along these lines, during high intensity large muscle mass exercise the increased sympathetic nerve activity may act to ensure optimal matching of perfusion and metabolism (Calbet et al. 2006). Thus, α-receptor blockade may limit this precise matching of blood flow and metabolism during hypoxic exercise by artificially increasing blood flow and decreasing mean capillary transit time, ‘diverting’ blood flow from the most metabolically active areas of the contracting muscles. During the conditions set in the current study (i.e. submaximal forearm exercise), a–v oxygen difference decreased during both 10% and 20% forearm exercise with α-receptor blockade, maintaining oxygen consumption. This decrease in a–v oxygen difference was probably due to the over-perfusion of exercising muscle during the submaximal hypoxic exercise with α-receptor blockade. It is important to note, that despite the over-perfusion of the exercising muscle, the β-mediated portion of the augmented hypoxic exercise hyperaemia remained evident at rest and low exercise intensities.
Adrenaline and augmented hypoxic vasodilatation
Rowell et al. (1986) reported significantly higher arterial adrenaline levels with incremental hypoxic leg exercise, including values obtained at maximal hypoxic exercise. In addition, we recently reported higher arterial adrenaline values during incremental hypoxic forearm exercise (Wilkins et al. 2006). To our knowledge, only the study of Mazzeo et al. (1995) investigated a role for the elevated arterial adrenaline concentration as a potential mechanism for the augmented hypoxic exercise hyperaemia. In their report, subjects receiving systemic β-adrenergic receptor blockade had lower leg blood flows, relative to control subjects, during cycling exercise at 50% peak oxygen consumption with acute altitude exposure. Since systemic β-adrenergic receptor blockade can lower blood pressure and cardiac output and evoke reflex increases in sympathetic outflow (Pawelczyk et al. 1992), an important strength of the current study was that we were able to determine the specific contribution of β-adrenergic receptor activation in the absence of potential systemic cardiovascular activation. Moreover, we isolated the effects of hypoxia, per se, without confounding factors associated with altitude (e.g. hypocapnia).
It should also be noted that both Rowell & Blackmon (1989) and Rowell & Seals (1990) observed that subjects becoming presyncopal during lower body negative pressure under hypoxic conditions were those that had a marked rise in arterial adrenaline. Dinenno et al. (2003) reported one subject with an exaggerated rise in plasma adrenaline, who demonstrated symptoms of vasovagal syncope during normocapnic hypoxia at rest. In the present study, we were able to obtain arterial catecholamine samples from one of the two subjects who did not complete the study due to symptoms of presyncope. In this subject, arterial adrenaline was 222 pg ml−1 in the sample obtained immediately before onset of symptoms, which occurred during 10% hypoxic exercise with phentolamine infusion. From this discussion, it appears that while adrenaline release during hypoxia contributes to augmented blood flow in skeletal muscle at rest and during mild exercise, in some subjects an exaggerated level of circulating adrenaline is associated with vasovagal responses.
Other potential contributors to augmented hypoxic vasodilatation
In the present study, β-adrenergic mechanisms contributed to approximately half of the augmented hypoxic blood flow response at rest and during 10% forearm exercise, suggesting a role for the contribution of other vasodilator substances. Under resting conditions, adenosine release (MacLean et al. 1998; Leuenberger et al. 1999) contributes to the augmented hypoxic vasodilatation in humans and rat models (Skinner & Marshall, 1996). However, the specific contribution of adenosine-mediated vasodilatation in the absence of overlying vasoconstrictor influence during hypoxia remains unknown. It is possible that adenosine and/or enhanced prostaglandin production via adenosine-mediated mechanisms (Ray et al. 2002), contribute to the augmented hypoxic exercise blood flow and will increase concurrently with exercise intensity. It also remains to be determined if NO release, via β-receptor activation (Blitzer et al. 1996; Weisbrod et al. 2001) or by some other mechanism, increases with exercise intensity, contributing to the augmented hypoxic exercise hyperaemia. Important to this discussion, deoxygenation of circulating erythrocytes can elicit ATP release (Bergfeld & Forrester, 1992; Jagger et al. 2001; Gonzalez-Alonso et al. 2002) and therefore may be an important signal for the augmented hypoxic vasodilatation. The combination of exercise and hypoxia may enhance erythrocyte ATP release, thereby increasing an ATP-mediated component for the augmented hypoxic exercise hyperaemia in a way that would optimize oxygen delivery and demand (Calbet et al. 2006).
