

Abstract
Reconciling ion channel α-subunit expression with native ionic currents and their pharmacological sensitivity in target organs has proved difficult. In native tissue, many K+ channel α-subunits co-assemble with ancillary subunits, which can profoundly affect physiological parameters including gating kinetics and pharmacological interactions. In this review, we examine the link between voltage-gated potassium ion channel pharmacology and the biophysics of ancillary subunits. We propose that ancillary subunits can modify the interaction between pore blockers and ion channels by three distinct mechanisms: changes in (1) binding site accessibility; (2) orientation of pore-lining residues; (3) the ability of the channel to undergo post-binding conformational changes. Each of these subunit-induced changes has implications for gating, drug affinity and use dependence of their respective channel complexes. A single subunit may modulate its associated α-subunit by more than one of these mechanisms. Voltage-gated potassium channels are the site of action of many therapeutic drugs. In addition, potassium channels interact with drugs whose primary target is another channel, e.g. the calcium channel blocker nifedipine, the sodium channel blocker quinidine, etc. Even when K+ channel block is the intended mode of action, block of related channels in non-target organs, e.g. the heart, can result in major and potentially lethal side-effects. Understanding factors that determine specificity, use dependence and other properties of K+ channel drug binding are therefore of vital clinical importance. Ancillary subunits play a key role in determining these properties in native tissue, and so understanding channel–subunit interactions is vital to understanding clinical pharmacology.
Although a single type of K+ channel α-subunit is often present in a variety of different organs, the kinetic behaviour and conformational changes of α-subunits are often modulated by co-assembly with ancillary subunits. The expression of ancillary subunits varies between organs, as well as between regions of an organ (Isom et al. 1994; McCrossan & Abbott, 2004; Birnbaum et al. 2004; Melnyk et al. 2005). This diversity of ancillary subunit expression therefore contributes to the diverse assortment of potassium currents recorded from native tissues. In addition, relative expression of K+ channels and their associated ancillary subunits can be affected by factors such as development, changes in hormonal state, ischaemic conditions, etc., which can also modulate the electrophysiology and pharmacology of native potassium currents (Soliven et al. 1989; Shimoni et al. 1997; Nerbonne, 1998; Liu et al. 2007). The importance of the role that subunits can play in regulating ion channel behaviour in native tissue is demonstrated by the number of mutations which are associated with arrhythmogenesis and periodic paralysis in humans (for reviews, see Chiang & Roden, 2000; Abbott & Goldstein, 2001; Shah et al. 2005). In addition, indirect alteration in subunit function can lead to epilepsy in humans (Schulte et al. 2006).
Compounds with K+ channel blocking properties are commonly employed as therapeutic agents for conditions such as arrhythmias (Tamargo et al. 2004), stroke (Surti & Jan, 2005), cancer (Conti, 2004), and neurological disorders such as psychoses, epilepsy, stroke and Alzheimer/s disease (Surti & Jan, 2005). For these therapeutic agents, block of specific K+ channels is the intended mechanism of action (e.g. class III anti-arrhythmic agents). However, there are a wide variety of therapeutic agents that are targeted to non-K+ channels, but result in unintended block of K+ channels. This K+ channel block can result in potentially serious and sometimes even fatal side-effects (e.g. cardiac arrhythmias). For example, Kv channels are blocked by calcium channel blockers including nifedipine and nicardipine (Grissmer et al. 1994; Zhang & Fedida, 1998; Hatano et al. 2003; Bett et al. 2006b), and sodium channel blockers including quinidine and flecainide (Yeola & Snyders, 1997; Caballero et al. 2003; Wang et al. 2003; Bett & Rasmusson, 2004). Even when K+ channel block is the intended mode of action (for example, anti-psychotics, anti-epileptics, etc. (Davis et al. 1996; Escande, 2000; Wickenden, 2002)), block of the same or related ion channels in non-target organs, e.g. the heart, can result in major and potentially lethal side-effects such as arrhythmogenesis. Side-effects are of particular importance in the HERG channel, which is the molecular basis of IKr. (see for example Sanguinetti & Tristani-Firouzi, 2006). The co-assembly of the core α-subunit of an ion channel with a variety of ancillary subunits results in channels which present a broad spectrum of potential targets and side-effects for drugs with channel blocking actions. Understanding the molecular basis of subunit–channel interactions and their impact on drug binding is therefore of critical importance in the development of safe pharmacological approaches for the treatment of arrhythmias, neurological disorders, and a multitude of other channel-related maladies.
This review focuses on the relationship between the ability of ancillary subunits to modify the interaction between voltage-gated K+ channels and drugs which bind to and block the intracellular vestibule of the open channel pore. Ancillary subunits alter the properties of the intracellular pore, which can result in direct changes in the interaction between the drug and the binding site, as well as changes in gating, which can indirectly affect channel pharmacology. We discuss three, non-mutually exclusive, biophysical models by which ancillary subunits could modify ion channel behaviour. These biophysical models form a framework for understanding the modification of drug–channel interactions by ancillary subunits, and are a necessary step in developing predictive state models of how drug binding to the open pore is modified by ancillary subunits.
1 Voltage-gated K+ channels and ancillary subunits
1.1 Voltage-gated K+ channels
Voltage-gated K+ channels are a large family of ion channels, which are found in almost every cell type, and play physiological roles in everything from electrical excitability to solute transport. A minimally functional K+ channel contains a tetramer of α-subunits, each of which contributes equally to the pore structure. In addition to the α-subunits, which form the core of the channel, many K+ channels co-assemble in native cells with ancillary subunits (e.g. KChIPs (K+ channel interacting proteins), KCNEs and Kvβs), which can result in dramatic changes in structure, behaviour, and function of the ion channel. Figure 1 shows the topology of a typical pore-forming α-subunit of a voltage-gated K+ channel. The N- and C-terminal domains are on the cytoplasmic side of the membrane. The channel has six transmembrane segments, S1–S6, thought to be α-helical in structure. A short lipophilic segment, the ‘H5 loop’, lies between S5 and S6. The H5 helix dips into the transmembrane domain, forms a small loop and returns to the extracellular side (Jan & Jan, 1992). H5 is thought to be the location of the selectivity filter of the channel (Yool & Schwarz, 1991; Lipkind et al. 1995; Doyle et al. 1998), and is important as a locus for extracellular channel blockers. These general structural elements established in KcsA (Doyle et al. 1998) are well conserved in voltage-gated K+ channels, and have been confirmed in crystal structures such as KvAP and Kv1.2 (Lee et al. 2005; Long et al. 2005a,b).
Figure 1. Cartoon representation of Kv channels and ancillary subunits.

The pore-forming component of Kv channels are formed from a tetramer of α-subunits. The α-subunits have six transmembrane segments, with intracellular N- and C-terminals. The fourth transmembrane segment has a net positive charge, and is thought to be the voltage sensor. Ancillary subunits interact with the core α-subunits in a variety of ways. The KCNE family are single transmembrane proteins, with an intracellular C-terminal and an extracellular N-terminal. The KChIP family of proteins are located intracellularly, and are thought to interact with the N-terminal. The Kvβ proteins interact with both N- and C-terminals.
1.2 K+ channel activation
K+ channel activation is a voltage-dependent event, i.e. channels open in response to a change in membrane voltage. Such voltage dependence requires that a net charge moves in the transmembrane electrical field in order to produce the physical work necessary to move the channel out of the stable closed conformation. Ca2+, Na+ and K+ channels share a similar structure and common voltage-sensing mechanism. The S4 transmembrane domain in K+ channels contains a highly conserved structure (Lee et al. 2005; Long et al. 2005a,b). Every third position is occupied by a lysine or arginine, which are positively charged in the physiological range of pH. At the resting membrane potential, many of these positive charges are on the intracellular face of the membrane. On membrane depolarization these residues move through the membrane due to the increased electrical driving force and reach the extracellular face (Mannuzzu et al. 1996). The transmembrane movement of S4 initiates large scale conformational changes that result in an open, conducting pore (Catterall, 1995; Yellen, 1998; Bezanilla, 2002; Lee et al. 2005; Long et al. 2005a,b).
1.3 The K+ channel open state
Channel opening involves a significant change in protein structure. One of the most important rearrangements is opening or widening of the intracellular pore. This widening is often referred to as the ‘intracellular gate’ (Holmgren et al. 1998). The entire pore volume has been measured to be ∼800–2000 Å3 for voltage-gated Na+ and K+ channels i.e. the volume of ∼50 water molecules (Zimmerberg et al. 1990; Rayner et al. 1992; Jiang et al. 2003). There are three general pictures of intracellular pore opening based on crystal structure measurements. The S6 transmembrane segment is thought to play a prominent role in lining the pore in the open channel models of both KcsA, Kv1.2 and MthK channels (Liu et al. 2001; Jiang et al. 2002; Cuello et al. 2004; Lee et al. 2005; Long et al. 2005a,b). In all voltage-gated channel models, activation involves a movement and deformation of S6 at critical hinge regions (Labro et al. 2003; Zhao et al. 2004; Webster et al. 2004) as the channel moves from the closed to the open state. However, the putative open conformation is very different for the KcsA and Kv1.2 versus MthK channels (Liu et al. 2001; Jiang et al. 2002; Cuello et al. 2004; Lee et al. 2005; Long et al. 2005a,b; Zimmer et al. 2006). This suggests there is tremendous physical diversity in the open channel conformation, even in related channels. Furthermore, ancillary subunits strongly modify gating properties and drug binding properties of the open channel, possibly by the substantial structural rearrangements that occur as a result of ancillary subunits co-assembling with their α-subunit partners (see Table 1 and Table 2).
Table 1.
Effect of subunits on Kv channel gating
| α-Subunit | Ancillary subunit family | Gating parameter modified by subunit |
|---|---|---|
| Kv1.1 | Kvβ | Introduces inactivation (Rettig et al. 1994; Heinemann et al. 1996) |
| Kv1.2 | Kvβ | Introduces inactivation (Heinemann et al. 1996; Wang et al. 1996) |
| Kv1.4 | Kvβ | Increases inactivation rate (Rettig et al. 1994; Morales et al. 1995,1996; Majumder et al. 1995) |
| Slows recovery from inactivation (Castellino et al. 1995; Accili et al. 1998). | ||
| Kv1.4 ΔN | Kvβ | Increases inactivation rate (Heinemann et al. 1995, 1996; Rasmusson et al. 1997) |
| Kv1.5 | Kvβ | Introduces inactivation (Majumder et al. 1995; England et al. 1995; Heinemann et al. 1996; De Biasi et al. 1997; Uebele et al. 1998; Gonzalez et al. 2002; Tipparaju et al. 2007) |
| Shifts activation kinetics (England et al. 1995; Uebele et al. 1996, 1998; Heinemann et al. 1996; De Biasi et al. 1997; Tipparaju et al. 2007) | ||
| Slows deactivation (De Biasi et al. 1997; Uebele et al. 1998) | ||
| Kv2 | KCNE3 | Slows activation and deactivation (McCrossan et al. 2003) |
| Kv3 | KCNE3 | Slows activation (McCrossan et al. 2003; Lewis et al. 2004) |
| Slows deactivation (Lewis et al. 2004) | ||
| Shifts activation and Increases rate of recovery from inactivation (Abbott et al. 2001) | ||
| Kv4 | KChIPs | Alters rate of inactivation (An et al. 2000; Bahring et al. 2001a; Beck et al. 2002; Deschenes & Tomaselli, 2002; Van Hoorick et al. 2003; Boland et al. 2003; Patel et al. 2004). |
| Speeds recovery from inactivation (An et al. 2000; Decher et al. 2001; Beck et al. 2002; Deschenes & Tomaselli, 2002; Patel et al. 2002; Boland et al. 2003). | ||
| KCNE1 | Slows activation and inactivation; slows recovery from inactivation (Deschenes & Tomaselli, 2002) | |
| KCNE2 | Slows activation, shifts activation and inactivation (Zhang et al. 2001) | |
| Slows inactivation (Zhang et al. 2001; Deschenes & Tomaselli, 2002) | ||
| KCNE3 | Slows and shifts activation; slows and shifts inactivation; Slows recovery from inactivation (Lundby & Olesen, 2006) | |
| Kvβ | Shifts inactivation; slows recovery from inactivation (Deschenes & Tomaselli, 2002) | |
| Kv7.1 | KCNE1 | Slows activation, abolishes inactivation (Barhanin et al. 1996; Sanguinetti et al. 1996) |
| (KCNQ1) | KCNE2 | Introduces Instantaneous activation; increases deactivation rate (Tinel et al. 2000) |
| KCNE3 | Channel constitutively open and voltage independent (Schroeder et al. 2000) | |
| KCNE4 | Slows and shifts activation (Bendahhou et al. 2005) | |
| KCNE5 | Slows and shifts activation (Angelo et al. 2002; Bendahhou et al. 2005) | |
| Speeds deactivation (Angelo et al. 2002) | ||
| Kv11.1 | KCNE1 | Shifts activation (McDonald et al. 1997) |
| (HERG) | KCNE2 | Slows and shifts activation (Abbott et al. 1999) |
| Speeds activation (Mazhari et al. 2001) | ||
| Increases the rate of deactivation (Abbott et al. 1999; Mazhari et al. 2001; Weerapura et al. 2002; Lu et al. 2003) |
Table 2.
