

Abstract
The rat α7 nicotinic acetylcholine receptor (nAChR) can undergo rapid onset of desensitization; however, the mechanisms of desensitization are largely unknown. The contribution of a tryptophan (W) residue at position 55 of the rat α7 nAChR subunit, which lies within the β2 strand, was studied by mutating it to other hydrophobic and/or aromatic amino acids, followed by voltage-clamp experiments in Xenopus oocytes. When mutated to alanine, the α7-W55A nAChR desensitized more slowly, and recovered from desensitization more rapidly, than wildtype α7 nAChRs. The contribution of desensitization was validated by kinetic modelling. Mutating W55 to other aromatic residues (phenylalanine or tyrosine) had no significant effect on the kinetics of desensitization, whereas mutation to various hydrophobic residues (alanine, cysteine or valine) significantly decreased the rate of onset and increased the rate of recovery from desensitization. To gain insight into possible structural rearrangements during desensitization, we probed the accessibility of W55 by mutating W55 to cysteine (α7-W55C) and testing the ability of various sulfhydryl reagents to react with this cysteine. Several positively charged sulfhydryl reagents blocked ACh-induced responses for α7-W55C nAChRs, whereas a neutral sulfhydryl reagent potentiated responses; residue C55 was not accessible for modification in the desensitized state. These data suggest that W55 plays an important role in both the onset and recovery from desensitization in the rat α7 nAChR, and that aromatic residues at position 55 are critical for maintaining rapid desensitization. Furthermore, these data suggest that W55 may be a potential target for modulatory agents operating via hydrophobic interactions.
Nicotinic acetylcholine receptors (nAChRs) are in the superfamily of Cys-loop ligand-gated ion channels that also include the serotonin 5-HT3, GABAA and GABAC, and glycine receptor channels. These channels are pentameric assemblies of five (either identical or similar) subunits, with each subunit arranged around the central ion-conducting pore (Unwin, 2005; Sine & Engel, 2006). The neuronal nAChRs are involved in a variety of physiological processes, such as mediating fast synaptic signalling and regulating neurotransmitter release, and normal brain functions including cognitive tasks, reward systems and neuronal development (Jones et al. 1999).
The binding of ACh to the extracellular interface between two nAChR subunits induces channel opening (Gay & Yakel, 2007). Major advances in the past few years have elucidated more detail about the structure of the binding pocket and channel, mostly due to the cloning and crystallization of a molluscan ACh-binding protein (AChBP; Brejc et al. 2001; Smit et al. 2001), and the 4 Å resolution of the Torpedo nAChR (Unwin, 2005). The AChBP is a soluble protein that has remarkable structural similarities to the extracellular binding domain of the nAChRs (Brejc et al. 2001; Smit et al. 2001; Celie et al. 2004; Hansen et al. 2005; Sine & Engel, 2006). Recently for both the muscle nAChR and the related 5-HT3 receptor channels, models have emerged to explain how the transition from the closed to open state may occur (Czajkowski, 2005; Lee & Sine, 2005; Lummis et al. 2005; Sine & Engel, 2006).
Tryptophan (W) residues in the binding site of the Cys-loop family of receptors have been implicated in ligand binding and channel function. One of these tryptophans lies approximately within the middle of the β2 strand of the complementary subunit at position 55 of the rat α7 nAChR subunit (Fig. 1). Mutation of this W residue (W55) within the Torpedo nAChR, chick α7 nAChR, 5-HT3 receptor or a chimeric α7–5-HT3 receptor alter ligand binding affinity or potency (Corringer et al. 1995; Spier & Lummis, 2000; Xie & Cohen, 2001; Fruchart-Gaillard et al. 2002). In addition, we have shown that apoE-derived peptides, which inhibit native and recombinant α7-containing nAChRs (Klein & Yakel, 2004; Gay et al. 2006), may be blocking the α7 nAChRs through hydrophobic interactions with W55 (Gay et al. 2007).
Figure 1. Molecular model of the rat α7 nAChR.

A, shown are two subunits of the pentameric α7 nAChR, as well as a close-up of the extracellular region of the α7 nAChR highlighting key structures involved in channel gating. Tryptophan 55 (W55) is within the β2 strand of the extracellular domain across from the C loop. The model was developed using the Schrodinger Prime protein homology modelling software package (Gay et al. 2007). B, sequence of the β2 strand of various nAChR subunits, AChBP and the 5-HT3A subunit. Numbering pertains to the rat and human α7 nAChR.
In the current study, the contribution of W55 to rat α7 nAChR function was studied by using site-directed mutagenesis of W55, followed by two-electrode voltage-clamp experiments in Xenopus oocytes. When mutated to alanine, the α7-W55A nAChR demonstrated a much slower rate of desensitization onset (greater than 14-fold slower half-time of desensitization) and faster recovery from desensitization. We developed a kinetic model to explain our experimental results, which adequately simulated the characteristics of the ACh-evoked current responses for wildtype and α7-W55A receptors by decreasing the rate of transition of the receptor from the open channel to the desensitized state, and increasing the rate of recovery from desensitization. In addition, we probed the accessibility of W55 using the substituted cysteine accessibility method (SCAM; Karlin & Akabas, 1998) by mutating W55 to cysteine and testing the ability of various sulfhydryl reagents to react with this cysteine. These data suggest that W55 may play an important role in both the onset and recovery from desensitization of the α7 nAChR. Since apoE-derived peptides appear to be blocking the α7 nAChRs through hydrophobic interactions with W55, this may be an important regulatory site for the regulation of function of the α7 nAChRs, playing a role both in the kinetics of desensitization, and perhaps as a site for interaction of hydrophobic ligands.