Experimental considerations
Our data highlight the concept that enhanced sympathetic activity (Saito et al. 1988; Rowell et al. 1989) can override compensatory vasodilator substances contributing to hypoxic exercise. In the current study, pharmacological inhibition of sympathetic vasoconstriction, via α-adrenoceptor blockade, revealed a substantial hypoxic exercise vasodilatation at both 10% and 20% MVC (Fig. 3). Thus, we have shown that the increased vasoconstrictor signal acts to limit a substantial hypoxic vasodilatation both at rest (Weisbrod et al. 2001) and during incremental hypoxic exercise. The present study could not differentiate between changes in muscle and skin circulation. However, while there does not seem to be substantial sympathetic vasoconstrictor restraint for hypoxic vasodilatation in the skin (Simmons et al. 2007), the contribution of the cutaneous circulation during hypoxia (Weisbrod et al. 2001) should be similar during both levels of hypoxic exercise. This discussion does not include the possible contribution of acral skin (hand), which has been shown to vasoconstrict during hypoxia (Kollai, 1983).
We are confident that we blocked both α- and β-adrenergic receptors. Similar doses of phentolamine block vasoconstriction during a sympathetic stimulus (Eklund & Kaijser, 1976) and agonist infusion (Dinenno et al. 2002). Further, a bolus propranolol dose of 0.5 mg (Eklund & Kaijser, 1976) blocks vasodilatation to agonist infusion (isoproterenol) for at least 30 min. In the current study, propranolol was administered throughout normoxic and hypoxic exercise. The observation of higher arterial adrenaline during combined α- and β-adrenoceptor blockade (Table 2) suggests β-blockade may be ineffective during hypoxic exercise at 20% MVC. Despite higher normoxic arterial adrenaline values during combined α- and β-adrenergic blockade, arterial adrenaline was essentially identical for 10% and 20% hypoxic exercise during combined phentolamine/propranolol infusion (Table 2). Our finding that the augmented hypoxic exercise vasodilatation was substantially decreased with β-blockade at 10% MVC yet statistically similar at 20% MVC (Fig. 3) only confirms the exercise intensity-dependent role for β-adrenergic vasodilatation during hypoxic exercise.
There is a potential for enhanced catecholamine release during α-receptor blockade (Saeed et al. 1982) and it is possible that noradrenaline-mediated β-receptor activation exists when α-receptors are blocked. If this were the case in the current study, we would be overestimating the contribution of β-receptor-mediated vasodilatation during hypoxic exercise. However, under normoxic conditions, we observed identical blood flow responses during incremental exercise relative to control conditions (Table 4; Fig. 3) despite substantially higher venous noradrenaline with α-receptor blockade (Table 3). Combined α-receptor and β-receptor blockade did not reduce normoxic exercise hyperaemia (Table 4; Fig. 3). In contrast, combined α- and β-receptor blockade reduced hypoxic vasodilatation at rest and hypoxic exercise hyperaemia at 10% MVC, despite similar venous noradrenaline values to normoxia. Thus, only under conditions when arterial adrenaline levels were high (i.e. hypoxia) was there significant β-mediated vasodilatation.