Effect of subunits on the interaction between open pore blockers and Kv channels.
| α-Subunit | Ancillary subunit | Drug parameter modified by subunit |
|---|---|---|
| Kv1.5 | Kvβ1.3 | Decreases sensitivity to bupivicaine, quinidine and S0100176 (Gonzalez et al. 2002; Decher et al. 2005) |
| Kv4 | KChIPs | Decreases sensitivity to nifedipine, nicardipine, bupivicaine and ropivicaine (Hatano et al. 2003; Friederich & Solth, 2004; Solth et al. 2005; Bett et al. 2006b). |
| Slows development of block by ropivicane and bupivacaine (Friederich & Solth, 2004; Solth et al. 2005) | ||
| KCNQ1 | KCNE1 | Increases sensitivity to chromanol 293B, HMR 1556, S5557, azimilide, XE991 and 17 β-oestradiol (Busch et al. (Kv7.1) 1997; Wang et al. 2000; Schroeder et al. 2000; Lerche et al. 2000; Bett et al. 2006a). |
| Affects use dependence (Seebohm et al. 2001; Bett et al. 2006a) | ||
| KCNE2 | Increases sensitivity to chromanol 293B (Tinel et al. 2000) | |
| KCNE3 | Increases sensitivity to chromanol 293B, azimilide, XE991 and 17 β-oestradiol (Schroeder et al. 2000; MacVinish et al. 2001; Bett et al. 2006a) | |
| HERG | KCNE2 | Increases sensitivity to E4031 and propranolol (Abbott et al. 1999; Dupuis et al. 2005) |
| (Kv11.1) Alters use dependence (Abbott et al. 1999) |
1.4 K+ channel inactivation
Inactivation is the process by which a channel becomes non-conducting in the presence of a continued stimulus. In general, the process of inactivation is slower than activation, although there are important exceptions to this observation, e.g. HERG (Trudeau et al. 1995; Sanguinetti et al. 1995). The molecular basis of K+ channel inactivation was originally based on data from Shaker K+ channels. Mutagenesis studies showed at least two mechanisms of inactivation, called ‘N-type’ and ‘C-type’ (Hoshi et al. 1991).
N-type inactivation
The best understood inactivation mechanism is N-type, which occurs on the order of ∼milliseconds to ∼tens of milliseconds. N-type inactivation is mediated by a ‘tethered ball’ mechanism in which a lipophilic segment of ∼20 amino acids in the N-terminus of the channel binds at the intracellular pore mouth (Zagotta et al. 1990; Hoshi et al. 1990, 1991). N-type inactivation has a number of well-defined properties: (1) N-type inactivation is removed after N-terminal deletion (Hoshi et al. 1990); (2) N-type inactivation is insensitive to extracellular [TEA+] but sensitive to intracellular [TEA+] (Choi et al. 1991; Demo & Yellen, 1991); (3) N-type inactivation is unaffected by increased [K+]o (Rasmusson et al. 1995); (4) The rate of N-type inactivation is voltage insensitive at positive potentials (Zagotta & Aldrich, 1990); (5) N-type inactivation is insensitive to point mutations at the outer mouth of the channel or the outer region of S6 (Hoshi et al. 1991; Rasmusson et al. 1995).
C-type inactivation
In K+ channels, C-type inactivation is defined as the inactivation mechanism that remains following the loss of N-type inactivation i.e. following N-terminal deletion (Hoshi et al. 1991; Hille, 2001). C-type inactivation involves small conformational changes at the extracellular side of the pore, and a substantial conformational change (on the same order as channel opening) at the intracellular side of the channel pore (Jiang et al. 2003; Li et al. 2003). This inactivation was labelled ‘C-type’ as it is dependent on the particular C-terminal splice variant of the Shaker K+ channel being studied (Pongs, 1992). The cytoplasmic portion of the C-terminal of Shaker does not mediate C-type inactivation through a second ball and chain mechanism, rather it alters permeation through the core domain pore region. Domains critical for C-type inactivation include the extracellular side of S6 (Hoshi et al. 1991), the intracellular side of S6 (Li et al. 2003; Bett & Rasmusson, 2004) and a specific residue in the extracellular H5–S6 loop (position 449 in Shaker B) (Lopez-Barneo et al. 1993; Wang et al. 2003). C-type inactivation is slowed by increasing external permeant ion concentrations (Busch et al. 1991; Lopez-Barneo et al. 1993) and by the application of extracellular, but not intracellular, TEA+ (Choi et al. 1991). Thus, C-type inactivation was determined to involve closure of the extracellular pore mouth and cooperative interactions between multiple core α-subunits (Ogielska et al. 1995; Kiss et al. 1999; Andalib et al. 2004). The movements associated with C-type inactivation involve more than just small regional changes on the extracellular face of the pore; changes in intracellular pore diameter (Jiang et al. 2003; Panyi & Deutsch, 2007), and transmembrane spanning domains (Hoshi et al. 1991; Adelman et al. 1995; Li et al. 2003; Bett & Rasmusson, 2004) have also been implicated, as have deeper regions of the selectivity filter (Kurata et al. 2005; Panyi & Deutsch, 2007; Cordero-Morales et al. 2007).
1.5 Drug binding to the open K+ channel
Potassium channels interact with a wide variety of drugs. For manageability, this review considers only the binding of drugs to the open pore of voltage-gated potassium channels. However, a similar analysis can be applied to conformational-specific drug binding to other states and channels. Drug binding to an intracellular pore site or vestibule is a well established concept (see Hille, 2001). More recently, site-directed mutagenesis has enabled identification of the key pore residues which form the binding site in the intracellular pore. Analysis of crystal structures of the pore region has shown the relative positions of these key residues, and the physical nature of the binding site.
The Kv1.5 S6 binding domains for quinidine were among the first to be mapped (Yeola et al. 1996). Other drug–channel combinations have subsequently been mapped against the putative crystal structure of this region (Seebohm et al. 2001, 2003; Chen et al. 2002). In some cases, a projection of the drug binding interactions in terms of individual Van der Waal/s interactions has been accomplished for high-affinity, high-specificity drugs (Chen et al. 2002). These mappings agree well with the putative closed state KcsA structure (Doyle et al. 1998), despite the drugs having the properties of open channel blockers. Analysis of quaternary ammonium binding has also been interpreted in the context of the putative closed structure of the KcsA channel (Doyle et al. 1998). Analysis of the crystal structure of this channel bound to the N-terminal inactivation ball showed some interesting characteristics. In addition to having structural orientation similar to the closed state of the channel, the N-terminal ‘ball’ bound with the N-terminal amino acids in an extended chain (Zhou et al. 2001), lining the pore, instead of the much more rounded structure predicted for these domains in solution using NMR (Wissmann et al. 1999). This structural information suggests that drug binding and N-terminal-inactivated states may involve considerable secondary changes in channel structure following initial binding to the open state.
For some drug–channel combinations, the degree of block observed depends on the stimulation frequency. When the block of an open channel blocking drug increases with stimulation frequency, this is called use dependence. Use dependence is thought to arise from the interplay of two kinetic parameters: the rate of drug binding/unbinding, and the transition rate of the channel between states (Starmer et al. 1984; Gintant & Hoffman, 1984; Hondeghem & Katzung, 1984). This occurs because some drugs bind only to, or preferentially to, specific open or closed states of the channel, i.e. some drugs display a variety of conformation-specific interactions with K+ channels. The amount of time a voltage-gated channel spends in any particular state depends in large part on the membrane potential. Conformation-specific interactions can therefore alter drug block of a given channel by orders of magnitude, depending on the pattern of electrical stimulation (Armstrong, 1969, 1971; Strichartz, 1973; Hondeghem & Katzung, 1977; Starmer et al. 1984). This remains a current topic of research in voltage-gated potassium channels (e.g. Spector et al. 1996; Zhang et al. 1999; Jo et al. 2000; Kushida et al. 2002; Persson et al. 2005; Bett et al. 2006a,b; Stork et al. 2007). These variations in patterns of use dependence can either be clinically detrimental (i.e. the drug has reduced effectiveness) or beneficial (e.g. the drug is only effective during periods of tachycardia) (Hondeghem & Snyders, 1990). Ancillary subunits can alter the conformation of K+ channels, as well as alter gating kinetics and the response of the channel to stimulation frequency. The presence of ancillary subunits can therefore modulate the relationship between drug and channel, in some cases quite dramatically.
2 Biophysical models of subunit modification of K+ channel behaviour
Ancillary subunits can strongly modulate drug–channel interactions (Tables 1 and 2). As more channel defects that affect drug–channel interactions are identified, understanding subunit–channel interactions will be critical in identifying the consequences of these defects. Drug–channel interactions are strongly influenced by channel gating, as well as the structure of the binding site. In an effort to develop a general framework for understanding how the relationship between ion channels and drug binding is affected by the presence of ancillary subunits, we will examine subunit–drug–channel interactions in terms of mechanisms of interaction between subunit in the pore/vestibule region. This biophysical classification may help form the basis for developing predictive state models of drug binding to the open pore, and how these models are modified in the presence of subunits.
We propose that the subunit-induced changes in drug–channel interactions can be separated into three discrete but related components with distinct molecular mechanisms:
1. Ancillary subunits can modulate the ability of drugs to access the binding site in the open pore.
2. Ancillary subunits can modulate the orientation of residues (primarily on S6) which confer specificity of drug binding.
3. Ancillary subunits can modulate the ability of the channel to undergo conformational changes (e.g. inactivation and closure) following drug block.
These three mechanisms are not necessarily exclusive but can, and probably do, co-exist in any combination in any given channel–subunit unit. Many features of gating and open pore drug binding are similar across the family of voltage-gated channels, so it is likely this biophysical model can be successfully applied to open pore drug binding modulation in other voltage-gated channels.
2.1 Ancillary subunits can modulate the ability of drugs to access the binding site in the open pore
In order for a drug to bind to the intracellular pore of a channel, the drug must be able to reach its binding site. For open-pore blockers, this requires that the channel is in the open state and the drug can reach the binding site. A simple state model of a drug binding to the open pore in a channel with closed (C), open (O), blocked (B) and blocked-inactivated (BI) states is given by:
| Scheme (1) |
A schematic representation of this model is shown in Fig. 2A. The simplest method by which a subunit can decrease the ability of a drug to reach the open pore binding site is by physical occlusion of the binding site. Figure 2B shows how an ancillary subunit that binds to the pore drug binding site will alter the interaction between drug and channel. If the subunit blocks access to the pore or competes at the open pore binding site (e.g. Rasmusson et al. 1997; De Biasi et al. 1997), then the subunit must unbind before the drug can gain access to the binding site. The equivalent state model is:
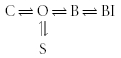 |
Scheme (2) |
where S is the state in which the subunit is bound to the pore of the channel. Electrophysiologically recorded current flow is only a measure of the time the channel spends in the open state. The effect of the addition of the subunit-bound state will therefore depend on the relative magnitude of the transition rates between the open and blocked states, and the open and subunit-bound states. The introduction of the subunit-bound state may result in a change in apparent affinity of the drug for the channel, even though the actual affinity of the channel has not changed. Changes in the apparent affinity are usually measured as reduction in peak current, but depending on model specifics, the actual changes in affinity can often be calculated from the changes in gating (Bett & Rasmusson, 2002).
Figure 2. Cartoon representation to indicate how an ancillary subunit can alter binding site accessibility.

A, a channel with a binding site in the intracellular pore has to open before the drug can bind. Drug binding may be followed by a post-binding conformational change in the channel. B, if the channel shown in A has a subunit associated with it that can bind to the open pore, then, following pore opening, the subunit can block access of the drug to the binding site, thus altering binding site accessibility.
In some cases, inactivation and drug block are competitive processes. A schematic depiction of the most simple case of competitive binding and inactivation is shown in Fig. 3A. The equivalent state model is given by:
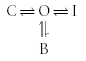 |
Scheme (3) |
Figure 3B shows the effect of adding a subunit-bound (occluding) state to the model. The equivalent state model is given by:
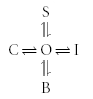 |
Scheme (4) |
Similar to Scheme (2), the net effect on the electrophysiologically measured current will depend on the relative magnitudes of the four transitions out of the open state. Although the rate of transition from the open to the drug-bound state is not changed by the presence of the ancillary subunit, there may be a change in the apparent affinity for the drug, due to the addition of the extra subunit-blocked state. Physical occlusion of the binding site that competes with drug binding is most easily understood in terms of the addition of an ‘N-terminal ball’, which Kvβ-subunits introduce to Kv channels (see Section 3.1).
Figure 3. Cartoon representation of physical block of a channel with competitive inactivation.

A, in some channels, inactivation and drug block are competitive processes. B, the presence of a subunit which can block access to the binding site is the equivalent of introducing an extra subunit-bound state to the model.
Physical occlusion of the pore by the ancillary subunit itself is not the only way in which access to the binding can be altered. In order for the binding site to be accessible, the channel must be in the open conformation, and the conformation of the intracellular pore must enable the drug to reach the binding site. Changes in channel gating can result in a change of accessibility to the binding site i.e. the amount of time the channel spends in the open state. Deactivated, closed and resting channels usually have an intracellular gate which precludes the entry of open channel blockers to their S6 binding site (Holmgren et al. 1998; del Camino & Yellen, 2001). Addition of extra closed states increases the complexity of the opening process, and can introduce a sigmoid delay between stimulus and channel opening. A reduction in the time the channel is in the open state can reduce the apparent binding affinity of a drug for the channel.
| Scheme (5) |
Alternatively, addition of extra open states, which have altered (or zero) affinity for the drug, will result in a change in apparent binding affinity. In the case where co-expression with an ancillary subunit results in additional open states with altered affinity for the drug, there cannot be an increase in the apparent affinity of the drug for the channel without re-orientation of the residues involved in creating the binding site.
| Scheme (6) |
Conversely, the presence of ancillary subunits can result in a reduction in the number of states, which can result in an increase in the ability of the drug to access the binding site. The most dramatic form of this is when the channel is not able to move in or out of the open state, and the binding site is always accessible, as is approximated by the case of KCNE3 interacting with KCNQ1 (see Section 3.3):
| Scheme (7) |
If accessibility to the pore binding site is changed by either physical block or changes in gating, there is a change in the number of states present in the equivalent state model. These changes will affect the rate at which the channel can return to the drug-unbound closed state, and so will usually affect the use dependence of the channel.
