

Abstract
Acute ethanol depresses respiration, but little is known about chronic ethanol exposure during gestation and breathing, while the deleterious effects of ethanol on CNS development have been clearly described. In a recent study we demonstrated that pre- and postnatal ethanol exposure induced low minute ventilation in juvenile rats. The present study analysed in juvenile rats the respiratory response to hypoxia in vivo by plethysmography and the phrenic (Phr) nerve response to ischaemia in situ. Glycinergic neurotransmission was assessed in situ with strychnine application and [3H]strychnine binding experiments performed in the medulla. After chronic ethanol exposure, hyperventilation during hypoxia was blunted in vivo. In situ Phr nerve response to ischaemia was also impaired, while gasping activity occurred earlier and recovery was delayed. Strychnine applications in situ (0.05–0.5 μm) demonstrated a higher sensitivity of expiratory duration in ethanol-exposed animals compared to control animals. Moreover, [3H]strychnine binding density was increased after ethanol and was associated with higher affinity. Furthermore, 0.2 μm strychnine in ethanol-exposed animals restored the low basal Phr nerve frequency, but also the Phr nerve response to ischaemia and the time to recovery, while gasping activity appeared even earlier with a higher frequency. Polycythaemia was present after ethanol exposure whereas lung and heart weights were not altered. We conclude that chronic ethanol exposure during rat brain development (i) induced polycythaemia to compensate for low minute ventilation at rest; (ii) impaired the respiratory network adaptive response to low oxygen because of an increase in central glycinergic tonic inhibitions, and (iii) did not affect gasping mechanisms. We suggest that ethanol exposure during early life can be a risk factor for the newborn respiratory adaptive mechanisms to a low oxygen environment.
Environmental conditions during pregnancy may have profound effects on the origin of certain adult diseases (Barker, 2004). Neonatal maternal separation, disturbances of fetal growth and exposure to drugs of abuse during pregnancy are examples of sources of developmental deficits affecting pulmonary or peripheral chemoreceptors and/or the central cardiorespiratory network (Barker, 2004; Kinkead et al. 2005; Cohen et al. 2005). Disturbances in the control of breathing may be observed during resting ventilation and/or during adaptive response to chemosensory challenges including induction of gasping activity (St-John, 1998a,b; Fewell et al. 2001; Gozal et al. 2003).
Chronic prenatal ethanol exposure induces several changes in the neurophysiology and the behaviour of offspring (Richardson et al. 2002; Iqbal et al. 2004). However, nothing is known about the effects of chronic ethanol exposure during gestation on the capacity of the respiratory network to adapt to chemosensory challenges. This is intriguing since in humans, 3–5% of women drink alcohol at some time during pregnancy (Burd & Wilson, 2004), increasing the risk of abortion, fetal death or premature delivery (Habbick et al. 1997; Rasch, 2003). Moreover, siblings of children with fetal alcohol syndrome have an increased risk of death due to sudden infant death syndrome (SIDS) (Burd et al. 2004), while SIDS is significantly associated with maternal binge drinking behaviour during pregnancy (Habbick et al. 1997; Kinney et al. 2001; Iyasu et al. 2002).
We recently reported (Dubois et al. 2006) that juvenile rats exposed to pre- and postnatal ethanol exposure presented a dramatic reduction in minute ventilation, suggesting that they were exposed to chronic hypoxia from birth. Moreover, the respiratory network subsequently became insensitive to application of muscimol, a GABAA agonist. Although ethanol is known to affect GABAA and NMDA receptor function (Costa et al. 2000), glycinergic inhibitions are also increased by acute ethanol in neurons studied in either brain slices or culture (Aguayo & Pancetti, 1994; Aguayo et al. 1996; Ye et al. 2001). Chronic ethanol treatment of glycinergic spinal interneurons maintained in culture also modified the glycine receptor sensitivity to strychnine and to Zn2+, a positive modulator of glycine receptors (van Zundert et al. 2000). It has also been shown that acute ethanol modulates glycinergic neurotransmission to a greater degree than GABAergic neurotransmission in hypoglossal motoneurons with more marked sensitivity in juvenile animals (Sebe et al. 2003; Eggers & Berger, 2004). Altogether, these findings suggest that glycine receptors can be an important target for ethanol during chronic exposure in early life. This could be particularly relevant for the respiratory network since glycinergic inhibition is important for respiratory rhythmogenesis in mature animals (Busselberg et al. 2001), coordination of reflex control of the upper airways (Paton & Dutschmann, 2002; Dutschmann & Paton, 2002), induction of gasping activity (St-John & Leiter, 2002) and embryonic maturation of the respiratory network (Fujii et al. 2007).