Methods
Oocyte preparation
All experiments were carried out in accordance with guidelines approved by the NIEHS Animal Care and Use Committee, which includes minimizing the number of animals used and their suffering. Female Xenopus laevis frogs were anaesthetized in cold water containing 0.2% metaaminobenzoate and the spinal cord severed. Oocytes were dissected and defolliculated by treatment with collagenase B (2 mg ml−1, Roche Diagnostics) and trypsin inhibitor (1 mg ml−1, Gibco) for 2 h. Oocytes were maintained in solution containing: 82.5 mm NaCl, 2.5 mm KCl, 1 mm Na2HPO4, 3 mm NaOH, 5 mm Hepes, 1 mm CaCl2, 1 mm MgCl2, 2.5 mm pyruvic acid, and 0.05 mg ml−1 gentamycin sulphate with constant rotation at 18°C. mRNA for each of the nAChR subunits (α7 wildtype and mutants) was transcribed from plasmids using mMessage mMachine 17 kit from Ambion (Austin, TX, USA) according to the manufacturer/s instructions. The total amount of RNA injected for each nAChR subunit was ∼50 ng. Recordings were made 2–7 days post RNA injection.
Oocyte electrophysiology
Current responses were obtained by two-electrode voltage-clamp recording at a holding potential of –60 mV using a Geneclamp 500 and pCLAMP 8 software. Electrodes contained 3 m KCl and had a resistance of < 1 MΩ. ACh and other compounds were prepared daily in bath solution (96 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 5 mm Hepes) from either frozen stocks or powder. The sulfhydryl reagents used were very labile, and therefore these reagents generally were diluted in bath solution no more than 15 min prior to application. The sulfhydryl reagents were purchased from Toronto Research Chemicals. ACh was applied for various time periods using a synthetic quartz perfusion tube (0.7 mm i.d.) operated by a computer-controlled valve, the tip of which was positioned within 1 mm from the oocyte. We estimated that the rate of solution exposure to the oocyte could be achieved in a half-time of ∼30 ms by measuring the change in liquid-junction potential at the tip of a pipette. Under these conditions, ACh-induced current responses in oocytes expressing wildtype α7 nAChRs activated with rise-times (10–90%) of 25 ± 2 ms (14 cells). Otherwise compounds were bath applied. Data were analysed using either pCLAMP 8, Excel (Microsoft), or Origin 6 (Microcal Software). Data for ACh dose–response curves were normalized to the peak current response at 1 mm ACh for each mutant receptor. Peak current responses to each dose of ACh were averaged, and then the mean ±s.e.m. were analysed by non-linear regression using a logistic equation (GraphPad Prism 4). ACh EC50 values were compared using a two-tailed t test with a Bonferroni correction for α inflation. Due to the inequality of variances for rates of onset and recovery from desensitization across various receptor types, data were analysed using the non-parametric Kruskal–Wallis test followed by a Dunn/s multiple comparison test. Otherwise, statistical analyses (e.g. t tests and fitting to exponential functions) were performed using Origin software (Microcal, Northampton, MA, USA). Averaged data are presented as mean ±s.e.m.
To measure the rate of onset of desensitization without activation of the endogenous Ca2+-activated chloride currents in Xenopus oocytes, oocytes were incubated in BAPTA-AM (100 μm) for 1–4 h. We confirmed that this treatment eliminated the Ca2+-activated chloride currents through current–voltage curves, and that there was no significant effect on peak nAChR-mediated current amplitudes (data not shown). We quantified the rate of desensitization onset by measuring the half-time of desensitization, which is the time required for the response to decay by 50% from peak amplitude during the continuous application of ACh.
Kinetic modelling; computer simulation method
We used the computer simulation method which is based on the solving of ordinary differential equations (Chretien & Chauvet, 1998) with the probability of occurrence of each receptor state given by the following equation:
where 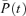 is the vector of the probability of existence of the receptor channel complex in different states at time t, and Q is the matrix of transitions between states. Our in-house-developed program was written in Pascal and used on an IBM-compatible PC to solve numerically this set of differential equations using the eight-order Runge–Kutta method (Baker et al. 1996). The membrane current can be calculated by the following equation:
is the vector of the probability of existence of the receptor channel complex in different states at time t, and Q is the matrix of transitions between states. Our in-house-developed program was written in Pascal and used on an IBM-compatible PC to solve numerically this set of differential equations using the eight-order Runge–Kutta method (Baker et al. 1996). The membrane current can be calculated by the following equation:
where V is the membrane potential, N is the total quantity of channels in the membrane, Popen(t) is the open probability at time t, and σ is the single channel conductance. If V, N and σ are constants, then the current at every moment is proportional to the total open channel probability. In this case, all parameters (rise-time, half-time, recovery rate, EC50 and Hill slope) can be found by Popen(t) analysis.
We used the following traditional cyclic scheme (Katz & Thesleff, 1957; Paradiso & Steinbach, 2003; Giniatullin et al. 2005) to develop our kinetic model:

where R is the receptor, A the agonist, R° the activated receptor and D the desensitized receptor. Our model was based on the previous model developed by Mike et al. (2000) for the α7 nAChR, which included three binding sites for ACh, and two open channel states of the receptor. For the model, a simulated pulse of agonist mimicked the experimentally determined solution exchange rate. According to the experimental data, solution flow to the oocyte in the model is 60 ms, and the washout of solution from the chamber in the model is 1.8 s.