Conclusions
The primary findings from the present study suggest that, during hypoxic exercise, the contribution of β-adrenergic receptor-mediated vasodilatation decreases with increasing exercise intensity. The present study highlights the importance of sympathetic vasoconstrictor inhibition when investigating augmented hypoxic muscle blood, and that enhanced sympathetic outflow can mask the role of vasodilator substances. Based on the work of others on issues related to hypoxic vasodilatation in resting muscle, adenosine-mediated vasodilatation alone or in combination with other substances seems likely to contribute to the augmented blood flow during hypoxic exercise. Moreover, an adenosine-mediated component to the augmented vasodilatation which increases concurrently with exercise intensity during hypoxia is an attractive hypothesis.
Acknowledgments
The authors are especially grateful to the subjects for their participation in this study. We also thank Shelly Roberts, Pam Engrav, Branton Walker, and Madhuri Somaraju for their technical assistance. This research was supported by the National Institutes of Health research grants HL-78019 (to B. W. Wilkins) and HL-46493 (to M. J. Joyner) and by CTSA RR-024150.
References
- Banzett RB, Garcia RT, Moosavi SH. Simple contrivance ‘clamps’ end-tidal PCO2 and PO2 despite rapid changes in ventilation. J Appl Physiol. 2000;88:1597–1600. doi: 10.1152/jappl.2000.88.5.1597. [DOI] [PubMed] [Google Scholar]
- Bergfeld GR, Forrester T. Release of ATP from human erythrocytes in response to a brief period of hypoxia and hypercapnia. Cardiovasc Res. 1992;26:40–47. doi: 10.1093/cvr/26.1.40. [DOI] [PubMed] [Google Scholar]
- Blauw GJ, Westendorp RG, Simons M, Chang PC, Frolich M, Meinders AE. β-Adrenergic receptors contribute to hypoxaemia induced vasodilation in man. Br J Clin Pharmacol. 1995;40:453–458. [PMC free article] [PubMed] [Google Scholar]
- Blitzer ML, Lee SD, Creager MA. Endothelium-derived nitric oxide mediates hypoxic vasodilation of resistance vessels in humans. Am J Physiol Heart Circ Physiol. 1996;271:H1182–H1185. doi: 10.1152/ajpheart.1996.271.3.H1182. [DOI] [PubMed] [Google Scholar]
- Calbet JA, Boushel R, Radegran G, Sondergaard H, Wagner PD, Saltin B. Determinants of maximal oxygen uptake in severe acute hypoxia. Am J Physiol Regul Integr Comp Physiol. 2003;284:R291–R303. doi: 10.1152/ajpregu.00155.2002. [DOI] [PubMed] [Google Scholar]
-
Calbet JA, Lundby C, Sander M, Robach P, Saltin B, Boushel R. Effects of ATP-induced leg vasodilation on
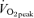 and leg O2 extraction during maximal exercise in humans. Am J Physiol Regul Integr Comp Physiol. 2006;291:R447–453. doi: 10.1152/ajpregu.00746.2005. [DOI] [PubMed] [Google Scholar]
and leg O2 extraction during maximal exercise in humans. Am J Physiol Regul Integr Comp Physiol. 2006;291:R447–453. doi: 10.1152/ajpregu.00746.2005. [DOI] [PubMed] [Google Scholar] - Dietz NM, Rivera JM, Eggener SE, Fix RT, Warner DO, Joyner MJ. Nitric oxide contributes to the rise in forearm blood flow during mental stress in humans. J Physiol. 1994;480:361–368. doi: 10.1113/jphysiol.1994.sp020366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinenno FA, Dietz NM, Joyner MJ. Aging and forearm postjunctional α-adrenergic vasoconstriction in healthy men. Circulation. 2002;106:1349–1354. doi: 10.1161/01.cir.0000028819.64790.be. [DOI] [PubMed] [Google Scholar]