In the present study, we tested whether chronic ethanol exposure during the developmental period of the rat brain disturbs the respiratory response to low oxygen of juvenile offspring and/or the genesis of gasping rhythmic activity. We assessed the role of glycinergic inhibition in the respiratory network and its involvement in the response to hypoxia after ethanol exposure. Binding experiments with [3H]strychnine were performed in the medulla. Finally, arterial blood samples were analysed under resting conditions after ethanol exposure to assess the animal's oxygenation status. The lungs and heart were also weighed to check for organ development.
Methods
Animal groups
Three- to four-week-old juvenile male Sprague–Dawley rats were used for the present study. Offspring of the control animal group were obtained from 18 different litters from dams never exposed to ethanol. The age-matched ethanol-exposed group was obtained from 22 different litters from dams who received a 10% (v/v) ethanol solution as unique drinking fluid for 4 weeks before mating and throughout the gestation and lactation periods (Naassila & Daoust, 2002; Dubois et al. 2006). Only male rats were used to avoid any sex difference in the response to hypoxia (Mortola & Saiki, 1996), which have also been observed after other prenatal chronic experimental procedures such as maternal separation (Kinkead et al. 2005).
Recording and analysis of breathing by plethysmography
Spontaneous respiratory activity and responses to hypoxia were recorded in awake and unrestrained animals by whole body plethysmography (Bartlett & Tenney, 1970) as previously described (Dubois et al. 2006). All animals (n = 12) were weighed before being introduced into a sealed Plexiglas chamber continuously flushed with humidified room air and connected to a reference chamber of the same size. The pressure difference between the two chambers was measured with a high gain differential pressure transducer (DP45, Validyne Engineering, Northridge, CA, USA). Barometric chamber temperature was maintained at 31°C by an external heat source. Rectal temperature was measured before and at the end of the experiment. After a 30 min acclimatization period in the chamber, measurements were performed for 60 s at rest (quiet waking state). Hypoxia was induced by switching the normoxic gas to a mixture of 11% O2 and 89% N2 for 5 min. Measurements were performed for 60 s at the end of the hypoxic episode. Analogue signals were continuously digitized (micro1401, CED electronics, UK), recorded with Spike2 software (CED electronics) and stored on a computer for off-line analysis.
Respiratory phase durations (inspiration, Ti and expiration, Te) as well as tidal volume (VT) and inspiratory inflow index (VT/Ti) were measured and averaged over 30 consecutive respiratory cycles before and at the end of hypoxia. Minute ventilation 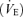 , and respiratory frequency (Rf, cycles min−1) were calculated. All comparisons between the two animal populations including body weight and animal temperature were performed with Student's unpaired t test with a limit of significance of P < 0.05.
, and respiratory frequency (Rf, cycles min−1) were calculated. All comparisons between the two animal populations including body weight and animal temperature were performed with Student's unpaired t test with a limit of significance of P < 0.05.