Results
W to A substitution at position 55 alters desensitization kinetics
The application of ACh (1 mm) to Xenopus oocytes expressing the rat wildtype α7 nAChRs induced currents that rapidly activated and then desensitized (with the continuous application of ACh) with a half-time of 111 ± 7.3 ms (18 cells; Fig. 2A; Table 1); rapid and complete desensitization is a common property of α7-containing nAChRs (Quick & Lester, 2002; Giniatullin et al. 2005). In order to explore the importance of W55 for receptor function, we substituted this residue for a number of hydrophobic and/or aromatic amino acids. Of all the mutations we tried (Table 1), the most dramatic affect on desensitization was obtained when W55 (Fig. 1) was mutated to the hydrophobic residue alanine (α7-W55A). In this case, the rate of onset of desensitization was dramatically slowed; the half-time of desensitization onset was 1590 ± 130 ms (12 cells; Fig. 2B). ACh dose–response curves for the α7-W55A nAChR demonstrated increased potency for ACh as compared with wildtype α7 nAChR (Table 1), which is expected with reduced desensitization of the α7 nAChR (Papke & Porter Papke, 2002). Consistent with this view, the average amplitude of current responses for the α7-W55A nAChRs (4420 ± 270 nA; 22 cells) was significantly larger than for wildtype α7 nAChRs (2330 ± 370 nA; 13 cells), when measured in the same batch of oocytes on the same day.
Figure 2. The rate of onset of desensitization is slowed by W55 mutations.

Representative inward current responses due to the rapid and sustained application of ACh (1 mm; indicated by the horizontal bar) for wildtype (A) and mutant (α7-W55A, B; α7-W55C, C) α7 nAChRs. The onset of desensitization half-time values are shown in Table 1. Oocytes were incubated in BAPTA-AM (100 μm) for 1–4 h to inhibit Ca2+-activated chloride currents.
Table 1.
Characterization of wildtype and mutant α7 nAChRs
| Receptor | Desen. half-time (ms) | Recovery percentage (5 s) | Recovery percentage (10 s) | ACh EC50 (μm) [95%CI] | Hill slope |
|---|---|---|---|---|---|
| α7 wt | 111 ± 7 (18) | 8.6 ± 2 (30) | 64 ± 3 (27) | 120 [100–140] | 1.7 ± 0.2 |
| α7-W55A | 1590 ± 130 (12)* | 61 ± 3 (28)* | 91 ± 1 (19)* | 58 [49–68]* | 3.1 ± 0.6 |
| α7-W55F | 126 ± 12 (10) | 9 ± 3 (9) | 57 ± 4 (9) | 130 [98–180] | 1.7 ± 0.4 |
| α7-W55Y | 110 ± 9 (11) | 2 ± 1 (7) | 42 ± 7 (7) | 51 [43–60]* | 2.5 ± 0.4 |
| α7-W55V | 191 ± 8 (12)* | 24 ± 3 (11)* | 75 ± 4 (11) | 200 [190–220]* | 2.3 ± 0.2 |
| α7-W55C | 339 ± 61 (10)* | 25 ± 4 (12)* | 72 ± 5 (5) | 320 [260–380]* | 2.1 ± 0.4 |
Significant difference (P < 0.05) from wildtype (wt) α7 receptor; for statistical analysis, see Methods. EC50 and Hill slope data have been presented previously (Gay et al. 2007).
The rate of recovery from desensitization was also significantly modified for the α7-W55A receptors. To induce near-complete (> 90%) desensitization, ACh was applied continuously for 8 s, while for wildtype α7 receptors, ACh was applied for 4 s (Fig. 3). After washout of ACh for either 5 or 10 s, a brief pulse of ACh was applied to assess the extent of recovery from the desensitized state. For α7-W55A receptors, the percentage recovery from desensitization at 5 s was 61 ± 3% (28 cells), which was significantly greater than for wildtype α7 receptors (8.6 ± 2%; 30 cells; P < 0.001). Similarly, the recovery at 10 s was 91 ± 1% (19 cells) for the α7-W55A receptors versus 64 ± 3% (27 cells; P < 0.001) for wildtype α7 receptors (Fig. 3; Table 1). Complete recovery time courses indicated that the rate of recovery for both wildtype (Fig. 3A) and α7-W55A (Fig. 3B) receptors were monophasic, with time constant values of 4.5 ± 0.4 s (9 cells) and 2.0 ± 0.2 s (10 cells; P < 0.001), respectively. All these data suggest that substitution of W55 to alanine dramatically reduced the probability that the α7 receptors would undergo and remain in the desensitized state.
Figure 3. The rate of recovery from desensitization is increased for the α7-W55A mutant receptor.

Representative traces showing the recovery from desensitization for wildtype α7 (A) and α7-W55A (B) receptors. ACh was applied continuously until the receptors were desensitized by > 90% (i.e. 4 s for wildtype α7 receptors, and 8 s for α7-W55A receptors; continuous horizontal bar), followed by the washout of ACh for either 5 or 10 s (dotted line), and then a brief pulse of ACh was applied (arrow) to assess the extent of desensitization. Complete recovery time courses are shown on the right. Values are the mean ±s.e.m. of 6–30 oocytes each, and the data have been fitted to a mono-exponential function.