- Dinenno FA, Joyner MJ, Halliwill JR. Failure of systemic hypoxia to blunt α-adrenergic vasoconstriction in the human forearm. J Physiol. 2003;549:985–994. doi: 10.1113/jphysiol.2003.042507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklund B, Kaijser L. Effect of regional α- and β-adrenergic blockade on blood flow in the resting forearm during contralateral isometric handgrip. J Physiol. 1976;262:39–50. doi: 10.1113/jphysiol.1976.sp011584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Alonso J, Olsen DB, Saltin B. Erythrocyte and the regulation of human skeletal muscle blood flow and oxygen delivery: role of circulating ATP. Circ Res. 2002;91:1046–1055. doi: 10.1161/01.res.0000044939.73286.e2. [DOI] [PubMed] [Google Scholar]
- Hartley LH, Vogel JA, Landowne M. Central, femoral, and brachial circulation during exercise in hypoxia. J Appl Physiol. 1973;34:87–90. doi: 10.1152/jappl.1973.34.1.87. [DOI] [PubMed] [Google Scholar]
- Jagger JE, Bateman RM, Ellsworth ML, Ellis CG. Role of erythrocyte in regulating local O2 delivery mediated by hemoglobin oxygenation. Am J Physiol Heart Circ Physiol. 2001;280:H2833–H2839. doi: 10.1152/ajpheart.2001.280.6.H2833. [DOI] [PubMed] [Google Scholar]
- Johnsson G. The effects of intra-arterially administered propranolol and H 56–28 on blood flow in the forearm – a comparative study of two β-adrenergic receptor antagonists. Acta Pharmacol Toxicol (Copenh) 1967;25:63–74. doi: 10.1111/j.1600-0773.1967.tb02997.x. [DOI] [PubMed] [Google Scholar]
- Joyner MJ, Nauss LA, Warner MA, Warner DO. Sympathetic modulation of blood flow and O2 uptake in rhythmically contracting human forearm muscles. Am J Physiol Heart Circ Physiol. 1992;263:H1078–H1083. doi: 10.1152/ajpheart.1992.263.4.H1078. [DOI] [PubMed] [Google Scholar]
- Kollai M. Responses in cutaneous vascular tone to transient hypoxia in man. J Auton Nerv Syst. 1983;9:497–512. doi: 10.1016/0165-1838(83)90009-7. [DOI] [PubMed] [Google Scholar]
- Leuenberger U, Gleeson K, Wroblewski K, Prophet S, Zelis R, Zwillich C, Sinoway L. Norepinephrine clearance is increased during acute hypoxemia in humans. Am J Physiol Heart Circ Physiol. 1991;261:H1659–H1664. doi: 10.1152/ajpheart.1991.261.5.H1659. [DOI] [PubMed] [Google Scholar]
- Leuenberger UA, Gray K, Herr MD. Adenosine contributes to hypoxia-induced forearm vasodilation in humans. J Appl Physiol. 1999;87:2218–2224. doi: 10.1152/jappl.1999.87.6.2218. [DOI] [PubMed] [Google Scholar]
- MacLean DA, Sinoway LI, Leuenberger U. Systemic hypoxia elevates skeletal muscle interstitial adenosine levels in humans. Circulation. 1998;98:1990–1992. doi: 10.1161/01.cir.98.19.1990. [DOI] [PubMed] [Google Scholar]
- Mazzeo RS, Brooks GA, Butterfield GE, Podolin DA, Wolfel EE, Reeves JT. Acclimatization to high altitude increase muscle sympathetic activity both at rest and during exercise. Am J Physiol Regul Integr Comp Physiol. 1995;269:R201–R207. doi: 10.1152/ajpregu.1995.269.1.R201. [DOI] [PubMed] [Google Scholar]
- Minson CT, Halliwill JR, Young TM, Joyner MJ. Influence of the menstrual cycle on sympathetic activity, baroreflex sensitivity, and vascular transduction in young women. Circulation. 2000a;101:862–868. doi: 10.1161/01.cir.101.8.862. [DOI] [PubMed] [Google Scholar]