In situ preparation
The surgical procedures were conducted in accordance with the guidelines for care and use of laboratory animals adopted by the European Community, law 86/609/EEC. In situ working-heart–brainstem preparations were performed as previously described (Leiter & St-John, 2004; Dubois et al. 2006). Briefly, under deep halothane anaesthesia, as assessed by lack of a withdrawal reflex to nociceptive pinch on a paw, the animal was decerebrated at the precollicular level and the forebrain was discarded. Subdiaphragmatic section was then performed and the rib cage and lungs were discarded. The phrenic nerve was isolated and cut distally before transferring the animal to the recording chamber. The descending aorta was perfused with Ringer solution maintained at 31°C containing (mm): MgSO4 1.25, KH2PO4 1.25, KCl 5, NaHCO3 25, NaCl 125, glucose 10, CaCl2 2.5 and 1.25% Ficoll70 and continuously equilibrated with carbogen (95% O2–5% CO2, pH 7.4). This Ringer solution was similar to one we used previously (Dubois et al. 2006). Although it is a hyperkaliemic perfusate, it has been shown that phrenic nerve patterns during ischaemia were not altered with [K+]o (St-John et al. 2005). Once the phrenic nerve discharge was spontaneously activated, the animal was subject to neuromuscular blockade by addition of pancuronium bromide (2–4 μg ml−1) into the perfusate. These experiments were conducted on a control group of 36 animals and ethanol-exposed group of 39 animals.
Protocols and measurements for in situ experiments
Phrenic (Phr) nerve activity was recorded extracellularly with glass suction electrodes. The raw signal was amplified (×50 000), filtered (0.3–3 kHz), integrated (τ= 100 ms) and stored in a computer using Spike2 acquisition software (Cambridge Electronic Design, Cambridge, UK). The experiments were started after stabilization of the amplitude and frequency of Phr nerve activity (about 20–30 min) with the presence of an augmenting ramp revealing an eupnoeic pattern (i.e. physiological) generated by the central respiratory network (St-John & Paton, 2000).
An ischaemic episode was induced once in each animal by completely stopping the perfusion pump for 2 min. This duration was chosen on the basis of a previous report demonstrating that it was sufficient to induce the three phases of the respiratory network response to hypoxia (i.e. fictive hyperventilation, apnoea and gasping; St-John & Paton, 2000) with recovery after restarting the pump (reoxygenation period).
Respiratory frequency (cycle min−1) and inspiratory burst amplitude were measured from the integrated form of the Phr nerve recording before (baseline), during the 2 min ischaemic episode and during the 15 min reoxygenation period. Baseline Phr nerve activity was defined as a three-phase respiratory cycle including an incrementing inspiratory burst (Ti) followed by a decrementing post-inspiratory burst (PI) and then a silence until the next burst, defining the second expiratory phase (or Te2). Total expiratory time was the summation of PI and Te2 durations (Fig. 1A). For measurements during ischaemia, the response was divided into (Fig. 1B): fictive hyperventilation (phase 1) defined as increased amplitude and frequency of Phr nerve activity, followed by measurement of the duration of complete arrest of rhythmic activity corresponding to apnoea (phase 2). Gasping discharge was then elicited and the time to onset of gasping (phase 3) from the onset of ischaemia was measured, together with the total duration of gasping activity until bell-shaped bursts were observed (phase 4). The total number of inspiratory gasps generated and the gasp frequency (cycle min−1) were also calculated. Finally, the time to the first burst from the onset of reoxygenation was measured with an incrementing ramp without the plateau phase (phase 5), and the frequency and amplitude were measured after 15 min of reoxygenation. Figure 1C illustrates the integrated traces of Phr nerve burst as recorded in situ with the various patterns recorded during the response to ischaemia. Bell-shaped bursts were defined as bursts with an incrementing slope followed by a decrementing slope and maximum amplitude close to 50% of burst duration. These bursts were encountered at the transition between apnoea and gasping activity and/or between the end of the gasping period and the first incrementing Phr burst.
Figure 1. Definition of measuring times during phrenic nerve response to ischaemia in situ.