Effect of other mutations at position 55
In order to test whether rapid desensitization was maintained by other aromatic residues, W55 was mutated to either phenylalanine (α7-W55F) or tyrosine (α7-W55Y). For the α7-W55F and α7-W55Y receptors, the onset of desensitization half-times were 126 ± 12 ms (10 cells) and 110 ± 9.4 ms (11 cells), respectively (Table 1). In addition, the percentage recovery from desensitization for the α7-W55F and α7-W55Y receptors at 5 s were 9 ± 3% (9 cells) and 2 ± 1% (7 cells), respectively; at 10 s these values were 57 ± 4% and 42 ± 7%, respectively (Table 1). None of these values were significantly different than for wildtype α7 receptors. Therefore mutating W55 to these other aromatic residues (either phenylalanine or tyrosine) had no significant effect on the kinetics of desensitization, suggesting that aromatic residues at position 55 have a key role in maintaining rapid desensitization of α7 nAChRs. The only significant difference was the increased potency of the α7-W55Y receptors for ACh (Table 1).
When W55 was mutated to the other hydrophobic residues (similar to alanine) valine (α7-W55V) or cysteine (α7-W55C), in contrast to the aromatic residues, the kinetics of desensitization onset were reduced and recovery (at 5 s) was accelerated. When W55 was mutated to leucine, the channels were non-functional. For the α7-W55V and α7-W55C receptors, the onset of desensitization half-times were 191 ± 8.2 ms (12 cells) and 339 ± 61 ms (10 cells), respectively (Table 1). In addition, the percentage recovery from desensitization for the α7-W55V and α7-W55C receptors at 5 s were 24 ± 3% (11 cells) and 25 ± 4% (12 cells), respectively; at 10 s these values were 75 ± 4% and 72 ± 5%, respectively (Table 1). Both α7-W55V and α7-W55C receptors had a decreased potency for ACh (Table 1). Lastly, when W55 was mutated to various other residues (arginine, threonine or lysine), the channels were non-functional.
We tested whether mutations in other β2 strand residues might alter the desensitization of the receptor. When the serine at position 59 was mutated to alanine (α7-S59A), the channels were functional and the rate of onset of desensitization was slightly faster than for wildtype α7 receptors; the half-time of desensitization onset was 79 ± 3 ms (19 cells). Furthermore the percentage recovery from desensitization at 5 and 10 s was 22 ± 4% and 78 ± 2% (14 cells), respectively, both values being significantly greater than for wildtype α7 receptors (P < 0.002). The substitution of S59 to alanine had different affects on the kinetics of desensitization than did mutation of W55, suggesting that mutations in the β2 strand do not necessarily produce global changes in receptor desensitization. Mutation of the threonine at position 51 to a cysteine resulted in a non-functional receptor.
A general model for α7 nAChR function
The increased ACh potency observed for the α7-W55A nAChR could be due to either the putative role of this residue in ligand binding, or it could be due to the reduced desensitization of the α7-W55A receptor (Papke & Porter Papke, 2002). To distinguish between these two possibilities and to validate our interpretation of the effects of the α7-W55A mutant receptor, we developed a kinetic model using the traditional cyclic scheme for nAChRs (Katz & Thesleff, 1957; Paradiso & Steinbach, 2003; Giniatullin et al. 2005; see Methods). Starting numerical values for the rate constants were taken from Mike et al. (2000), who had developed a kinetic model for α7 nAChR function which included three binding sites for ACh, and two open channel states of the receptor. We reduced some rate constants in the model for conformity of the modelled traces to our experimental results. To refine the model for our experimental conditions, we took into account (1) the rise-time, desensitization onset and recovery of ACh-induced currents, and (2) the EC50 and Hill slope values of ACh dose–response curves for the wildtype and α7-W55A receptors. The final values of the kinetic rate constants are shown in Table 2, with values estimated from the model shown in Table 3.
Table 2.
Kinetic model rate constants
| Transition | Rate constant | Transition | Rate constant |
|---|---|---|---|
| Wildtype and W55A | |||
| A + R→AR | *300000 mm−1·s−1 | AR→A + R | *10000 s−1 |
| A + AR→A2R | *200000 mm−1·s−1 | A2R→A + AR | *20000 s−1 |
| A + A2R→A3R | *100000 mm−1·s−1 | A3R→A + A2R | *30000 s−1 |
| A2R→A2R° | 6 s−1 | A2R°→A2R | 1 s−1 |
| A3R→A3R° | 30 s−1 | A3R°→A3R | 1 s−1 |
| A2D→A2R° | 0.01 s−1 | A3D→A3R° | 0.05 s−1 |
| R→D | *0.01 s−1 | D→R | *500 s−1 |
| A + D→AD | 30 mm−1·s−1 | A + AD→A2D | 20 mm−1·s−1 |
| A + A2D→A3D | 10 mm−1·s−1 | ||
| Wildtype | |||
| A2R°→A2D | 25 s−1 | A3R°→A3D | 25 s−1 |
| AD→A + D | 0.25 s−1 | A2D→A + AD | 0.5 s−1 |
| A3D→A + A2D | 0.75 s−1 | ||
| W55A | |||
| A2R°→A2D | 1.25 s−1 | A3R°→A3D | 0.45 s−1 |
| AD→A + D | 0.6 s−1 | A2D→A + AD | 1.2 s−1 |
| A3D→A + A2D | 1.8 s−1 | ||
Rate constant transitions pertain to kinetic scheme in Methods. In the upper part are the rate constants that are identical for both wildtype and W55A receptors, and the bottom part for the rate constants that varied between the two. *denotes rate constants taken from Mike et al. (2000).
Table 3.