In summary, if the subunit modulates ion channel–drug interactions by altering the ability of the drug to access the open pore binding site, there will be:
1. A change in the number of states in the Markov model of the channel.
2. A change in the apparent affinity of the drug, but not the actual affinity.
3. Changes in use dependence
2.2 Ancillary subunits can modulate the orientation of residues (primarily on S6) which confer specificity of drug binding
The traditional view of drug binding is often presented in terms of a ‘lock and key’ type of interaction. The lock and key hypothesis suggests that specific epitopes on a drug (the key) interact with specific side chains of pore-lining residues (the lock). This requires that the lock, i.e. the pore-lining residues, is in a particular physical orientation. However, the transmembrane segments of a channel are composed of a mixture of rigid and hinged structures which can adopt a number of different physical orientations and therefore the pore-lining side chains are able to adopt different physical conformations which still form an open vestibule capable of conducting ions. This is shown in Fig. 4A, which demonstrates how movement (e.g. rotation) of the side chains on the S6 segment orients the side chain residues to form the binding site in the open state. If an ancillary subunit interferes with rotation of S6 during gating in such a situation, then the side chains which form the lock can end up in altered relative orientations (Fig. 4B). Changing the orientation of the binding site residues can result in a binding site with either an increased or decreased affinity for the drug. Examples of subunits that increase affinity of a drug for the channel are the KCNEs and KCNQ1 (see Section 3.3).
Figure 4. The binding site depends on residues on S6.

A, channel opening involves rotation of S6, which aligns residues to form the binding site. B, ancillary subunits can alter the energetic favourability of some conformations, and so rotation of S6 may result in an alteration of the relative positions of the side chains which form the binding site. Such a mechanism has been demonstrated in HERG by Chen et al. (2002).
A schematic representation of ancillary subunits altering the binding site is shown in Fig. 5. There is no change in the ability of the drug to access the binding site, only a difference in the interaction between the drug and the side chains forming the binding site. The only transition affected is therefore that between the open and the drug-bound state. The equivalent state model is:
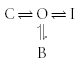 |
Scheme (8) |
Figure 5. Cartoon representation of alteration in binding kinetics.

If an ancillary subunit changes the ‘shape’ of the binding site, this will affect the transition rates between the open and drug-bound states.
For a channel in which inactivation and drug binding are competitive, the schematic depiction is given in Fig. 6. Once again, the only transition affected is open to blocked states. The equivalent state model is:
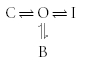 |
Scheme (9) |
For the state model described in Schemes (8) and (9), there are no changes in the number of states in the model, only a change in the rate at which the drug binds and unbinds to the channel. This change can result in either an increase or a decrease in the affinity of the drug for the channel.
Figure 6. Alteration in binding kinetics in a competitive model.

The ancillary subunit changes only the transition rates between the open and drug-bound states.
Use dependence depends on the transition and drug binding rates of the drug–channel combination relative to the stimulation frequency. Changing the conformation of the binding site can result in a dramatic increase or decrease in the rate at which the drug binds and unbinds to the channel. A subunit that increases the rate at which a drug unbinds has the potential to reduce or eliminate use dependence. Conversely, a subunit that decreases the rate at which a drug unbinds from a channel has the potential to increase use dependence. Experimental examples of this include KChIP2b, which eliminates use dependence from Kv4.3, and KCNE1, which introduces use dependence to KCNQ1 (see Sections 3.2 and 3.3).
In summary, a change in the orientation of the residues lining the pore results in:
A change (either an increase or decrease) in the actual affinity of the drug for the channel
No changes in the number of states
Any changes in use dependence will follow changes in the on-rate and/or off-rate of drug binding
2.3 Ancillary subunits can modulate the ability of the channel to undergo conformational changes (e.g. inactivation and closure) following drug block
When a drug binds to the residues lining the pore, there will be a release of energy as the drug and channel interact. This drug–binding site interaction can result in deformation of the channel, which can either promote or inhibit the normal conformational changes associated with channel gating. In addition, the physical presence of the drug can alter the ability of the channel to adopt certain conformational changes it would otherwise undergo. For example, the mere physical presence of a blocker in the pore can prevent deactivation and normal closure of the pore (for reviews see Rasmusson et al. 1994; Bett & Rasmusson, 2002). Alternatively, drug binding to the open pore can compete with the N-terminal domain ‘ball and chain’ binding, and result in a slowing of the development of N-type inactivation (Rasmusson et al. 1994). These ‘foot in the door’ type of mechanisms of drug–channel interactions (Armstrong, 1966) are relatively easy to understand, and result in the relatively simple kinetic predictions (Bett & Rasmusson, 2002). If a channel cannot deactivate, close or inactivate in the presence of an open channel blocking drug, then the drug-bound site becomes an additional blocked state which only has transitions to and from the open state.
For a growing number of channels and drugs it is becoming clear that the presence of a drug bound to an intracellular binding site can promote further conformational changes that confer many of the time- and use-dependent properties of drug action (Baukrowitz & Yellen, 1996; Balser et al. 1996; Rasmusson et al. 1998; Bett & Rasmusson, 2002; Sandtner et al. 2004). Lipophilic open channel blocking compounds can increase the rate of development and stability of the C-type inactivated state (Baukrowitz & Yellen, 1996; Wang et al. 2003; Bett & Rasmusson, 2004). Stabilization of the C-type inactivated state can result in a very slowly recovering open channel block, if the α-subunit has slowly recovering C-type inactivation. Promotion of a stable C-type inactivation mechanism in Kv1.4 is critically dependent upon lipophilic interactions and the movement of S6 (Bett & Rasmusson, 2004). Drug binding that causes allosteric interactions involving the pore mouth and intra- and extracellular domains of S6 can create a long-lived, combined drug-bound and C-type-inactivated state which governs recovery and use dependence. A general picture of binding to the open pore is emerging in which drug binding properties are strongly dependent on the rotational mobility and stability of S6. Movement of S6 links key elements that govern the accessibility and orientation of the high-affinity binding site in response to both activation and inactivation. In addition, the induced conformational changes that follow initial drug binding are critically dependent upon the flexibility and movement of S6.
Any intervention which limits the ability of the channel to adopt certain conformations following drug block will therefore be predicted to alter drug–channel relationships. An ancillary subunit can block post-binding conformational changes either by physically preventing the channel from adopting the conformation or by making transitions less energetically favourable.
Figure 7 shows a schematic representation of the effects of a subunit altering the post-binding conformational changes following drug binding. There are no additional states, and the affinity of the channel for the drug is not affected. Only the transition from the drug-bound to the drug-bound inactivated state is affected. The equivalent state model is given by:
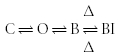 |
Scheme (10) |
This model applies equally well to channels that can close with the drug bound, i.e. exhibit trapping block:
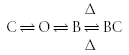 |
Scheme (11) |
where BC is a blocked-closed (deactivated) state.
Figure 7. Changes in post-binding transitions.

Following drug binding, some channels undergo significant conformational changes. If the presence of a subunit alters the ability of the channel to undergo these changes, there will be a change in the transition rates between the drug-bound and drug-bound-inactivated states.
The changes to the transition rates shown in Schemes (10) and (11) will affect the stability of the blocked-inactivated or blocked-closed states. These changes can affect properties such as apparent threshold for activation, recovery from inactivation, and use dependence. Channels that bind drugs which become trapped in the deactivated or closed channel may require a higher applied voltage to activate as the closed/trapped state can become more stable relative to the open state. This would appear as an apparent shift in the voltage of activation. If the presence of a subunit makes the transition to the drug-bound inactivated state less energetically favourable, the inactivated state may be less stable, and recovery from inactivation may be more rapid than without the subunit.
The rate at which the channel recovers from the open state is a key determinant of use dependence. If the subunit creates a relatively unstable inactivated state with rapid recovery from inactivation, this would have the potential to decrease use dependence. Conversely, a subunit that promotes a stable slowly recovering inactivated state has the potential to increase use dependence.
In summary, a change in the ability of the channel to undergo conformational changes following drug binding: (1) Does not affect actual initial binding/affinity; (2) Can affect activation voltage; (3) Affects the stability of the inactivated state; and (4) Affects use dependence
3 Experimental observations of modulation of K+ channel gating and pharmacology by ancillary subunits
The three distinct mechanisms detailed above can co-exist with each other in any channel–subunit pair. In this section we review some experimental data on how drug–channel interactions are modified by the presence of ancillary subunits and test the applicability of the three components of our biophysical model. Ancillary subunits interact with their core α-subunits in a variety of ways including altering cell surface expression, acting as redox sensors, intracellular calcium concentration sensors, etc. (for a review see Patel & Campbell, 2005). For the analysis of the biophysical model, we are only interested in how ancillary subunits modify open channel drug binding.
3.1 Kvβ-subunits and Kv1.X channels
The Kvβs were the first family of voltage-gated K+ channels ancillary subunits to be isolated (Rettig et al. 1994; Scott et al. 1994; Morales et al. 1995; McCormack et al. 1995). Kvβ-subunits are relatively large protein structures, with two functional domains: a core C-terminal domain, which associates with the α-subunit domain, and an N-terminal domain, which varies in length.
Kvβs interact with the Kv1.X family of voltage-gated ion channels, which are homologs of the Shaker channels from Drosophila. Kv1.5 and Kv1.4 α-subunits are thought to be the molecular basis of the ultrarapid delayed rectifier current in human atrium (IKur) (Wang et al. 1994; Li et al. 1996; Yue et al. 1996; Feng et al. 1998) and a slowly recovering transient outward K+ current in mammalian endocardium (Ito,s) (Brahmajothi et al. 1999), respectively. In addition, Kv1.4 and Kv1.5 are thought to be the molecular basis of A-type currents in tissues including brain (Sheng et al. 1993; Chung et al. 2001) and smooth muscle (Lu et al. 2001; Fergus et al. 2003; McGahon et al. 2007).
Kvβ-subunits assemble on the intracellular side of Kv1.X channels, and interact with both N- and C-terminal domains. Most Kvβ-subunits induce a ‘ball and chain’ binding to the open pore mechanism, reminiscent of N-type inactivation in shaker-related channels (Heinemann et al. 1994; Rettig et al. 1994; Morales et al. 1995, 1996; England et al. 1995; Heinemann et al. 1995, 1996). The lipophilic domain of the N-terminus of the β-subunit binds to the same region of the pore as lipophilic channel blockers (Rettig et al. 1994; De Biasi et al. 1997; Rasmusson et al. 1997; Zhou et al. 2001). The addition of rapid N-type inactivation suggests that Kvβs modulate drug binding via a competitive limitation of open channel accessibility in a manner similar to Fig. 2 and Scheme (2).
The introduction of the extra subunit-bound state to the Kv1.X channels predicts that the rate of development of apparent current inactivation/block in the presence of the Kvβ-subunits should be additive in nature, but of a lower apparent affinity. This has been demonstrated for internal block by bupivicaine, quinidine and the anthranilic acid S0100176 in Kv1.5 channels with Kvβ1.3 (Gonzalez et al. 2002; Decher et al. 2005).
There may be additional interactions between Kv1 channels and Kvβ-subunits. The presence of Kvβ slows the rate of recovery from inactivation of Kv1.4, which has an intrinsic N-terminal ‘ball and chain’ inactivation mechanism, but does not affect recovery from inactivation of Kv1.4 ΔN, which is a Kv1.4 construct with the N-terminal removed (Castellino et al. 1995; Accili et al. 1998). This suggests that subunits may have some effect on the inactivated state. The core domain of the β-subunit may interact with the α-subunit in an allosteric way resulting in an alteration of the pore structure, and the arrangement of the drug binding site. Such a possibility is suggested by the ability of the core domain to shift the activation kinetics of Kv1.5 channels (Uebele et al. 1996) and by recent reports of the effects of the core domain of Kvβ1 on inactivation rates of mutant Kv1 channels (Weng et al. 2006 but see Bahring et al. 2001b). However, the dominant mechanism of modulation of drug binding seems to be via competitive interaction in those β-subunits possessing an intact ‘ball and chain’ mechanism.
In summary, Kvβ-subunits may have some mild affect on the orientation of residues forming the binding site and the stability of the bound-inactivated state in Kv1 channels, but the primary mechanism of Kvβ regulation of Kv1 drug–channel interactions appears to be through changes in the accessibility of the drug to the binding site (see Section 2.1).
3.2 KChIPs and Kv4.X channels
Kv channel interacting proteins or KChIPs, are a diverse group of proteins that are part of the large neuronal intracellular calcium-sensing protein family (An et al. 2000; Bahring et al. 2001a; Burgoyne & Weiss, 2001; Nakamura et al. 2001; Jerng et al. 2004; Patel & Campbell, 2005). KChIPs are thought to bind to the N-terminal of the Kv4 family of K+ channels (An et al. 2000).