A, definition of the three phases of the respiratory cycle as measured on phrenic nerve discharge in situ during baseline activity. Ti: inspiration; Te: expiration; PI: post-inspiration; Te2: second phase of expiration. B, top trace is the time course of incrementing burst phrenic (Phr) nerve frequency before, during and after ischaemia. Bottom trace is the corresponding integrated Phr nerve activity. The measuring times are defined as follows: (1) hyperventilation with increased amplitude and frequency of incrementing Phr bursts; (2) apnoea duration; (3) time to first gasping burst; (4) duration of gasping activity; (5) recovery time to first ramping Phr burst after reoxygenation. C, Phr burst pattern showing the difference in ramp slope according to the type of bursting activity. Incrementing ramp discharges defined eupnoea. Gasping was characterized by decrementing ramp discharge. Bell-shaped discharges are observed at the transition between eupnoeic and gasping rhythms or at the beginning of recovery. Rf: respiratory frequency (cycles min−1); ∫Phr: integrated phrenic nerve activity.
For experiments on the role of glycinergic neurotransmission, eight preparations from each group of animals (control and ethanol-exposed) were used. The dose–response curve to strychnine (0.05, 0.1, 0.2 and 0.5 μm) applied directly into the perfusate was determined and respiratory phase durations were measured (inspiration, Ti; post-inspiration, PI; and second phase of expiration, Te2; Fig. 1A) as well as Phr burst amplitude at each dose. Responses to ischaemia were tested in the presence of 0.2 μm strychnine in eight preparations (4 in each group). The same measurements were performed during the response to ischaemia as with control perfusate solution. In four other preparations from early ethanol-exposed animals, the response to ischaemia was tested after increasing the extracellular potassium concentration to 8 mm in the perfusate.
Phrenic nerve activity data analysis
All results are presented as means ±s.e.m. and expressed as either raw data or percentage change of control values recorded prior to hypoxia. Measurements of in situ Phr nerve activity were averaged over 5–15 consecutive respiratory cycles during the control period (before ischaemia) and during the previously defined periods of Phr nerve response to ischaemia. The spontaneous (baseline value) Phr nerve rhythmic activity was compared between control and ethanol-exposed animals by Student's unpaired t test. A possible effect of chronic ethanol exposure on the animal's body weight was also compared between the two populations using an unpaired t test. Statistical analysis of the strychnine concentration–response curve was performed on raw data with one- or two-way repeated-measures ANOVA, with drug treatment (ethanol versus water) as independent variables and nerve amplitude and phase duration as dependent variables. When ANOVA was significant, pairwise comparisons were performed with the Student–Neumann–Keuls post hoc test. For all tests, P-values less than 0.05 were considered significant.
Membrane preparation and [3H]strychnine binding
Binding experiments were performed as previously described (Naassila & Daoust, 2002). Medulla from juvenile rats (n = 10 in each group) was removed on ice and thawed in five volumes of 0.32 m sucrose using an Elvejhem-type potter. After a first centrifugation (3000 g, 4°C, 15 min), the supernatant was further centrifuged (48 000 g, 4°C, 15 min). The pellet was rinsed at least 10 times with 5 volumes of Tris-HCl buffer (pH 7.4, 20°C). The final pellet was resuspended in 1 ml buffer, frozen (−18°C) until use and a 10 μl aliquot was used for protein measurement by the method of Lowry et al. (1951). [3H]Strychnine binding assays were performed using well-washed membranes. Then, 0.15 mg of protein was incubated in 0.5 ml of 50 mm Tris-HCl buffer (pH 7.4, 20°C) containing 2.5–120 nm[3H]strychnine at room temperature for 10 min. Non-specific binding was determined using 100 μm unlabelled strychnine. The contents of the tubes were filtered on Whatman glass fibre filters (GF/B, 45 μm pore size) and rinsed with 2 × 5 ml of cold Tris buffer. Radioactivity was determined using 5 ml of ACS scintillation fluid and counted in a Wallac 1414 Winspectral liquid scintillation counter (Perkin Elmer, Courtaboeuf, France, 50% efficiency for 3H). Binding parameters (Kd, Bmax) were evaluated using MultiCalc Software (Perkin Elmer).