Characterization of α7 nAChR model values
| Receptor | Desens. half-time (ms) | Recovery percentage (5 s) | Recovery percentage (10 s) | ACh EC50 (μm) | Hill slope |
|---|---|---|---|---|---|
| α7 wt | 103 | 15 | 64 | 150 | 1.2 |
| α7-W55A | 1680 | 59 | 97 | 49 | 1.5 |
As shown in Fig. 4, our optimized model effectively simulated the basic characteristics of the ACh-induced current responses for wildtype α7 nAChRs, including fast desensitization onset and slow recovery (Fig. 4A). Furthermore, simply by decreasing the rate of transition from the open (A2R° and A3R°) to the desensitized states (A2D and A3D; Table 2), and by increasing the rate of recovery from desensitization (i.e. for each of the following transitions, A3D to A2D, A2D to AD and AD to D), this simulated the properties of the α7-W55A receptor; this included slower desensitization onset and faster recovery, changes in EC50 and Hill slope values, as well as the increase in maximal amplitude of current responses (Fig. 4B; Table 3).
Figure 4. Kinetic model of the rat α7 nAChR.

Modelled traces showing desensitization onset and recovery for wildtype α7 (A) and α7-W55A (B) receptors. ACh was applied continuously for 4 s for wildtype α7 receptors, and 8 s for α7-W55A receptors (continuous horizontal bar), followed by the washout of ACh for 5 s, and then a brief pulse (0.2 s) of ACh was applied (arrow) to assess the extent of desensitization. The y-axis is the probability of existence of the receptor in the open state. Complete recovery time courses are shown on the right.
SCAM; probing the accessibility of position 55
Since the α7-W55C receptor was functional, to gain insight into possible structural rearrangements involving W55 during channel gating and/or desensitization, we probed the water-surface accessibility of residue 55 by using the ability of various methanethiosulphonate (MTS) compounds to react with this cysteine (C55; Karlin & Akabas, 1998). Major determinants of accessibility include both steric and electrostatic factors, therefore we tested if MTS accessibility to C55 could be altered by varying the charge and size of the MTS compound. In addition, we altered the receptor environment around C55 by testing MTS compounds in both the closed and desensitized receptor states.
We initially tested various positively charged MTS compounds on α7-W55C and wildtype α7 receptors: 2-aminoethyl methanethiosulphonate hydrobromide (MTSEA+), 2-(trimethylammonium)ethyl methanethiosulphonate bromide (MTSET+), and 3-(triethylammonium)propyl methanethiosulphonate bromide (MTS-PTrEA+). After obtaining a stable ACh-induced current response, oocytes expressing α7-W55C receptors were pre-incubated with MTSEA+ (1 mm for 1 min); after the complete washout of MTSEA+ (> 5 min), ACh-induced currents were irreversibly reduced by 99 ± 0% (10 cells; Fig. 5). Pre-incubation with MTSET+ (1 mm for 5 min) reduced ACh-induced currents by 93 ± 2% (15 cells; Figs 5A and C). Incubation with MTS-PTrEA+ (1 mm for 1 min) reduced ACh-induced currents by 97 ± 1% (5 cells; Fig. 5C). MTSEA+, MTSET+ nor MTS-PTrEA+ had any significant effect on wildtype α7 nAChR-mediated currents (data not shown). These data suggest that residue C55 of the closed α7-W55C receptor is accessible to these positively charged MTS compounds, and that covalent modification with these compounds prevents ACh from opening the channels.
Figure 5. Positively charged MTS compounds inhibit α7-W55C receptors in the closed but not desensitized receptor state.

A, inward current responses in an oocyte expressing α7-W55C receptors in response to ACh (1 mm; indicated by the horizontal bar) prior to (left) and after pre-incubation (right) with MTSET+ (1 mm for 5 min). The various MTS compounds were added in the absence of ACh, followed by washout for > 5 min prior to the application of ACh. B, representative traces of ACh-induced currents in an oocyte expressing α7-W55C receptors in control (left), after co-application of ACh and MTSET+ (middle), and then after application of MTSET+ alone subsequently (right). C, bar graph of averaged data for percentage block of ACh-induced currents after pre-incubation with either MTSEA+, MTSET+ or MTS-PTrEA+ when applied alone (left bars), and in the same oocytes during co-application of ACh and the various MTS reagents (middle bars), and for the MTS reagent alone subsequently (right bars). Values are the mean ±s.e.m. of 3–4 oocytes each.
Since our previous data suggested a key role of residue 55 in desensitization, we next tested whether in the continuous presence of agonist, C55 was still accessible. ACh was applied continuously for 1 min; then the MTS compound was co-applied with ACh, followed by ACh alone for another minute. After a 5 min washout period, a brief (200 ms) application of ACh was given. In this manner, we were able to test whether ACh responses were altered by the MTS compounds when the receptor was in the desensitized state. Under these conditions, neither MTSET+ (1 mm for 5 min; Fig. 5B and C), MTSEA+ nor MTS-PTrEA+ (1 mm for 1 min; Fig. 5C), had any significant effect on the ACh-induced current of the α7-W55C receptors. However, when these MTS compounds were applied alone subsequently, the ACh-induced responses were completely blocked (Fig. 5C). These data suggest that C55 of the α7-W55C receptor is accessible for modification by these positively charged MTS compounds only in the closed (but not desensitized) receptor state.
Potentiation of ACh responses by the neutral MTS compound, MMTS
We tested whether neutral MTS compounds, such as methyl methanethiosulphonate (MMTS), had any effect on the function of the α7-W55C receptors. In contrast to the effects of the positively charged MTS compounds, MMTS application (1 mm for 1 min) potentiated the ACh-induced responses by 57 ± 7% (6 cells; Fig. 6). There was no significant difference in either the rate of activation or half-time of decay of ACh responses after potentiation by MMTS (data not shown). For wildtype α7 receptors, MMTS increased responses by only 8 ± 3% (5 cells). These data suggest that C55 of the closed α7-W55C receptor is accessible to MMTS, and that covalent modification with this compound enhanced ACh-induced responses.