The Kv4.X family of channels mediate a rapidly recovering transient outward current (Ito,f) in the myocardium (Brahmajothi et al. 1999; Tseng, 1999; Oudit et al. 2001) and an A-type current (ISA) in the brain (Jerng et al. 2004). The Kv4 channel family has been less extensively studied than the Kv1 family in part because of the complex gating of this channel, which remains the subject of debate. Kv4 channels have a complex multi-exponential inactivation process, the molecular basis of which is unclear (Jerng & Covarrubias, 1997; Jerng et al. 1999; Nakamura et al. 2001; Wang et al. 2005). The core α-subunits of the Kv4.X channels recapitulate many of the properties of native Ito,f and ISA, but the gating of the heterologously expressed cloned channels does not match the native currents. Many of the differences in inactivation and recovery between the Kv4s and the cardiac Ito,f can be reconciled by co-expression of Kv4.3 with KChIP2b (Kuo et al. 2001; Patel et al. 2002a,b; Shibata et al. 2003). In the brain, the co-localization of KChIPs and Kv4 channels has heterogeneous regional and cellular distribution (An et al. 2000; Rhodes et al. 2004).
The gating mechanisms underlying Kv4.X channel activity remain highly controversial, as the molecular basis for the components of Kv4.X inactivation do not match the criteria for either the N- or C-type inactivation seen in Kv channels (Rasmusson et al. 1998; Wang et al. 2005). The effect of KChIPs on the inactivation rate of Kv4.3 channels is modest, usually resulting in a mild decrease in the rate of inactivation, depending on the particular KChIP splice variant and the voltage (An et al. 2000; Beck et al. 2002; Deschenes & Tomaselli, 2002; Van Hoorick et al. 2003; Patel et al. 2004).
KChIPs also affect transitions from the inactivated state, dramatically increasing the speed of recovery from inactivation (An et al. 2000; Decher et al. 2001; Patel et al. 2002a; Deschenes & Tomaselli, 2002; Beck et al. 2002; Boland et al. 2003). This suggests that the inactivated state which the channel enters is less stable when the KChIP is present. When recovery from inactivation is measured in the presence of nifedipine, Kv4.3 channels recover more slowly than without the drug. However, when KChIPs are present, nifedipine has no effect on the rate of recovery from inactivation (Bett et al. 2006b). This suggests that not only do KChIPs make the inactivated state less stable, but they also prevent the channel from entering the longer lived drug-bound inactivated state. In other words, KChIPs have altered the ability of Kv4 channels to undergo conformational changes subsequent to drug binding. On a structural level, this could be interpreted as KChIPs reducing the flexibility or ‘stiffening’ the channel, which inhibits the channel from developing the slowly recovering inactivated and inactivated/blocked states which Kv4.3 channels usually enter.
KChIPs affect the ability of several open channel blockers to bind to Kv4 channels. KChIP co-expression results in a decreased sensitivity to Kv4 block by nifedipine, nicardipine, bupivicaine and ropivicaine (Hatano et al. 2003; Friederich & Solth, 2004; Solth et al. 2005; Bett et al. 2006b). If KChIPs modulate drug binding by altering accessibility to the binding site, the fact that KChIPs reduce the speed of inactivation would suggest that drugs have more time to access the binding site, and so should modestly increase the apparent affinity of the drug for the channel. However, the decreased affinity for some drugs suggest that changing the accessibility due to altered gating is not the primary mechanism by which KChIPs modulate drug–channel affinity.
Further evidence for KChIPs having a direct effect on the binding site is the fact that although KChIP2.2 reduces the rate of inactivation of Kv4.3 channels, with the result that accessibility to the binding site is increased, the rate of development of block by ropivicane and bupivacaine are both slowed in the presence of KChIP2.2 (Friederich & Solth, 2004; Solth et al. 2005). These changes to the rate at which the drug binds are opposite to the changes predicted by slowed inactivation gating, which suggests they are a direct result of changes in the affinity of the drug for the open pore binding site, as a consequence of a change in the conformation of the binding site residues.
In summary, KChIPs appear to interact with Kv4 channels by two of the mechanisms in the biophysical model: KChIPs appear to change the orientation of pore residues, resulting in a change in the interaction between drug and channel (see Section 2.2). In addition, KChIPs may affect the stability of the inactivated state, and prevent conformational changes of the channel subsequent to drug binding (see Section 2.3).
3.3 KCNEs and KCNQ1
Although the KCNE subunit proteins are relatively small, they have dramatic effects on activation of voltage-gated channels. KCNE subunits are composed of 57–150 amino acids that cross the membrane, with an intracellular C-terminal and an extracellular N-terminal (McCrossan & Abbott, 2004). KCNE subunits interact with KCNQ1 α-subunits.
KCNQ1 (Kv7.1 or KvLQT1) is a member of the KCNQ K+ channel gene family (Barhanin et al. 1996; Sanguinetti et al. 1996; Wang et al. 1996; Yang & Sigworth, 1998). KCNQ1 is found in an exceedingly diverse range of tissues including the heart, neuronal tissue, kidney, colonic crypt cells, pituitary, the stria vascularis and the vestibular dark cells of the cochlea, stomach, small intestine, liver, thymus, exocrine pancreas, prostate, skeletal muscle, airway epithelia, ovarian tissue, testis, uterus and placenta (Vetter et al. 1996; Wang et al. 1996; Brahmajothi et al. 1996; Chouabe et al. 1997; Neyroud et al. 1997; Yang et al. 1997; Gould & Pfeifer, 1998; Jentsch, 2000; Schroeder et al. 2000; Wulfsen et al. 2000; Demolombe et al. 2001; Mason et al. 2002; Tsevi et al. 2005; Lan et al. 2005).
The tissue-specific characteristics of electrophysiologically recorded KCNQ1 current are diverse, due to strong modification of KCNQ1 by ancillary subunits. In cardiac myocytes, KCNQ1 is co-expressed with a small single transmembrane domain ancillary subunit, KCNE1 (minK). KvLQT1–KCNE1 forms a K+ channel that reproduces the slowly activating delayed-rectifier current (IKs), responsible for repolarization of the cardiac action potential (Sanguinetti et al. 1996). Mutations in KCNQ1 and KCNE1 have been linked to prolonged QT of the cardiac EKG, and fatal arrhythmias (long QT syndrome). KCNQ1–KCNE1 results in currents with greatly slowed kinetics of activation and deactivation, and no voltage-dependent inactivation. In epithelial tissues, KCNQ1 associates with the single transmembrane domain subunit KCNE3 (Schroeder et al. 2000), which produces a constitutively open, voltage-independent K+ current.
Chromanol 293B is a lipophilic drug that is thought to bind to the open pore of KCNQ1 (Seebohm et al. 2001). KCNE subunits have a dramatic effect on the affinity and use dependence of drugs for KCNQ1 (Seebohm et al. 2001; Bett et al. 2006a). The apparent affinities of chromanols 293B, HMR 1556, S5557, as well as azimilide, XE991 and 17 β-oestradiol are increased by over an order of magnitude depending on subunit composition (Busch et al. 1997; Wang et al. 2000; Lerche et al. 2000; Tinel et al. 2000; Schroeder et al. 2000; MacVinish et al. 2001; Bett et al. 2006a).
KCNE3 appears to ‘lock’ the channel in an open position, resulting in a constitutively open channel (Schroeder et al. 2000; Melman et al. 2001). This suggests that part of the increased affinity for KCNQ1–KCNE3 may be caused, at least in part, by increased access time to the binding site when KCNE3 is present. Conversely, KCNE1 greatly increases the sigmoid delay in activation of KCNQ1, and appears to prevent the channel from entering the inactivated state (Seebohm et al. 2005). The net result is that KCNE1 decreases the amount of time the channel spends in the open state, at most physiologically important potentials relative to KCNQ1 expressed alone. However, in both cases, the presence of the subunit greatly increases drug affinity. Therefore, the changes in gating cannot account for changes in affinity. The KCNE subunits probably also modify the orientation of the residues which form the drug binding site. The presence of this mechanism is further supported by mutagenesis results which demonstrate that the kinetic and affinity effects of KCNE3 and KCNE1 can be changed independently (Bett et al. 2006a). When KCNQ1 channels are co-expressed with KCNE3 with the valine at position 72 mutated to a threonine, the channels are no longer constitutively open, but have voltage-dependent gating (Melman et al. 2002). Although the KCNQ1–KCNE3[V72T] channel spends much less time in the open state than the KCNQ1–KCNE3 channels, the high affinity phenotype is retained. This suggests that it is a structural change in the binding site rather than the change in gating which is the dominant factor in determining chromanol 293B affinity. KCNQ1 has been reported to have multiple open states (Pusch et al. 2001), so KCNE subunits may work by modifying the number and/or stability of KCNQ1 open and closed states and the ability of drugs to bind to these states. KCNQ1 alone has a fast early activation which reaches a stable steady state. In contrast, KCNQ1–KCNE1 activation is relatively complex, with a relatively slow sigmoid time course, and a second very slowly developing component. The time-course of open channel block in KCNQ1–KCNE1 channels occurs relatively slowly following channel opening, and may actually reflect a balance between multiple open states such as those reported for the KCNQ1 channel alone (Pusch et al. 2001). These multiple open states may be modulated by KCNE subunits, and may have different affinities for open channel blockers such as chromanol 293B (Bett et al. 2006a).
Rapid repetitive stimulation of KCNQ1–KCNE channels results in a significant increase in channel block, which is not seen in KCNQ1 channels (Seebohm et al. 2001; Bett et al. 2006a). The ability of KCNE1 to introduce use dependence to KCNQ1 channels, even though KCNE1 removes the KCNQ1 inactivated state is consistent with the hypothesis that the KCNE1 subunit alters the conformational changes that the channel undergoes once the drug has bound.
In summary, data currently available on KCNQ1 support the notion that KCNE1 and KCNE3 appear to modify the drug binding properties of KCNQ1 channel by three distinct mechanisms: (1) changing binding site accessibility (see Section 2.1); (2) changing the orientation of pore lining residues thus modifying the binding site (see Section 2.2); and (3) limiting the ability of the channel to undergo gating conformational changes secondary to drug binding (see Section 2.3). However, the relationship between these components and specific drug–channel–subunit interactions remains a current topic of research.
3.4 KCNEs and HERG
In addition to interacting with the KCNQ1 channel, KCNE subunits have been seen to interact with other Kv channels including the HERG channel, or Kv11.1 (McDonald et al. 1997; Abbott et al. 1999; Schroeder et al. 2000; Mazhari et al. 2001; Ohyama et al. 2001; Weerapura et al. 2002; Lu et al. 2003). The HERG α-subunit is thought to be the molecular basis of the voltage-gated K+ channel underlying cardiac IKr (Warmke & Ganetzky, 1994; Trudeau et al. 1995; Sanguinetti et al. 1995). The drug binding site in the intracellular pore has been well described in the HERG channel. In particular, two aromatic residues (tyrosine Y652 and phenylalanine F656) on S6 are thought to play a critical role in drug–channel interactions for several HERG open channel blockers (Mitcheson et al. 2000). Drug binding to the open HERG pore is thought to be highly conformation dependent (see Sanguinetti & Tristani-Firouzi, 2006).
The HERG channel gating has some unique features such as the rate of inactivation being faster than activation at positive potentials. This makes state-dependent analysis of channel activity difficult, and results can be influenced by voltage protocols used to obtain them and the analysis used to interpret the data. Data on the effect of KCNE2 (MiRP1) on HERG gating is therefore understandably mixed. KCNE2 has been suggested to slow or speed the rate at which HERG activates and speeds deactivation, or have little effect at all (Abbott et al. 1999; Mazhari et al. 2001; Weerapura et al. 2002; Lu et al. 2003). The data on the effects of KCNE2 on drug binding are equally mixed. KCNE2 increases the sensitivity of HERG for propranolol (Dupuis et al. 2005). In addition, KCNE2 has been reported to increase HERG sensitivity to E4031, and increase the rate at which use dependence develops (Abbott et al. 1999). Conversely, using a different voltage clamp protocol, KCNE2 was shown to have no effect on E4031 binding (Weerapura et al. 2002). The differences in the voltage protocols used to study E4031 sensitivity suggest that KCNE2 alters HERG conformations. When the protocol involves holding the channel mainly in the open state (0 mV) then briefly stepped to −110 mV for 25 ms, there is no difference in the rate of development or the degree of E4031 block whether KCNE2 is present or not (Weerapura et al. 2002). In contrast, when the holding potential is −80 mV (i.e. the channel is held in the closed state) and stepped to +20 then −40 mV then there is a significant difference in the rate of development and the affinity of E4031 for HERG in the presence of KCNE2 (Abbott et al. 1999). This suggests that KCNE2 does not affect E4031 access to the binding site in the open state, but does modify access of E4031 to the drug binding site in the closed state. The orientation of the binding site residues will be different in the open and closed states of the channels, so the affinity for closed and open states should be different.
Not all HERG drug affinities appear to be affected by the presence of KCNE2. There are several binding sites in the HERG channels, and KCNE2 may not enable access to all of this sites. In addition, some drugs have more restrictive requirements for a binding site. Drugs which are reported to have no change in affinity by KCNE2 include local anaesthetics (Friederich et al. 2004), sulfamethoxazole (Park et al. 2003), terfenadine (Scherer et al. 2002) and vesnarinone (Kamiya et al. 2001). Terfenadine interacts with Y652 and F656, but not the binding sites deeper within the pore V625, G648 and T623 (Mitcheson et al. 2000). Vesnarinone interacts with G648, F656 and V659 located in the S6 domain and T623, S624 and V625 located at the base of the pore helix (Kamiya et al. 2001).