Arterial blood sample analysis
Twelve control and 12 ethanol-exposed juvenile rats were used for blood sample analysis. They were deeply anaesthetized with pentobarbital sodium (Nembutal, 50 mg kg−1, i.p.) and arterial blood was collected from the common carotid artery in an Eppendorf vial containing 5 IU of heparin. Blood samples were analysed immediately with an ABL725 device (Radiometer, Copenhagen, Denmark). Statistical analysis was performed with an unpaired t test with P < 0.05 as the limit of significance. After blood collection, the animal was killed with a large bolus of Nembutal until cardiac arrest and the neck was then dislocated.
Lung and heart tissues
Animals were weighed and then received an overdose of Nembutal i.p. (100 mg kg−1, n = 10). After death, confirmed by respiratory and cardiac arrest, lungs and heart were quickly removed from the thoracic cage. The organs were weighed both wet and after drying for 24 h at 75°C, as previously described (Inselman et al. 1985; Gleed & Mortola, 1991). Mean weights were expressed with respect to body weight (g (100 g bw)−1) ±s.e.m. Statistical analyses were performed by Student's unpaired t test with P < 0.05 as the limit of significance.
Results
Response to hypoxia in vivo
Body weight (controls, 65.2 ± 4.8 g; ethanol, 68 ± 4.9 g; n = 5 and 7, respectively; P > 0.05) and body temperature (controls, 36.2 ± 1.5°C; ethanol; 35.2 ± 0.6°C, P > 0.05) were similar between groups. During normoxia, ethanol-exposed animals had a slower respiratory frequency (controls, 144 ± 13 cycles min−1; ethanol, 113 ± 6 cycles min−1, Fig. 2A and B, P < 0.01), as previously reported (Dubois et al. 2006). Inspiratory time was not affected by ethanol (controls, 0.16 ± 0.01 s; ethanol, 0.19 ± 0.02 s, P > 0.05), while expiratory time was significantly increased (controls, 0.28 ± 0.02 s; ethanol, 0.35 ± 0.03 s, P < 0.05). Tidal volume (VT) was lower in ethanol-exposed animals (control, 0.75 ± 0.07 ml (100 g)−1; ethanol, 0.56 ± 0.05 ml (100 g)−1, P > 0.05; Fig. 2B) although not enough to reach statistical significance and minute ventilation was significantly reduced (controls, 104.8 ± 8.3 ml min−1; ethanol, 63.4 ± 7.3 ml min−1, P < 0.001; Fig. 2B). The inspiratory inflow index (VT/Ti) was significantly lower in ethanol-exposed than in control animals (controls, 5.45 ± 0.2 ml (100 g)−1 min−1; ethanol, 3.78 ± 0.32 ml (100 g)−1 min−1, P < 0.05; Fig. 2B).
Figure 2. Response to hypoxia in vivo.

A, recordings of spontaneous breathing activity in the two animal populations under normoxic conditions and at the end of hypoxia. Inspiration is upwards. B, analysis of ventilatory parameters shows that respiratory frequency was significantly increased in both populations (upper left panel), while tidal volume increased only in control animals (upper right panel). Minute ventilation and inspiratory inflow index were both increased only in the control population (lower left and right panels, respectively). **P < 0.01, ***P < 0.001, compared to baseline values; ##P < 0.01, ###P < 0.001, comparison between populations.