Figure 6. Neutral MTS compound, MMTS, potentiates α7-W55C receptors.

Representative traces in an oocyte expressing α7-W55C receptors of ACh-induced current responses before (A) and after (B) pre-incubation with MMTS (1 mm for 1 min). C, bar graph of averaged data showing the relative change in amplitude after potentiation by MMTS when applied alone (left). In another batch of oocytes, there was no change in amplitude after co-application of ACh and MMTS (middle); however, after the washout of ACh, MMTS alone still potentiated the ACh-induced responses (right). Values are the mean ±s.e.m. of 6–9 oocytes each.
Similar to the positively charged MTS compounds, MMTS was unable to potentiate ACh-induced responses when the receptor was in the desensitized state (Fig. 6C). When MMTS was applied alone subsequently to these oocytes, the ACh-induced responses were potentiated by 51 ± 15% (9 cells; Fig. 6C). Again these data suggest that C55 of the α7-W55C receptor may be accessible for modification by MMTS only in the closed (but not desensitized) receptor state.
Discussion
By combining voltage-clamp electrophysiological recordings in Xenopus oocytes, site-directed mutagenesis, kinetic and molecular modelling, along with probing with various sulfhydryl reagents, we have shown here that for rat α7 nAChRs, a tryptophan residue at position 55 (W55), that lies within the β2 strand, plays an important role in both the onset and recovery from desensitization of these receptors. When mutating W55 to other aromatic residues (either phenylalanine or tyrosine), the α7 nAChRs still rapidly desensitized. However, mutations of W55 to small hydrophobic amino acids (alanine, cysteine or valine) appears to stabilize the open (versus desensitized) state since the rate of onset of desensitization significantly decreased, and the rate of recovery from desensitization increased. Charge does not appear to be well tolerated at this position since W55 mutations to either arginine or lysine resulted in non-functional channels. The role of this residue in desensitization was validated by kinetic modelling, and probing with sulfhydryl reagents using SCAM. Although the mechanism of desensitization is not completely understood, it might be important in controlling cholinergic signalling, and perhaps in certain nAChR-related diseases (Giniatullin et al. 2005). These data provide new insights into the mechanism of desensitization of α7-containing nAChRs.
In order to explain the changes in the experimental data between wildtype and α7-W55A receptors, we developed a kinetic model, based on the previous model by Mike et al. (2000) for the α7 nAChR. Using this model, we were able to effectively simulate the effects of the α7-W55A mutant receptor by slowing the desensitization onset and accelerating the desensitization recovery, without changes in the activation kinetics. These changes to the model also predicted the observed change to the EC50 value, as well as the increase in receptor efficacy. Although traditionally changes in EC50 values could indicate changes in receptor affinity to ACh, our mathematical simulations have shown that dose–response curves can be significantly altered by the rapid desensitization present in wildtype α7 nAChRs (see also Papke & Porter Papke, 2002). Therefore, by decreasing the extent of desensitization (e.g. with the mutant α7-W55A receptor), it is likely that the experimentally determined EC50 value for the α7-W55A receptor will be more accurate than for the fast-desensitizing wildtype α7 receptors. Furthermore, changes to the model in the kinetics of either binding or gating did not reproduce the desensitization properties of the α7-W55A receptor, although at this point we cannot rule out that such changes in either have not occurred.
We have shown that the engineered cysteine at position 55 of the α7 nAChR is accessible for covalent modification in the closed, but not desensitized, receptor state, for both positively charged methanethiosulphonate (MTS) compounds and the neutral MMTS. The fact that these particular MTS compounds have access to the closed but not desensitized states means that for the desensitized receptor, either there were structural changes in the vicinity around C55 unfavourable for MTS accessibility, or perhaps the presence of ACh in the binding pocket proved unfavourable for MTS access due to steric hindrance.
Based on alignment with the AChBP, the W55 residue in the α7 nAChR was proposed to be in or near the ligand binding pocket (Corringer et al. 2000; Brejc et al. 2001; Celie et al. 2004; Young et al. 2007). Mutation of this W residue within similar receptors causes a decrease in agonist binding and ACh potency (Torpedo nAChR), a decrease in antagonist binding (chick α7 nAChR), a small decrease in agonist and antagonist affinity and a decrease in 5-HT potency (5-HT3 receptor), and a decrease in binding affinity and potency for both agonists and antagonists (chimeric α7–5-HT3 receptor) (Corringer et al. 1995; Spier & Lummis, 2000; Xie & Cohen, 2001; Fruchart-Gaillard et al. 2002). However, this is the first demonstration that the mutation of W55 can have a dramatic affect on the kinetics of desensitization.
Interestingly for the α3β4 nAChR, it was recently reported (Young et al. 2007) that a novel nAChR agonist (TMAQ) was able to selectively activate human rather than rat β4-containing receptors, and this ability was due to two adjacent amino acids just upstream from the analogous residue at W55 (i.e. residues 53–54). Furthermore, we have shown that apoE-derived peptides may be blocking the α7 nAChRs through hydrophobic interactions with W55 (Gay et al. 2007). These data further suggest the importance of this region as a possible site for regulating channel function via hydrophobic interactions.