In summary, the interaction between HERG and KCNE2 remains a subject of current research. However, KCNE2 appears to alter accessibility to the binding site for some drugs, by enabling binding to the closed state.
Conclusions
Ancillary subunits strongly modulate open channel block by drugs as well as modifying basic α-subunit gating. This subunit-specific pharmacology suggests that drugs that are targeted to specific α-subunits in specific organs can be made more specific by considering the characteristics of associated subunits found in these tissues. Furthermore, the kinetic differences in gating and the corresponding differences in drug use dependence suggest that the response of channels to a drug will depend on the patterns of electrical activity of a particular organ or cell type, providing the opportunity for additional specificity of action or reduction in side-effects. However, the drug-modifying properties of some subunits may be very limited. Drugs which change the structure of the binding site may alter the pharmacological profile of a channel in a qualitative as well as quantitative fashion. Conversely, subunits which act primarily by changing binding site access time, may not confer altered specificity, but may just raise or lower the apparent degree of channel inhibition at a specific concentration or pattern of electrical activity. Changing the relationship between drug binding and conformational changes should strongly alter use-dependent properties and may confer specific inhibitory action to abnormal electrical behaviour, such as seizures or arrhythmias. Better understanding of the molecular properties of drug–subunit interactions will help in the development of organ-specific pharmacological therapies, and the reduction of adverse side-effects associated with current pharmacological interventions.
References
- Abbott GW, Goldstein SA. Potassium channel subunits encoded by the KCNE gene family: physiology and pathophysiology of the MinK-related peptides (MiRPs) Mol Interv. 2001;1:95–107. [PubMed] [Google Scholar]
- Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SA. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–187. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- Abbott GW, Butler MH, Bendahhou S, Dalakas MC, Ptacek LJ, Goldstein SA. MiRP2 forms potassium channels in skeletal muscle with Kv3.4 and is associated with periodic paralysis. Cell. 2001;104:217–231. doi: 10.1016/s0092-8674(01)00207-0. [DOI] [PubMed] [Google Scholar]
- Accili EA, Kuryshev YA, Wible BA, Brown AM. Separable effects of human Kvβ1.2 N- and C-termini on inactivation and expression of human Kv1.4. J Physiol. 1998;512:325–336. doi: 10.1111/j.1469-7793.1998.325be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelman JP, Bond CT, Pessia M, Maylie J. Episodic ataxia results from voltage-dependent potassium channels with altered functions. Neuron. 1995;15:1449–1454. doi: 10.1016/0896-6273(95)90022-5. [DOI] [PubMed] [Google Scholar]
- An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KI, Strassle BW, Trimmer JS, Rhodes KJ. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 2000;403:553–556. doi: 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- Andalib P, Consiglio JF, Trapani JG, Korn SJ. The external TEA binding site and C-type inactivation in voltage-gated potassium channels. Biophys J. 2004;87:3148–3161. doi: 10.1529/biophysj.104.046664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelo K, Jespersen T, Grunnet M, Nielsen MS, Klaerke DA, Olesen SP. KCNE5 induces time- and voltage-dependent modulation of the KCNQ1 current. Biophys J. 2002;83:1997–2006. doi: 10.1016/S0006-3495(02)73961-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM. Time course of TEA+-induced anomalous recification in squid giant axons. J Gen Physiol. 1966;50:491–503. doi: 10.1085/jgp.50.2.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM. Inactivation of the potassium conductance and related phenomena caused by quaternary ammonium ion injection in squid axons. J Gen Physiol. 1969;54:553–575. doi: 10.1085/jgp.54.5.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J Gen Physiol. 1971;58:413–437. doi: 10.1085/jgp.58.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahring R, Dannenberg J, Peters HC, Leicher T, Pongs O, Isbrandt D. Conserved Kv4 N-terminal domain critical for effects of Kv channel-interacting protein 2.2 on channel expression and gating. J Biol Chem. 2001a;276:23888–23894. doi: 10.1074/jbc.M101320200. [DOI] [PubMed] [Google Scholar]
- Bahring R, Milligan CJ, Vardanyan V, Engeland B, Young BA, Dannenberg J, Waldschutz R, Edwards JP, Wray D, Pongs O. Coupling of voltage-dependent potassium channel inactivation and oxidoreductase active site of Kvβ subunits. J Biol Chem. 2001b;276:22923–22929. doi: 10.1074/jbc.M100483200. [DOI] [PubMed] [Google Scholar]
- Balser JR, Nuss HB, Orias DW, Johns DC, Marban E, Tomaselli GF, Lawrence JH. Local anesthetics as effectors of allosteric gating. Lidocaine effects on inactivation-deficient rat skeletal muscle Na channels. J Clin Invest. 1996;98:2874–2886. doi: 10.1172/JCI119116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. KVLQT1 and lsK (minK) proteins associate to form the IKs cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Yellen G. Use-dependent blockers and exit rate of the last ion from the multi-ion pore of a K+ channel. Science. 1996;271:653–656. doi: 10.1126/science.271.5249.653. [DOI] [PubMed] [Google Scholar]
- Beck EJ, Bowlby M, An WF, Rhodes KJ, Covarrubias M. Remodelling inactivation gating of Kv4 channels by KChIP1, a small-molecular-weight calcium-binding protein. J Physiol. 2002;538:691–706. doi: 10.1113/jphysiol.2001.013127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendahhou S, Marionneau C, Haurogne K, Larroque MM, Derand R, Szuts V, Escande D, Demolombe S, Barhanin J. In vitro molecular interactions and distribution of KCNE family with KCNQ1 in the human heart. Cardiovasc Res. 2005;67:529–538. doi: 10.1016/j.cardiores.2005.02.014. [DOI] [PubMed] [Google Scholar]
- Bett GCL, Morales MJ, Beahm DL, Duffey ME, Rasmusson RL. Ancillary subunits and stimulation frequency determine the potency of chromanol 293B block of the KCNQ1 potassium channel. J Physiol. 2006a;576:755–767. doi: 10.1113/jphysiol.2006.116012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett GCL, Morales MJ, Strauss HC, Rasmusson RL. KChIP2b modulates the affinity and use-dependent block of Kv4.3 by nifedipine. Biochem Biophys Res Commun. 2006b;340:1167–1177. doi: 10.1016/j.bbrc.2005.12.135. [DOI] [PubMed] [Google Scholar]
- Bett GCL, Rasmusson RL. Models of cardiac ion channels. In: Cabo C, Rosenbaum DS, editors. Quantitative Cardiac Electrophysiology. New York, NY: Marcel Dekker, Inc.; 2002. pp. 1–60. [Google Scholar]
- Bett GCL, Rasmusson RL. Inactivation and recovery in Kv1.4 K+ channels: lipophilic interactions at the intracellular mouth of the pore. J Physiol. 2004;556:109–120. doi: 10.1113/jphysiol.2003.055012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F. Voltage sensor movements. J Gen Physiol. 2002;120:465–473. doi: 10.1085/jgp.20028660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum SG, Varga AW, Yuan LL, Anderson AE, Sweatt JD, Schrader LA. Structure and function of Kv4-family transient potassium channels. Physiol Rev. 2004;84:803–833. doi: 10.1152/physrev.00039.2003. [DOI] [PubMed] [Google Scholar]
- Boland LM, Jiang M, Lee SY, Fahrenkrug SC, Harnett MT, O/Grady SM. Functional properties of a brain-specific NH2-terminally spliced modulator of Kv4 channels. Am J Physiol Cell Physiol. 2003;285:C161–C170. doi: 10.1152/ajpcell.00416.2002. [DOI] [PubMed] [Google Scholar]
- Brahmajothi MV, Campbell DL, Rasmusson RL, Morales MJ, Trimmer JS, Nerbonne JM, Strauss HC. Distinct transient outward potassium current (Ito) phenotypes and distribution of fast-inactivating potassium channel alpha subunits in ferret left ventricular myocytes. J Gen Physiol. 1999;113:581–600. doi: 10.1085/jgp.113.4.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmajothi MV, Morales MJ, Liu S, Rasmusson RL, Campbell DL, Strauss HC. In situ hybridization reveals extensive diversity of K+ channel mRNA in isolated ferret cardiac myocytes. Circ Res. 1996;78:1083–1089. doi: 10.1161/01.res.78.6.1083. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD, Weiss JL. The neuronal calcium sensor family of Ca2+-binding proteins. Biochem J. 2001;353:1–12. [PMC free article] [PubMed] [Google Scholar]
- Busch AE, Busch GL, Ford E, Suessbrich H, Lang HJ, Greger R, Kunzelmann K, Attali B, Stuhmer W. The role of the IsK protein in the specific pharmacological properties of the IKs channel complex. Br J Pharmacol. 1997;122:187–189. doi: 10.1038/sj.bjp.0701434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch AE, Hurst RS, North RA, Adelman JP, Kavanaugh MP. Current inactivation involves a histidine residue in the pore of the rat lymphocyte potassium channel RGK5. Biochem Biophys Res Comms. 1991;179:1384–1390. doi: 10.1016/0006-291x(91)91726-s. [DOI] [PubMed] [Google Scholar]
- Caballero R, Pourrier M, Schram G, Delpon E, Tamargo J, Nattel S. Effects of flecainide and quinidine on Kv4.2 currents: voltage dependence and role of S6 valines. Br J Pharmacol. 2003;138:1475–1484. doi: 10.1038/sj.bjp.0705199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellino RC, Morales MJ, Strauss HC, Rasmusson RL. Time- and voltage-dependent modulation of a Kv1.4 channel by a β-subunit (Kvβ3) cloned from ferret ventricle. Am J Physiol Heart Circ Physiol. 1995;269:H385–H391. doi: 10.1152/ajpheart.1995.269.1.H385. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Structure and function of voltage-gated ion channels. Annu Rev Biochem. 1995;64:493–531. doi: 10.1146/annurev.bi.64.070195.002425. [DOI] [PubMed] [Google Scholar]
- Chen J, Seebohm G, Sanguinetti MC. Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. Proc Natl Acad Sci U S A. 2002;99:12461–12466. doi: 10.1073/pnas.192367299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CE, Roden DM. The long QT syndromes: genetic basis and clinical implications. J Am College Cardiol. 2000;36:1–12. doi: 10.1016/s0735-1097(00)00716-6. [DOI] [PubMed] [Google Scholar]
- Choi KL, Aldrich RW, Yellen G. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc Natl Acad Sci U S A. 1991;88:5092–5095. doi: 10.1073/pnas.88.12.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouabe C, Neyroud N, Guicheney P, Lazdunski M, Romey G, Barhanin J. Properties of KvLQT1 K+ channel mutations in Romano-Ward and Jervell and Lange-Nielsen inherited cardiac arrhythmias. EMBO J. 1997;16:5472–5479. doi: 10.1093/emboj/16.17.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung YH, Shin C, Kim MJ, Lee BK, Cha CI. Immunohistochemical study on the distribution of six members of the Kv1 channel subunits in the rat cerebellum. Brain Res. 2001;895:173–177. doi: 10.1016/s0006-8993(01)02068-6. [DOI] [PubMed] [Google Scholar]
- Conti M. Targeting K+ channels for cancer therapy. J Exp Ther Oncol. 2004;4:161–166. [PubMed] [Google Scholar]
- Cordero-Morales JF, Jogini V, Lewis A, Vasquez V, Cortes DM, Roux B, Perozo E. Molecular driving forces determining potassium channel slow inactivation. Nat Struct Mol Biol. 2007;14:1062–1069. doi: 10.1038/nsmb1309. [DOI] [PubMed] [Google Scholar]
- Cuello LG, Cortes DM, Perozo E. Molecular architecture of the KvAP voltage-dependent K+ channel in a lipid bilayer. Science. 2004;306:491–495. doi: 10.1126/science.1101373. [DOI] [PubMed] [Google Scholar]
- Davis TM, Dembo LG, Kaye-Eddie SA, Hewitt BJ, Hislop RG, Batty KT. Neurological, cardiovascular and metabolic effects of mefloquine in healthy volunteers: a double-blind, placebo-controlled trial. Br J Clin Pharmacol. 1996;42:415–421. doi: 10.1046/j.1365-2125.1996.04745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Biasi M, Wang Z, Accili E, Wible B, Fedida D. Open channel block of human heart hKv1.5 by the β-subunit hKvβ1.2. Am J Physiol Heart Circ Physiol. 1997;272:H2932–H2941. doi: 10.1152/ajpheart.1997.272.6.H2932. [DOI] [PubMed] [Google Scholar]