During hypoxia, both populations responded with a significant and comparable increase in respiratory frequency (controls, from 144 ± 13 to 179 ± 7 cycles min−1, P < 0.01; ethanol, from 113 ± 6 to 139 ± 7 cycles min−1, P < 0.05, Fig. 2A and B). However, the respiratory frequency remained lower in ethanol-exposed animals (P < 0.01, Fig. 2B). In contrast with respiratory frequency, only control animals exhibited a significant increase in VT during hyperventilation (controls, from 0.75 ± 0.07 to 1.41 ± 0.24 ml (100 g)−1, P < 0.05; ethanol, from 0.56 ± 0.05 to 0.77 ± 0.05 ml (100 g)−1, P > 0.05, Fig. 2B). Consequently, only the control group showed an increase in minute ventilation 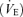 during hypoxia (controls, from 104.8 ± 8.3 to 255.1 ± 5.2, P < 0.05; ethanol, from 63.4 ± 7.3 to 106.1 ± 8.8 ml min−1, P > 0.05, Fig. 2B). Inspiratory time during hyperventilation was shortened but not significantly in both groups (about −10%, P > 0.05) and VT/Ti was increased only in control animals (controls, from 4.78 ± 0.46 to 10.04 ± 1.83 ml (100 g)−1 s−1, P < 0.05; ethanol, from 3.04 ± 0.34 to 4.9 ± 0.39 ml (100 g)−1 s−1, P > 0.05; Fig. 2B) and remained significantly lower in ethanol-exposed animals (P < 0.001). Expiratory time was significantly reduced in both groups (controls, −28%, P < 0.05; ethanol, −20%; P < 0.05, Fig. 2A). After 15 min of reoxygenation, both groups presented similar ventilatory parameters as before hypoxia (data not shown).
during hypoxia (controls, from 104.8 ± 8.3 to 255.1 ± 5.2, P < 0.05; ethanol, from 63.4 ± 7.3 to 106.1 ± 8.8 ml min−1, P > 0.05, Fig. 2B). Inspiratory time during hyperventilation was shortened but not significantly in both groups (about −10%, P > 0.05) and VT/Ti was increased only in control animals (controls, from 4.78 ± 0.46 to 10.04 ± 1.83 ml (100 g)−1 s−1, P < 0.05; ethanol, from 3.04 ± 0.34 to 4.9 ± 0.39 ml (100 g)−1 s−1, P > 0.05; Fig. 2B) and remained significantly lower in ethanol-exposed animals (P < 0.001). Expiratory time was significantly reduced in both groups (controls, −28%, P < 0.05; ethanol, −20%; P < 0.05, Fig. 2A). After 15 min of reoxygenation, both groups presented similar ventilatory parameters as before hypoxia (data not shown).
These results demonstrated that ethanol-exposure during the early period of development significantly reduced respiratory responses to hypoxia in vivo in juvenile rats.
Response to ischaemia in situ
In order to more clearly understand the mechanisms underlying the blunted response to hypoxia in vivo, in situ perfused juvenile rat brainstem preparations (Paton, 1996) were exposed to ischaemia, and temporal changes in the phrenic (Phr) activity pattern until the induction of gasping activity and recovery were analysed.
With baseline extracellular potassium concentration (6.25 mm), spontaneous Phr nerve frequency was 19.8 ± 2.6 cycles min−1 in the control group (n = 12) and 12.5 ± 3.1 cycles min−1 in the ethanol-exposed group (n = 9; Fig. 3A and B). This 37% decrease in frequency in situ after early and chronic ethanol exposure (P < 0.01) was consistent with the findings of our previous study (Dubois et al. 2006). In both groups, ischaemia induced a three-phase response followed by recovery when oxygen was reintroduced (i.e. fictive hyperventilation, eupnoea and gasping activity; see also Methods and Fig. 1B for definitions of phase measurements).
Figure 3. Phrenic nerve response to ischaemia in control and ethanol-exposed populations in situ.

A, recordings of integrated Phr nerve activity illustrating the absence of hyperventilation in ethanol-exposed animal and the earlier onset of gasping activity (single vertical arrows) due to shorter apnoea duration (horizontal arrows). During reoxygenation, recovery of eupnoeic activity was significantly delayed in the ethanol-exposed group (double arrows indicate first incrementing Phr burst). B, ethanol-exposed animals did not present a significant increase in Phr burst frequency and amplitude. ∫Phr: integrated phrenic nerve activity. Same statistical symbols as in Fig. 2. The simple arrow points to the first gasping burst and the double arrow to the first incrementing burst. Horizontal arrow delineates apnoea.