Within the Cys-loop ligand-gated ion channel family, recent models have emerged to suggest a mechanism for translating ligand binding to channel gating, which include a major rearrangement of the C-loop within the ligand binding pocket, the interaction of a conserved tyrosine residue in the C-loop and a conserved lysine residue in the β7 strand, and the subsequent disruption of a salt bridge between an arginine residue at the end of the β10 strand and a glutamate residue in the β1–β2 linker (Corringer et al. 2000; Absalom et al. 2003; Kash et al. 2003, 2004; Schofield et al. 2004; Xiu et al. 2005; Mercado & Czajkowski, 2006). However, how ligand binding leads to channel desensitization remains to be resolved. Residue W55 in the α7 nAChR could be involved in this process because of its close proximity to the β1–β2 linker. Thus, for the 5-HT3 receptor, Reeves et al. (2005) proposed that ligand binding induced a global conformational change whereby movements of the β1–β2 linker result in a rotation of M2 away from the channel axis, thereby inducing channel opening. An additional conformational change induces desensitization, and recovery from desensitization may require reformation of the interaction between the β1–β2 linker and the M2–M3 loop (Reeves et al. 2005). Therefore, changes to the α7 nAChR structure that interfere with the breaking or reformation of this β1–β2 linker/M2–M3 loop interaction might be projected to have an affect on the kinetics of desensitization.
In conclusion, these data suggest that position 55 of the rat α7 nAChR plays an important role in both onset and recovery from desensitization, and therefore may be a structural determinant of receptor inactivation during the continual presence of agonist. The presence of an aromatic amino acid residue at position 55 preserved normal receptor desensitization, suggesting that the aromatic moiety may play an important role in maintaining the rapid desensitization of α7 nAChRs. In addition, since apoE-derived peptides appear to be blocking the α7 nAChRs through hydrophobic interactions with W55 (Gay et al. 2007), this position may be key for the interaction of particular ligands with the receptor. Therefore, W55 may be an important regulatory site for the modulation of α7 nAChR function, playing a pivotal role not only in the ligand/receptor interactions, but also in the kinetics of desensitization.
Acknowledgments
We would like to thank Patricia Lamb for her assistance in preparing the mRNA and oocytes, C. Erxleben and S. Gentile for advice in preparing the manuscript, and R. Bienstock for the molecular model of the α7 nAChR. Research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences. A.S. would like to thank the Russian Foundation for Basic Research and the Russian government for a Leading Scientific School grant.
References
- Absalom NL, Lewis TM, Kaplan W, Pierce KD, Schofield PR. Role of charged residues in coupling ligand binding and channel activation in the extracellular domain of the glycine receptor. J Biol Chem. 2003;278:50151–50157. doi: 10.1074/jbc.M305357200. [DOI] [PubMed] [Google Scholar]
- Baker TS, Dormand JR, Gilmore JP, Prince PJ. Continuous approximation with embedded Runge-Kutta methods. Appl Numl Math. 1996;22:51–62. [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- Celie PH, van Rossum-Fikkert SE, van Dijk WJ, Brejc K, Smit AB, Sixma TK. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron. 2004;41:907–914. doi: 10.1016/s0896-6273(04)00115-1. [DOI] [PubMed] [Google Scholar]
- Chretien JM, Chauvet GA. An algorithmic method for determining the kinetic system of receptor-channel complex. Math Biosci. 1998;147:227–257. doi: 10.1016/s0025-5564(97)00102-8. [DOI] [PubMed] [Google Scholar]
- Corringer PJ, Galzi JL, Eisele JL, Bertrand S, Changeux JP, Bertrand D. Identification of a new component of the agonist binding site of the nicotinic α7 homooligomeric receptor. J Biol Chem. 1995;270:11749–11752. doi: 10.1074/jbc.270.20.11749. [DOI] [PubMed] [Google Scholar]
- Corringer PJ, Le Novere N, Changeux JP. Nicotinic receptors at the amino acid level. Annu Rev Pharmacol Toxicol. 2000;40:431–458. doi: 10.1146/annurev.pharmtox.40.1.431. [DOI] [PubMed] [Google Scholar]
- Czajkowski C. Neurobiology: triggers for channel opening. Nature. 2005;438:167–168. doi: 10.1038/438167a. [DOI] [PubMed] [Google Scholar]
- Fruchart-Gaillard C, Gilquin B, Antil-Delbeke S, Le Novere N, Tamiya T, Corringer PJ, Changeux JP, Menez A, Servent D. Experimentally based model of a complex between a snake toxin and the α7 nicotinic receptor. Proc Natl Acad Sci U S A. 2002;99:3216–3221. doi: 10.1073/pnas.042699899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay EA, Bienstock RJ, Lamb PW, Yakel JL. Structural determinates for apolipoprotein E-derived peptide interaction with the α7 nicotinic acetylcholine receptor. Mol Pharm. 2007;72:838–849. doi: 10.1124/mol.107.035527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay EA, Klein RC, Yakel JL. Apolipoprotein E-derived peptides block α7 neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Pharm Exp Ther. 2006;316:835–842. doi: 10.1124/jpet.105.095505. [DOI] [PubMed] [Google Scholar]