- Decher N, Kumar P, Gonzalez T, Renigunta V, Sanguinetti MC. Structural basis for competition between drug binding and Kvβ 1.3 accessory subunit-induced N-type inactivation of Kv1.5 channels. Mol Pharmacol. 2005;68:995–1005. doi: 10.1124/mol.105.011668. [DOI] [PubMed] [Google Scholar]
- Decher N, Uyguner O, Scherer CR, Karaman B, Yuksel-Apak M, Busch AE, Steinmeyer K, Wollnik B. hKChIP2 is a functional modifier of hKv4.3 potassium channels: cloning and expression of a short hKChIP2 splice variant. Cardiovasc Res. 2001;52:255–264. doi: 10.1016/s0008-6363(01)00374-1. [DOI] [PubMed] [Google Scholar]
- del Camino D, Yellen G. Tight steric closure at the intracellular activation gate of a voltage-gated K+ channel. Neuron. 2001;32:649–656. doi: 10.1016/s0896-6273(01)00487-1. [DOI] [PubMed] [Google Scholar]
- Demo SD, Yellen G. The inactivation gate of the Shaker K+ channel behaves like an open-channel blocker. Neuron. 1991;7:743–753. doi: 10.1016/0896-6273(91)90277-7. [DOI] [PubMed] [Google Scholar]
- Demolombe S, Franco D, de Boer P, Kuperschmidt S, Roden D, Pereon Y, Jarry A, Moorman AF, Escande D. Differential expression of KvLQT1 and its regulator IsK in mouse epithelia. Am J Physiol Cell Physiol. 2001;280:C359–C372. doi: 10.1152/ajpcell.2001.280.2.C359. [DOI] [PubMed] [Google Scholar]
- Deschenes I, Tomaselli GF. Modulation of Kv4.3 current by accessory subunits. FEBS Lett. 2002;528:183–188. doi: 10.1016/s0014-5793(02)03296-9. [DOI] [PubMed] [Google Scholar]
- Doyle DA, Morais CJ, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Dupuis DS, Klaerke DA, Olesen SP. Effect of β-adrenoceptor blockers on human ether-a-go-go-related gene (HERG) potassium channels. Basic Clin Pharmacol Toxicol. 2005;96:123–130. doi: 10.1111/j.1742-7843.2005.pto960206.x. [DOI] [PubMed] [Google Scholar]
- England SK, Uebele VN, Kodali J, Bennett PB, Tamkun MM. A novel K+ channel β-subunit (hKvβ1.3) is produced via alternative mRNA splicing. J Biol Chem. 1995;270:28531–28534. doi: 10.1074/jbc.270.48.28531. [DOI] [PubMed] [Google Scholar]
- England SK, Uebele VN, Shear H, Kodali J, Bennett PB, Tamkun MM. Characterization of a voltage-gated K+ channel beta subunit expressed in human heart. Proc Natl Acad Sci USA. 1995;92:6309–6313. doi: 10.1073/pnas.92.14.6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escande D. Pharmacogenetics of cardiac K+ channels. Eur J Pharmacol. 2000;410:281–287. doi: 10.1016/s0014-2999(00)00821-9. [DOI] [PubMed] [Google Scholar]
- Feng J, Xu D, Wang Z, Nattel S. Ultrarapid delayed rectifier current inactivation in human atrial myocytes: properties and consequences. Am J Physiol Heart Circ Physiol. 1998;275:H1717–H1725. doi: 10.1152/ajpheart.1998.275.5.H1717. [DOI] [PubMed] [Google Scholar]
- Fergus DJ, Martens JR, England SK. Kv channel subunits that contribute to voltage-gated K+ current in renal vascular smooth muscle. Pflugers Arch. 2003;445:697–704. doi: 10.1007/s00424-002-0994-7. [DOI] [PubMed] [Google Scholar]
- Friederich P, Solth A. Interaction of ropivacaine with cloned cardiac Kv4.3/KChIP2.2 complexes. Anesthesiology. 2004;101:1347–1356. doi: 10.1097/00000542-200412000-00015. [DOI] [PubMed] [Google Scholar]
- Friederich P, Solth A, Schillemeit S, Isbrandt D. Local anaesthetic sensitivities of cloned HERG channels from human heart: comparison with HERG/MiRP1 and HERG/MiRP1 T8A. Br J Anaesth. 2004;92:93–101. doi: 10.1093/bja/aeh026. [DOI] [PubMed] [Google Scholar]
- Gintant GA, Hoffman BF. Use-dependent block of cardiac sodium channels by quaternary derivatives of lidocaine. Pflugers Arch. 1984;400:121–129. doi: 10.1007/BF00585029. [DOI] [PubMed] [Google Scholar]
- Gonzalez T, Navarro-Polanco R, Arias C, Caballero R, Moreno I, Delpon E, Tamargo J, Tamkun MM, Valenzuela C. Assembly with the Kvβ1.3 subunit modulates drug block of hKv1.5 channels. Mol Pharmacol. 2002;62:1456–1463. doi: 10.1124/mol.62.6.1456. [DOI] [PubMed] [Google Scholar]
- Gould TD, Pfeifer K. Imprinting of mouse Kvlqt1 is developmentally regulated. Hum Mol Genet. 1998;7:483–487. doi: 10.1093/hmg/7.3.483. [DOI] [PubMed] [Google Scholar]
- Grissmer S, Nguyen AN, Aiyar J, Hanson DC, Mather RJ, Gutman GA, Karmilowicz MJ, Auperin DD, Chandy KG. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol. 1994;45:1227–1234. [PubMed] [Google Scholar]
- Hatano N, Ohya S, Muraki K, Giles W, Imaizumi Y. Dihydropyridine Ca2+ channel antagonists and agonists block Kv4.2, Kv4.3 and Kv1.4 K+ channels expressed in HEK293 cells. Br J Pharmacol. 2003;139:533–544. doi: 10.1038/sj.bjp.0705281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann S, Rettig J, Scott V, Parcej DN, Lorra C, Dolly J, Pongs O. The inactivation behaviour of voltage-gated K-channels may be determined by association of α- and β-subunits. J Physiol (Paris) 1994;88:173–180. doi: 10.1016/0928-4257(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Heinemann SH, Rettig J, Graack HR, Pongs O. Functional characterization of Kv channel β-subunits from rat brain. J Physiol. 1996;493:625–633. doi: 10.1113/jphysiol.1996.sp021409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann SH, Rettig J, Wunder F, Pongs O. Molecular and functional characterization of a rat brain Kvβ3 potassium channel subunit. FEBS Lett. 1995;377:383–389. doi: 10.1016/0014-5793(95)01377-6. [DOI] [PubMed] [Google Scholar]
- Hille B. Ion Channels of Excitable Membranes. 3. Sunderland, Massachusetts: Sinauer Associates Inc.; 2001. [Google Scholar]
- Holmgren M, Shin KS, Yellen G. The activation gate of a voltage-gated K+ channel can be trapped in the open state by an intersubunit metal bridge. Neuron. 1998;21:617–621. doi: 10.1016/s0896-6273(00)80571-1. [DOI] [PubMed] [Google Scholar]
- Hondeghem LM, Katzung BG. Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim Biophys Acta. 1977;472:373–398. doi: 10.1016/0304-4157(77)90003-x. [DOI] [PubMed] [Google Scholar]
- Hondeghem LM, Katzung BG. Antiarrhythmic agents: the modulated receptor mechanism of action of sodium and calcium channel-blocking drugs. Annu Rev Pharmacol Toxicol. 1984;24:387–423. doi: 10.1146/annurev.pa.24.040184.002131. [DOI] [PubMed] [Google Scholar]
- Hondeghem LM, Snyders DJ. Class III antiarrhythmic agents have a lot of potential but a long way to go. Reduced effectiveness and dangers of reverse use dependence. Circulation. 1990;81:686–690. doi: 10.1161/01.cir.81.2.686. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Isom LL, De Jongh KS, Catterall WA. Auxiliary subunits of voltage-gated ion channels. Neuron. 1994;12:1183–1194. doi: 10.1016/0896-6273(94)90436-7. [DOI] [PubMed] [Google Scholar]
- Jan LY, Jan YN. Structural elements involved in specific K+ channel functions. Annu Rev Physiol. 1992;54:537–555. doi: 10.1146/annurev.ph.54.030192.002541. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- Jerng HH, Covarrubias M. K+ channel inactivation mediated by the concerted action of the cytoplasmic N- and C-terminal domains. Biophys J. 1997;72:163–174. doi: 10.1016/S0006-3495(97)78655-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng HH, Pfaffinger PJ, Covarrubias M. Molecular physiology and modulation of somatodendritic A-type potassium channels. Mol Cell Neurosci. 2004;27:343–369. doi: 10.1016/j.mcn.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Jerng HH, Shahidullah M, Covarrubias M. Inactivation gating of Kv4 potassium channels: molecular interactions involving the inner vestibule of the pore. J Gen Physiol. 1999;113:641–660. doi: 10.1085/jgp.113.5.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Bett GC, Li X, Bondarenko VE, Rasmusson RL. C-type inactivation involves a significant decrease in the intracellular aqueous pore volume of Kv1.4 K+ channels expressed in Xenopus oocytes. J Physiol. 2003;549:683–695. doi: 10.1113/jphysiol.2002.034660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. The open pore conformation of potassium channels. Nature. 2002;417:523–526. doi: 10.1038/417523a. [DOI] [PubMed] [Google Scholar]
- Jo SH, Youm JB, Lee CO, Earm YE, Ho WK. Blockade of the HERG human cardiac K+ channel by the antidepressant drug amitriptyline. Br J Pharmacol. 2000;129:1474–1480. doi: 10.1038/sj.bjp.0703222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya K, Mitcheson JS, Yasui K, Kodama I, Sanguinetti MC. Open channel block of HERG K+ channels by vesnarinone. Mol Pharmacol. 2001;60:244–253. doi: 10.1124/mol.60.2.244. [DOI] [PubMed] [Google Scholar]
- Kiss L, LoTurco J, Korn SJ. Contribution of the selectivity filter to inactivation in potassium channels. Biophys J. 1999;76:253–263. doi: 10.1016/S0006-3495(99)77194-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo HC, Cheng CF, Clark RB, Lin JJ, Lin JL, Hoshijima M, Nguyen-Tran VT, Gu Y, Ikeda Y, Chu PH, Ross J, Giles WR, Chien KR. A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of Ito and confers susceptibility to ventricular tachycardia. Cell. 2001;107:801–813. doi: 10.1016/s0092-8674(01)00588-8. [DOI] [PubMed] [Google Scholar]
- Kurata HT, Doerksen KW, Eldstrom JR, Rezazadeh S, Fedida D. Separation of P/C- and U-type inactivation pathways in Kv1.5 potassium channels. J Physiol. 2005;568:31–46. doi: 10.1113/jphysiol.2005.087148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushida S, Ogura T, Komuro I, Nakaya H. Inhibitory effect of the class III antiarrhythmic drug nifekalant on HERG channels: mode of action. Eur J Pharmacol. 2002;457:19–27. doi: 10.1016/s0014-2999(02)02666-3. [DOI] [PubMed] [Google Scholar]
- Labro AJ, Raes AL, Bellens I, Ottschytsch N, Snyders DJ. Gating of shaker-type channels requires the flexibility of S6 caused by prolines. J Biol Chem. 2003;278:50724–50731. doi: 10.1074/jbc.M306097200. [DOI] [PubMed] [Google Scholar]
- Lan WZ, Abbas H, Lemay AM, Briggs MM, Hill CE. Electrophysiological and molecular identification of hepatocellular volume-activated K+ channels. Biochim Biophys Acta. 2005;1668:223–233. doi: 10.1016/j.bbamem.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Lee SY, Lee A, Chen J, MacKinnon R. Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc Natl Acad Sci U S A. 2005;102:15441–15446. doi: 10.1073/pnas.0507651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerche C, Seebohm G, Wagner CI, Scherer CR, Dehmelt L, Abitbol I, Gerlach U, Brendel J, Attali B, Busch AE. Molecular impact of MinK on the enantiospecific block of IKs by chromanols. Br J Pharmacol. 2000;131:1503–1506. doi: 10.1038/sj.bjp.0703734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis A, McCrossan ZA, Abbott GW. MinK, MiRP1, and MiRP2 diversify Kv3.1 and Kv3.2 potassium channel gating. J Biol Chem. 2004;279:7884–7892. doi: 10.1074/jbc.M310501200. [DOI] [PubMed] [Google Scholar]
- Li GR, Feng J, Wang Z, Fermini B, Nattel S. Adrenergic modulation of ultrarapid delayed rectifier K+ current in human atrial myocytes. Circulation Res. 1996;78:903–915. doi: 10.1161/01.res.78.5.903. [DOI] [PubMed] [Google Scholar]
- Li X, Bett GC, Jiang X, Bondarenko VE, Morales MJ, Rasmusson RL. Regulation of N- and C-type inactivation of Kv1.4 by pHo and K+: evidence for transmembrane communication. Am J Physiol Heart Circ Physiol. 2003;284:H71–H80. doi: 10.1152/ajpheart.00392.2002. [DOI] [PubMed] [Google Scholar]
- Lipkind GM, Hanck DA, Fozzard HA. A structural motif for the voltage-gated potassium channel pore. Proc Natl Acad Sci U S A. 1995;92:9215–9219. doi: 10.1073/pnas.92.20.9215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XS, Jiang M, Zhang M, Tang D, Clemo HF, Higgins RS, Tseng GN. Electrical remodeling in a canine model of ischemic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2007;292:H560–H571. doi: 10.1152/ajpheart.00616.2006. [DOI] [PubMed] [Google Scholar]
- Liu YS, Sompornpisut P, Perozo E. Structure of the KcsA channel intracellular gate in the open state. Nat Struct Biol. 2001;8:883–887. doi: 10.1038/nsb1001-883. [DOI] [PubMed] [Google Scholar]