- Minson CT, Halliwill JR, Young TM, Joyner MJ. Sympathetic activity and baroreflex sensitivity in young women taking oral contraceptives. Circulation. 2000b;102:1473–1476. doi: 10.1161/01.cir.102.13.1473. [DOI] [PubMed] [Google Scholar]
- Pawelczyk JA, Hanel B, Pawelczyk RA, Warberg J, Secher NH. Leg vasoconstriction during dynamic exercise with reduced cardiac output. J Appl Physiol. 1992;73:1838–1846. doi: 10.1152/jappl.1992.73.5.1838. [DOI] [PubMed] [Google Scholar]
- Ray CJ, Abbas MR, Coney AM, Marshall JM. Interactions of adenosine, prostaglandins and nitric oxide in hypoxia-induced vasodilatation: in vivo and in vitro studies. J Physiol. 2002;544:195–209. doi: 10.1113/jphysiol.2002.023440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowell LB, Blackmon JR. Hypotension induced by central hypovolaemia and hypoxaemia. Clin Physiol. 1989;9:269–277. doi: 10.1111/j.1475-097x.1989.tb00979.x. [DOI] [PubMed] [Google Scholar]
- Rowell LB, Johnson DG, Chase PB, Comess KA, Seals DR. Hypoxemia raises muscle sympathetic activity but not norepinephrine in resting humans. J Appl Physiol. 1989;66:1736–1743. doi: 10.1152/jappl.1989.66.4.1736. [DOI] [PubMed] [Google Scholar]
- Rowell LB, Saltin B, Kiens B, Christensen NJ. Is peak quadriceps blood flow in humans even higher during exercise with hypoxemia? Am J Physiol Heart Circ Physiol. 1986;251:H1038–H1044. doi: 10.1152/ajpheart.1986.251.5.H1038. [DOI] [PubMed] [Google Scholar]
- Rowell LB, Seals DR. Sympathetic activity during graded central hypovolemia in hypoxemic humans. Am J Physiol Heart Circ Physiol. 1990;259:H1197–H1206. doi: 10.1152/ajpheart.1990.259.4.H1197. [DOI] [PubMed] [Google Scholar]
- Saeed M, Sommer O, Holtz J, Bassenge E. α-Adrenoceptor blockade by phentolamine causes β-adrenergic vasodilation by increased catecholamine release due to presynaptic α-blockade. J Cardiovasc Pharmacol. 1982;4:44–52. doi: 10.1097/00005344-198201000-00008. [DOI] [PubMed] [Google Scholar]
- Saito M, Mano T, Iwase S, Koga K, Abe H, Yamazaki Y. Responses in muscle sympathetic activity to acute hypoxia in humans. J Appl Physiol. 1988;65:1548–1552. doi: 10.1152/jappl.1988.65.4.1548. [DOI] [PubMed] [Google Scholar]
- Simmons GH, Minson CT, Cracowski JL, Halliwill JR. Systemic hypoxia causes cutaneous vasodilation in healthy humans. J Appl Physiol. 2007;103:608–615. doi: 10.1152/japplphysiol.01443.2006. [DOI] [PubMed] [Google Scholar]
- Skinner MR, Marshall JM. Studies on the roles of ATP, adenosine and nitric oxide in mediating muscle vasodilatation induced in the rat by acute systemic hypoxia. J Physiol. 1996;495:553–560. doi: 10.1113/jphysiol.1996.sp021615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers VK, Mark AL, Zavala DC, Abboud FM. Influence of ventilation and hypocapnia on sympathetic nerve responses to hypoxia in normal humans. J Appl Physiol. 1989;67:2095–2100. doi: 10.1152/jappl.1989.67.5.2095. [DOI] [PubMed] [Google Scholar]
- Weisbrod CJ, Minson CT, Joyner MJ, Halliwill JR. Effects of regional phentolamine on hypoxic vasodilatation in healthy humans. J Physiol. 2001;537:613–621. doi: 10.1111/j.1469-7793.2001.00613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins BW, Schrage WG, Liu Z, Hancock KC, Joyner MJ. Systemic hypoxia and vasoconstrictor responsiveness in exercising human muscle. J Appl Physiol. 2006;101:1343–1350. doi: 10.1152/japplphysiol.00487.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]