- Gay EA, Yakel JL. Gating of nicotinic ACh receptors; new insights into structural transitions triggered by agonist binding that induce channel opening. J Physiol. 2007;584:727–733. doi: 10.1113/jphysiol.2007.142554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giniatullin R, Nistri A, Yakel JL. Desensitization of nicotinic ACh receptors: shaping cholinergic signaling. Trends Neurosci. 2005;28:371–378. doi: 10.1016/j.tins.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Hansen SB, Sulzenbacher G, Huxford T, Marchot P, Taylor P, Bourne Y. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 2005;24:3635–3646. doi: 10.1038/sj.emboj.7600828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Sudweeks S, Yakel JL. Nicotinic receptors in the brain: correlating physiology with function. Trends Neurosci. 1999;22:555–561. doi: 10.1016/s0166-2236(99)01471-x. [DOI] [PubMed] [Google Scholar]
- Karlin A, Akabas MH. Substituted-cysteine accessibility method. Methods Enzymol. 1998;293:123–145. doi: 10.1016/s0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
- Kash TL, Jenkins A, Kelley JC, Trudell JR, Harrison NL. Coupling of agonist binding to channel gating in the GABAA receptor. Nature. 2003;421:272–275. doi: 10.1038/nature01280. [DOI] [PubMed] [Google Scholar]
- Kash TL, Kim T, Trudell JR, Harrison NL. Evaluation of a proposed mechanism of ligand-gated ion channel activation in the GABAA and glycine receptors. Neurosci Lett. 2004;371:230–234. doi: 10.1016/j.neulet.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Katz B, Thesleff S. A study of the desensitization produced by acetylcholine at the motor end-plate. J Physiol. 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RC, Yakel JL. Inhibition of nicotinic acetylcholine receptors by Apolipoprotein E-derived peptides in rat hippocampal slices. Neuroscience. 2004;127:563–567. doi: 10.1016/j.neuroscience.2004.05.045. [DOI] [PubMed] [Google Scholar]
- Lee WY, Sine SM. Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature. 2005;438:243–247. doi: 10.1038/nature04156. [DOI] [PubMed] [Google Scholar]
- Lummis SC, Beene DL, Lee LW, Lester HA, Broadhurst RW, Dougherty DA. Cis-trans isomerization at a proline opens the pore of a neurotransmitter-gated ion channel. Nature. 2005;438:248–252. doi: 10.1038/nature04130. [DOI] [PubMed] [Google Scholar]
- Mercado J, Czajkowski C. Charged residues in the α1 and β2 pre-M1 regions involved in GABAA receptor activation. J Neurosci. 2006;26:2031–2040. doi: 10.1523/JNEUROSCI.4555-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mike A, Castro NG, Albuquerque EX. Choline and acetylcholine have similar kinetic properties of activation and desensitization on the α7 nicotinic receptors in rat hippocampal neurons. Brain Res. 2000;882:155–168. doi: 10.1016/s0006-8993(00)02863-8. [DOI] [PubMed] [Google Scholar]
- Papke RL, Porter Papke JK. Comparative pharmacology of rat and human α7 nAChR conducted with net charge analysis. Br J Pharmacol. 2002;137:49–61. doi: 10.1038/sj.bjp.0704833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradiso KG, Steinbach JH. Nicotine is highly effective at producing desensitization of rat α4β2 neuronal nicotinic receptors. J Physiol. 2003;553:857–871. doi: 10.1113/jphysiol.2003.053447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick MW, Lester RA. Desensitization of neuronal nicotinic receptors. J Neurobiol. 2002;53:457–478. doi: 10.1002/neu.10109. [DOI] [PubMed] [Google Scholar]
- Reeves DC, Jansen M, Bali M, Lemster T, Akabas MH. A role for the β1-β2 loop in the gating of 5-HT3 receptors. J Neurosci. 2005;25:9358–9366. doi: 10.1523/JNEUROSCI.1045-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield CM, Trudell JR, Harrison NL. Alanine-scanning mutagenesis in the signature disulfide loop of the glycine receptor α1 subunit: critical residues for activation and modulation. Biochemistry. 2004;43:10058–10063. doi: 10.1021/bi036159g. [DOI] [PubMed] [Google Scholar]
- Sine SM, Engel AG. Recent advances in Cys-loop receptor structure and function. Nature. 2006;440:448–455. doi: 10.1038/nature04708. [DOI] [PubMed] [Google Scholar]
- Smit AB, Syed NI, Schaap D, van Minnen J, Klumperman J, Kits KS, Lodder H, van der Schors RC, van Elk R, Sorgedrager B, Brejc K, Sixma TK, Geraerts WP. A glia-derived acetylcholine-binding protein that modulates synaptic transmission. Nature. 2001;411:261–268. doi: 10.1038/35077000. [DOI] [PubMed] [Google Scholar]
- Spier AD, Lummis SC. The role of tryptophan residues in the 5-hydroxytryptamine3 receptor ligand binding domain. J Biol Chem. 2000;275:5620–5625. doi: 10.1074/jbc.275.8.5620. [DOI] [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Xie Y, Cohen JB. Contributions of Torpedo nicotinic acetylcholine receptor γTrp-55 and δTrp-57 to agonist and competitive antagonist function. J Biol Chem. 2001;276:2417–2426. doi: 10.1074/jbc.M009085200. [DOI] [PubMed] [Google Scholar]
- Xiu X, Hanek AP, Wang J, Lester HA, Dougherty DA. A unified view of the role of electrostatic interactions in modulating the gating of Cys loop receptors. J Biol Chem. 2005;280:41655–41666. doi: 10.1074/jbc.M508635200. [DOI] [PubMed] [Google Scholar]
- Young GT, Broad LM, Zwart R, Astles PC, Bodkin M, Sher E, Millar NS. Species selectivity of a nicotinic acetylcholine receptor agonist is conferred by two adjacent extracellular β4 amino acids that are implicated in the coupling of binding to channel gating. Mol Pharmacol. 2007;71:389–397. doi: 10.1124/mol.106.030809. [DOI] [PubMed] [Google Scholar]