- Long SB, Campbell EB, MacKinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005a;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- Long SB, Campbell EB, MacKinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005b;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Lu Y, Mahaut-Smith MP, Huang CL, Vandenberg JI. Mutant MiRP1 subunits modulate HERG K+ channel gating: a mechanism for pro-arrhythmia in long QT syndrome type 6. J Physiol. 2003;551:253–262. doi: 10.1113/jphysiol.2003.046045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhang J, Tang G, Wang R. Modulation of voltage-dependent K+ channel current in vascular smooth muscle cells from rat mesenteric arteries. J Membr Biol. 2001;180:163–175. doi: 10.1007/s002320010067. [DOI] [PubMed] [Google Scholar]
- Lundby A, Olesen SP. KCNE3 is an inhibitory subunit of the Kv4.3 potassium channel. Biochem Biophys Res Commun. 2006;346:958–967. doi: 10.1016/j.bbrc.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Majumder K, De Biasi M, Wang Z, Wible BA. Molecular cloning and functional expression of a novel potassium channel beta-subunit from human atrium. FEBS Lett. 1995;361:13–16. doi: 10.1016/0014-5793(95)00120-x. [DOI] [PubMed] [Google Scholar]
- McCormack K, McCormack T, Tanouye M, Rudy B, Stuhmer W. Alternative splicing of the human Shaker K+ channel β1 gene and functional expression of the β2 gene product. FEBS Lett. 1995;370:32–36. doi: 10.1016/0014-5793(95)00785-8. [DOI] [PubMed] [Google Scholar]
- McCrossan ZA, Abbott GW. The MinK-related peptides. Neuropharmacology. 2004;47:787–821. doi: 10.1016/j.neuropharm.2004.06.018. [DOI] [PubMed] [Google Scholar]
- McCrossan ZA, Lewis A, Panaghie G, Jordan PN, Christini DJ, Lerner DJ, Abbott GW. MinK-related peptide 2 modulates Kv2.1 and Kv3.1 potassium channels in mammalian brain. J Neurosci. 2003;23:8077–8091. doi: 10.1523/JNEUROSCI.23-22-08077.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald TV, Yu Z, Ming Z, Palma E, Meyers MB, Wang KW, Goldstein SA, Fishman GI. A minK-HERG complex regulates the cardiac potassium current IKr. Nature. 1997;388:289–292. doi: 10.1038/40882. [DOI] [PubMed] [Google Scholar]
- McGahon MK, Dawicki JM, Arora A, Simpson DA, Gardiner TA, Stitt A, Scholfield CN, McGeown G, Curtis T. Kv1.5 is a major component underlying the A-type potassium current in retinal arteriolar smooth muscle. Am J Physiol Heart Circ Physiol. 2007;292:H1001–H1008. doi: 10.1152/ajpheart.01003.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacVinish LJ, Guo Y, Dixon AK, Murrell-Lagnado RD, Cuthbert AW. Xe991 reveals differences in K+ channels regulating chloride secretion in murine airway and colonic epithelium. Mol Pharmacol. 2001;60:753–760. [PubMed] [Google Scholar]
- Mannuzzu LM, Moronne MM, Isacoff EY. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 1996;271:213–216. doi: 10.1126/science.271.5246.213. [DOI] [PubMed] [Google Scholar]
- Mason DE, Mitchell KE, Li Y, Finley MR, Freeman LC. Molecular basis of voltage-dependent potassium currents in porcine granulosa cells. Mol Pharmacol. 2002;61:201–213. doi: 10.1124/mol.61.1.201. [DOI] [PubMed] [Google Scholar]
- Mazhari R, Greenstein JL, Winslow RL, Marban E, Nuss HB. Molecular interactions between two long-QT syndrome gene products, HERG and KCNE2, rationalized by in vitro and in silico analysis. Circulation Res. 2001;89:33–38. doi: 10.1161/hh1301.093633. [DOI] [PubMed] [Google Scholar]
- Melman YF, Domenech A, de la Luna S, McDonald TV. Structural determinants of KvLQT1 control by the KCNE family of proteins. J Biol Chem. 2001;276:6439–6444. doi: 10.1074/jbc.M010713200. [DOI] [PubMed] [Google Scholar]
- Melman YF, Krumerman A, McDonald TV. A single transmembrane site in the KCNE-encoded proteins controls the specificity of KvLQT1 channel gating. J Biol Chem. 2002;277:25187–25194. doi: 10.1074/jbc.M200564200. [DOI] [PubMed] [Google Scholar]
- Melnyk P, Ehrlich JR, Pourrier M, Villeneuve L, Cha TJ, Nattel S. Comparison of ion channel distribution and expression in cardiomyocytes of canine pulmonary veins versus left atrium. Cardiovasc Res. 2005;65:104–116. doi: 10.1016/j.cardiores.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci U S A. 2000;97:12329–12333. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales MJ, Castellino RC, Crews AL, Rasmusson RL, Strauss HC. A novel β subunit increases rate of inactivation of specific voltage-gated potassium channel α subunits. J Biol Chem. 1995;270:6272–6277. doi: 10.1074/jbc.270.11.6272. [DOI] [PubMed] [Google Scholar]
- Morales MJ, Wee JO, Wang S, Strauss HC, Rasmusson RL. The N-terminal domain of a K+ channel β subunit increases the rate of C-type inactivation from the cytoplasmic side of the channel. Proc Natl Acad Sci U S A. 1996;93:15119–15123. doi: 10.1073/pnas.93.26.15119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura TY, Pountney DJ, Ozaita A, Nandi S, Ueda S, Rudy B, Coetzee WA. A role for frequenin, a Ca2+-binding protein, as a regulator of Kv4 K+-currents. Proc Natl Acad Sci U S A. 2001;98:12808–12813. doi: 10.1073/pnas.221168498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne JM. Regulation of voltage-gated K+ channel expression in the developing mammalian myocardium. J Neurobiol. 1998;37:37–59. doi: 10.1002/(sici)1097-4695(199810)37:1<37::aid-neu4>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, Faure S, Gary F, Coumel P, Petit C, Schwartz K, Guicheney P. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange–Nielsen cardioauditory syndrome. Nat Genet. 1997;15:186–189. doi: 10.1038/ng0297-186. [DOI] [PubMed] [Google Scholar]
- Ogielska EM, Zagotta WN, Hoshi T, Heinemann SH, Haab J, Aldrich RW. Cooperative subunit interactions in C-type inactivation of K channels. Biophys J. 1995;69:2449–2457. doi: 10.1016/S0006-3495(95)80114-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyama H, Kajita H, Omori K, Takumi T, Hiramoto N, Iwasaka T, Matsuda H. Inhibition of cardiac delayed rectifier K+ currents by an antisense oligodeoxynucleotide against IsK (minK) and over-expression of IsK mutant D77N in neonatal mouse hearts. Pflugers Arch. 2001;442:329–335. doi: 10.1007/s004240100547. [DOI] [PubMed] [Google Scholar]
- Oudit GY, Kassiri Z, Sah R, Ramirez RJ, Zobel C, Backx PH. The molecular physiology of the cardiac transient outward potassium current (Ito) in normal and diseased myocardium. J Mol Cellular Cardiol. 2001;33:851–872. doi: 10.1006/jmcc.2001.1376. [DOI] [PubMed] [Google Scholar]
- Panyi G, Deutsch C. Probing the cavity of the slow inactivated conformation of shaker potassium channels. J Gen Physiol. 2007;129:403–418. doi: 10.1085/jgp.200709758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KH, Kwok SM, Sharon C, Berga R, Sesti F. N-Glycosylation-dependent block is a novel mechanism for drug-induced cardiac arrhythmia. FASEB J. 2003;17:2308–2309. doi: 10.1096/fj.03-0577fje. [DOI] [PubMed] [Google Scholar]
- Patel SP, Campbell DL. Transient outward potassium current, ‘Ito’, phenotypes in the mammalian left ventricle: underlying molecular, cellular and biophysical mechanisms. J Physiol. 2005;569:7–39. doi: 10.1113/jphysiol.2005.086223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Campbell DL, Morales MJ, Strauss HC. Heterogeneous expression of KChIP2 isoforms in the ferret heart. J Physiol. 2002a;539:649–656. doi: 10.1113/jphysiol.2001.015156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Campbell DL, Strauss HC. Elucidating KChIP effects on Kv4.3 inactivation and recovery kinetics with a minimal KChIP2 isoform. J Physiol. 2002b;545:5–11. doi: 10.1113/jphysiol.2002.031856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Parai R, Parai R, Campbell DL. Regulation of Kv4.3 voltage-dependent gating kinetics by KChIP2 isoforms. J Physiol. 2004;557:19–41. doi: 10.1113/jphysiol.2003.058172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson F, Carlsson L, Duker G, Jacobson I. Blocking characteristics of hERG, hNav1.5, and hKvLQT1/hminK after administration of the novel anti-arrhythmic compound AZD7009. J Cardiovasc Electrophysiol. 2005;16:329–341. doi: 10.1046/j.1540-8167.2005.40427.x. [DOI] [PubMed] [Google Scholar]
- Pongs O. Molecular biology of voltage-dependent potassium channels. Physiol Rev. 1992;72:S69–S88. doi: 10.1152/physrev.1992.72.suppl_4.S69. [DOI] [PubMed] [Google Scholar]
- Pusch M, Ferrera L, Friedrich T. Two open states and rate-limiting gating steps revealed by intracellular Na+ block of human KCNQ1 and KCNQ1/KCNE1 K+ channels. J Physiol. 2001;533:135–143. doi: 10.1111/j.1469-7793.2001.0135b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmusson RL, Campbell DL, Qu Y, Strauss HC. Conformation-dependent drug binding to cardiac potassium channels. In: Spooner PM, Brown AM, Catterall WA, Kaczorowski GJ, Strauss HC, editors. Ion Channels in the Cardiovascular System: Function and Dysfunction. Armonk, NY: Futura Publishing Co., Inc.; 1994. pp. 387–414. [Google Scholar]
- Rasmusson RL, Morales MJ, Castellino RC, Zhang Y, Campbell DL, Strauss HC. C-type inactivation controls recovery in a fast inactivating cardiac K+ channel (Kv1.4) expressed in Xenopus oocytes. J Physiol. 1995;489:709–721. doi: 10.1113/jphysiol.1995.sp021085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmusson RL, Morales MJ, Wang S, Liu S, Campbell DL, Brahmajothi MV, Strauss HC. Inactivation of voltage-gated cardiac K+ channels. Circ Res. 1998;82:739–750. doi: 10.1161/01.res.82.7.739. [DOI] [PubMed] [Google Scholar]
- Rasmusson RL, Wang S, Castellino RC, Morales MJ, Strauss HC. The β subunit, Kvb1.2, acts as a rapid open channel blocker of NH2-terminal deleted Kv1.4 α-subunits. Adv Exp Med Biol. 1997;430:29–37. doi: 10.1007/978-1-4615-5959-7_3. [DOI] [PubMed] [Google Scholar]
- Rayner MD, Starkus JG, Ruben PC, Alicata DA. Voltage-sensitive and solvent-sensitive processes in ion channel gating. Kinetic effects of hyperosmolar media on activation and deactivation of sodium channels. Biophys J. 1992;61:96–108. doi: 10.1016/S0006-3495(92)81819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig J, Heinemann SH, Wunder F, Lorra C, Parcej DN, Dolly JO, Pongs O. Inactivation properties of voltage-gated K+ channels altered by presence of β-subunit. Nature. 1994;369:289–294. doi: 10.1038/369289a0. [DOI] [PubMed] [Google Scholar]
- Rhodes KJ, Carroll KI, Sung MA, Doliveira LC, Monaghan MM, Burke SL, Strassle BW, Buchwalder L, Menegola M, Cao J, An WF, Trimmer JS. KChIPs and Kv4 α subunits as integral components of A-type potassium channels in mammalian brain. J Neurosci. 2004;24:7903–7915. doi: 10.1523/JNEUROSCI.0776-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandtner W, Szendroedi J, Zarrabi T, Zebedin E, Hilber K, Glaaser I, Fozzard HA, Dudley SC, Todt H. Lidocaine: a foot in the door of the inner vestibule prevents ultra-slow inactivation of a voltage-gated sodium channel. Mol Pharmacol. 2004;66:648–657. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- Scherer CR, Lerche C, Decher N, Dennis AT, Maier P, Ficker E, Busch AE, Wollnik B, Steinmeyer K. The antihistamine fexofenadine does not affect IKr currents in a case report of drug-induced cardiac arrhythmia. Br J Pharmacol. 2002;137:892–900. doi: 10.1038/sj.bjp.0704873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BC, Waldegger S, Fehr S, Bleich M, Warth R, Greger R, Jentsch TJ. A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature. 2000;403:196–199. doi: 10.1038/35003200. [DOI] [PubMed] [Google Scholar]
- Schulte U, Thumfart JO, Klocker N, Sailer CA, Bildl W, Biniossek M, Dehn D, Deller T, Eble S, Abbass K, Wangler T, Knaus HG, Fakler B. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvβ1. Neuron. 2006;49:697–706. doi: 10.1016/j.neuron.2006.01.033. [DOI] [PubMed] [Google Scholar]
- Scott VE, Rettig J, Parcej DN, Keen JN, Findlay JB, Pongs O, Dolly JO. Primary structure of a β subunit of α-dendrotoxin-sensitive K+ channels from bovine brain. Proc Natl Acad Sci U S A. 1994;91:1637–1641. doi: 10.1073/pnas.91.5.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebohm G, Chen J, Strutz N, Culberson C, Lerche C, Sanguinetti MC. Molecular determinants of KCNQ1 channel block by a benzodiazepine. Mol Pharmacol. 2003;64:70–77. doi: 10.1124/mol.64.1.70. [DOI] [PubMed] [Google Scholar]
- Seebohm G, Lerche C, Pusch M, Steinmeyer K, Bruggemann A, Busch AE. A kinetic study on the stereospecific inhibition of KCNQ1 and IKs by the chromanol 293B. Br J Pharmacol. 2001;134:1647–1654. doi: 10.1038/sj.bjp.0704421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebohm G, Westenskow P, Lang F, Sanguinetti MC. Mutation of colocalized residues of the pore helix and transmembrane segments S5 and S6 disrupt deactivation and modify inactivation of KCNQ1 K+ channels. J Physiol. 2005;563:359–368. doi: 10.1113/jphysiol.2004.080887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah M, Akar FG, Tomaselli GF. Molecular basis of arrhythmias. Circulation. 2005;112:2517–2529. doi: 10.1161/CIRCULATIONAHA.104.494476. [DOI] [PubMed] [Google Scholar]
- Sheng M, Liao YJ, Jan YN, Jan LY-group. Presynaptic A-current based on heteromultimeric K+ channels detected in vivo. Nature. 1993;365:72–75. doi: 10.1038/365072a0. [DOI] [PubMed] [Google Scholar]
- Shibata R, Misonou H, Campomanes CR, Anderson AE, Schrader LA, Doliveira LC, Carroll KI, Sweatt JD, Rhodes KJ, Trimmer JS. A fundamental role for KChIPs in determining the molecular properties and trafficking of Kv4.2 potassium channels. J Biol Chem. 2003;278:36445–36454. doi: 10.1074/jbc.M306142200. [DOI] [PubMed] [Google Scholar]
- Shimoni Y, Fiset C, Clark RB, Dixon JE, McKinnon D, Giles WR. Thyroid hormone regulates postnatal expression of transient K+ channel isoforms in rat ventricle. J Physiol. 1997;500:65–73. doi: 10.1113/jphysiol.1997.sp021999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliven B, Szuchet S, Arnason BG, Nelson DJ. Expression and modulation of K+ currents in oligodendrocytes: possible role in myelinogenesis. Dev Neurosci. 1989;11:118–131. doi: 10.1159/000111893. [DOI] [PubMed] [Google Scholar]
- Solth A, Siebrands CC, Friederich P. Inhibition of Kv4.3/KChIP2.2 channels by bupivacaine and its modulation by the pore mutation Kv4.3V401I. Anesthesiology. 2005;103:796–804. doi: 10.1097/00000542-200510000-00018. [DOI] [PubMed] [Google Scholar]
- Spector PS, Curran ME, Keating MT, Sanguinetti MC. Class III antiarrhythmic drugs block HERG, a human cardiac delayed rectifier K+ channel. Open-channel block by methanesulfonanilides. Circ Res. 1996;78:499–503. doi: 10.1161/01.res.78.3.499. [DOI] [PubMed] [Google Scholar]
- Starmer CF, Grant AO, Strauss HC. Mechanisms of use-dependent block of sodium channels in excitable membranes by local anesthetics. Biophys J. 1984;46:15–27. doi: 10.1016/S0006-3495(84)83994-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork D, Timin EN, Berjukow S, Huber C, Hohaus A, Auer M, Hering S. State dependent dissociation of HERG channel inhibitors. Br J Pharmacol. 2007;151:1368–1376. doi: 10.1038/sj.bjp.0707356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strichartz GR. The inhibition of sodium currents in myelinated nerve by quaternary derivatives of lidocaine. J Gen Physiol. 1973;62:37–57. doi: 10.1085/jgp.62.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surti TS, Jan LY. A potassium channel, the M-channel, as a therapeutic target. Curr Opin Invest Drugs. 2005;6:704–711. [PubMed] [Google Scholar]
- Tamargo J, Caballero R, Gomez R, Valenzuela C, Delpon E. Pharmacology of cardiac potassium channels. Cardiovasc Res. 2004;62:9–33. doi: 10.1016/j.cardiores.2003.12.026. [DOI] [PubMed] [Google Scholar]
- Tinel N, Diochot S, Borsotto M, Lazdunski M, Barhanin J. KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. EMBO J. 2000;19:6326–6330. doi: 10.1093/emboj/19.23.6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipparaju SM, Liu SQ, Barski OA, Bhatnagar A. NADPH binding to beta-subunit regulates inactivation of voltage-gated K(+) channels. Biochem Biophys Res Commun. 2007;359:269–276. doi: 10.1016/j.bbrc.2007.05.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- Tseng GN. Molecular structure of cardiac Ito channels: Kv4.2, Kv4.3, and other possibilities? Cardiovasc Res. 1999;41:16–18. doi: 10.1016/s0008-6363(98)00282-x. [DOI] [PubMed] [Google Scholar]
- Tsevi I, Vicente R, Grande M, Lopez-Iglesias C, Figueras A, Capella G, Condom E, Felipe A. KCNQ1/KCNE1 channels during germ-cell differentiation in the rat: expression associated with testis pathologies. J Cell Physiol. 2005;202:400–410. doi: 10.1002/jcp.20132. [DOI] [PubMed] [Google Scholar]
- Uebele VN, England SK, Chaudhary A, Tamkun MM, Snyders DJ. Functional differences in Kv1.5 currents expressed in mammalian cell lines are due to the presence of endogenous Kvβ2.1 subunits. J Biol Chem. 1996;271:2406–2412. doi: 10.1074/jbc.271.5.2406. [DOI] [PubMed] [Google Scholar]
- Uebele VN, England SK, Gallagher DJ, Snyders DJ, Bennett PB, Tamkun MM. Distinct domains of the voltage-gated K+ channel Kv beta 1.3 beta-subunit affect voltage-dependent gating. American Journal of Physiology. 1998;274:C1485–C1495. doi: 10.1152/ajpcell.1998.274.6.C1485. [DOI] [PubMed] [Google Scholar]
- Van Hoorick D, Raes A, Keysers W, Mayeur E, Snyders DJ. Differential modulation of Kv4 kinetics by KCHIP1 splice variants. Mol Cell Neurosci. 2003;24:357–366. doi: 10.1016/s1044-7431(03)00174-x. [DOI] [PubMed] [Google Scholar]
- Vetter DE, Mann JR, Wangemann P, Liu J, McLaughlin KJ, Lesage F, Marcus DC, Lazdunski M, Heinemann SF, Barhanin J. Inner ear defects induced by null mutation of the isk gene. Neuron. 1996;17:1251–1264. doi: 10.1016/s0896-6273(00)80255-x. [DOI] [PubMed] [Google Scholar]
- Wang HS, Brown BS, McKinnon D, Cohen IS. Molecular basis for differential sensitivity of KCNQ and IKs channels to the cognitive enhancer XE991. Mol Pharmacol. 2000;57:1218–1223. [PubMed] [Google Scholar]
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- Wang S, Bondarenko VE, Qu YJ, Bett GC, Morales MJ, Rasmusson RL, Strauss HC. Time- and voltage-dependent components of Kv4.3 inactivation. Biophys J. 2005;89:3026–3041. doi: 10.1529/biophysj.105.059378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Morales MJ, Qu YJ, Bett GC, Strauss HC, Rasmusson RL. Kv1.4 channel block by quinidine: evidence for a drug-induced allosteric effect. J Physiol. 2003;546:387–401. doi: 10.1113/jphysiol.2002.029512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Fermini B, Nattel S. Rapid and slow components of delayed rectifier current in human atrial myocytes. Cardiovasc Res. 1994;28:1540–1546. doi: 10.1093/cvr/28.10.1540. [DOI] [PubMed] [Google Scholar]
- Wang Z, Kiehn J, Yang Q, Brown AM, Wible BA. Comparison of binding and block produced by alternatively spliced Kvbeta1 subunits. J Biol Chem. 1996;271:28311–28317. doi: 10.1074/jbc.271.45.28311. [DOI] [PubMed] [Google Scholar]
- Warmke JW, Ganetzky B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci U S A. 1994;91:3438–3442. doi: 10.1073/pnas.91.8.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster SM, del Camino D, Dekker JP, Yellen G. Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature. 2004;428:864–868. doi: 10.1038/nature02468. [DOI] [PubMed] [Google Scholar]
- Weerapura M, Nattel S, Chartier D, Caballero R, Hebert TE. A comparison of currents carried by HERG, with and without coexpression of MiRP1, and the native rapid delayed rectifier current. Is MiRP1 the missing link? J Physiol. 2002;540:15–27. doi: 10.1113/jphysiol.2001.013296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng J, Cao Y, Moss N, Zhou M. Modulation of voltage-dependent Shaker family potassium channels by an aldo-keto reductase. J Biol Chem. 2006;281:15194–15200. doi: 10.1074/jbc.M513809200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickenden AD. Potassium channels as anti-epileptic drug targets. Neuropharmacol. 2002;43:1055–1060. doi: 10.1016/s0028-3908(02)00237-x. [DOI] [PubMed] [Google Scholar]
- Wissmann R, Baukrowitz T, Kalbacher H, Kalbitzer HR, Ruppersberg JP, Pongs O, Antz C, Fakler B. NMR structure and functional characteristics of the hydrophilic N terminus of the potassium channel β-subunit Kvβ1.1. J Biol Chem. 1999;274:35521–35525. doi: 10.1074/jbc.274.50.35521. [DOI] [PubMed] [Google Scholar]
- Wulfsen I, Hauber HP, Schiemann D, Bauer CK, Schwarz JR. Expression of mRNA for voltage-dependent and inward-rectifying K channels in GH3/B6 cells and rat pituitary. J Neuroendocrinol. 2000;12:263–272. doi: 10.1046/j.1365-2826.2000.00447.x. [DOI] [PubMed] [Google Scholar]
- Yang WP, Levesque PC, Little WA, Conder ML, Shalaby FY, Blanar MA. KvLQT1, a voltage-gated potassium channel responsible for human cardiac arrhythmias. Proc Natl Acad Sci U S A. 1997;94:4017–4021. doi: 10.1073/pnas.94.8.4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Sigworth FJ. Single-channel properties of IKs potassium channels. J Gen Physiol. 1998;112:665–678. doi: 10.1085/jgp.112.6.665. [erratum appears in J Gen Physiol113, 505] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G. The moving parts of voltage-gated ion channels. Q Rev Biophysics. 1998;31:239–295. doi: 10.1017/s0033583598003448. [DOI] [PubMed] [Google Scholar]
- Yeola SW, Rich TC, Uebele VN, Tamkun MM, Snyders DJ. Molecular analysis of a binding site for quinidine in a human cardiac delayed rectifier K+ channel. Role of S6 in antiarrhythmic drug binding. Circ Res. 1996;78:1105–1114. doi: 10.1161/01.res.78.6.1105. [DOI] [PubMed] [Google Scholar]
- Yeola SW, Snyders DJ. Electrophysiological and pharmacological correspondence between Kv4.2 current and rat cardiac transient outward current. Cardiovasc Res. 1997;33:540–547. doi: 10.1016/s0008-6363(96)00221-0. [DOI] [PubMed] [Google Scholar]
- Yool AJ, Schwarz TL. Alteration of ionic selectivity of a K+ channel by mutation of the H5 region. Nature. 1991;349:700–704. doi: 10.1038/349700a0. [DOI] [PubMed] [Google Scholar]
- Yue L, Feng J, Li GR, Nattel S. Characterization of an ultrarapid delayed rectifier potassium channel involved in canine atrial repolarization. J Physiol. 1996;496:647–662. doi: 10.1113/jphysiol.1996.sp021716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Aldrich RW. Voltage-dependent gating of Shaker A-type potassium channels in Drosophila muscle. J Gen Physiol. 1990;95:29–60. doi: 10.1085/jgp.95.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]
- Zhang X, Fedida D. Potassium channel-blocking actions of nifedipine: a cause for morbidity at high doses? Circulation. 1998;97:2098. doi: 10.1161/01.cir.97.20.2098. [DOI] [PubMed] [Google Scholar]
- Zhang S, Zhou Z, Gong Q, Makielski JC, January CT. Mechanism of block and identification of the verapamil binding domain to HERG potassium channels. Circ Res. 1999;84:989–998. doi: 10.1161/01.res.84.9.989. [DOI] [PubMed] [Google Scholar]
- Zhang M, Jiang M, Tseng GN. minK-related peptide 1 associates with Kv4.2 and modulates its gating function: potential role as beta subunit of cardiac transient outward channel? Circulation Research. 2001;88:1012–1019. doi: 10.1161/hh1001.090839. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Yarov-Yarovoy V, Scheuer T, Catterall WA. A gating hinge in Na+ channels; a molecular switch for electrical signaling. Neuron. 2004;41:859–865. doi: 10.1016/s0896-6273(04)00116-3. [DOI] [PubMed] [Google Scholar]
- Zhou M, Morais-Cabral JH, Mann S, MacKinnon R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 2001;411:657–661. doi: 10.1038/35079500. [DOI] [PubMed] [Google Scholar]
- Zimmer J, Doyle DA, Grossmann JG. Structural characterization and pH-induced conformational transition of full-length KcsA. Biophys J. 2006;90:1752–1766. doi: 10.1529/biophysj.105.071175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerberg J, Bezanilla F, Parsegian VA. Solute inaccessible aqueous volume changes during opening of the potassium channel of the squid giant axon. Biophys J. 1990;57:1049–1064. doi: 10.1016/S0006-3495(90)82623-0. [DOI] [PMC free article] [PubMed] [Google Scholar]