Ischaemia induced an increase in Phr nerve frequency to 29 ± 2.6 cycles min−1 in the control group and to 17.2 ± 3.1 cycles min−1 in the ethanol-exposed group. This increase was significant for the control group (P < 0.05, Fig. 3A and B), but not for the ethanol-exposed group (P > 0.05, Fig. 3A and B). The difference in frequency between the two populations during fictive hyperventilation was still significant (P < 0.05). The increase in Phr burst amplitude was significant only in the control population (controls, P < 0.001; ethanol, P > 0.05; Fig. 3B). Apnoea duration was shorter in ethanol-exposed animals (P < 0.05, Fig. 3A and Table 1‘strychnine 0’ part) and was terminated by gasping activity in both populations. Due to the shorter apnoea duration, gasping appeared earlier in ethanol-exposed animals than in control animals (Fig. 3A, Table 1, P < 0.05). Despite this earlier onset of gasping, the number of gasps generated during ischaemia, the total number of gasps generated and total gasp frequency were not different between the two groups (Table 1). Finally, after oxygen was reintroduced, the time to the first burst with an incrementing ramp discharge was significantly increased in the ethanol group compared to the control group (P < 0.05, Fig. 3A, Table 1). After 15 min of reoxygenation, Phr nerve burst frequency was still lower in the ethanol group (controls, 19.5 ± 2.6 cycles min−1; ethanol, 16 ± 3.3 cycles min−1; P > 0.05). These values were not different from baseline values. Phr nerve amplitude also recovered after 15 min for the two populations (controls, 92.4 ± 8.9% of prehypoxia burst amplitude, P > 0.05; ethanol-exposed, 114 ± 11.3%, P > 0.05).
Table 1.
Phrenic nerve response to ischaemia in absence and in presence of 0.2 μm strychnine in control and ethanol-exposed animals
| 0 μm Strychnine | 0.2 μm Strychnine | |||
|---|---|---|---|---|
| Control group (n = 12) | Ethanol exposed group (n = 9) | Control group (n = 4) | Ethanol exposed group (n = 4) | |
| Apnoea duration (s) | 26.9 ± 3.2 | 16.4 ± 2.4 (#) | 3.4 ± 0.2 (***) | 5.8 ± 0.6 (*) |
| Delay to first gasp (s) | 92.0 ± 11.8 | 60.8 ± 5.6 (#) | 43.7 ± 1.3 (**) | 40.2 ± 2.3 (*) |
| Total gasp number | 9 ± 2 | 8 ± 2 | 7 ± 1 | 6 ± 1 |
| Gasp number during anoxia | 5.1 ± 0.8 | 5.7 ± 1.6 | 7 ± 1 | 6 ± 1 |
| Gasp frequency (burst.min−1) | 12.8 ± 2.1 | 11.8 ± 1 | 21.5 ± 2.9 (*) | 21.7 ± 2.1 (*) |
| Delay to first incrementing | 76.1 ± 17 | 174 ± 51.4 (#) | 77.7 ± 10.8 | 84.5 ± 13.3 (*) |
| burst after reoxygenation (s) | ||||
Data are expressed as mean ±s.e.m. For strychnine 0 μm (#) P < 0.05, express a significant difference between control group and ethanol-exposed group. In presence of strychnine, no differences were obtained between populations. However (*) P < 0.05 (**) P < 0.01 (***) P < 0.001 express a significant difference from values without strychnine in the respective population.
These results demonstrated that early chronic ethanol exposure abolished ischaemia-induced fictive hyperventilation as seen in Phr nerve activity, reduced apnoea duration, did not modify gasping activity and delayed recovery after reoxygenation.
Effects of strychnine after early ethanol exposure
The in situ preparation was used to test the possibility that ethanol exposure modifies glycinergic inhibitions in the network during both spontaneous activity and response to ischaemia.
Strychnine dose–response curve during baseline activity
Inspiratory time (Fig. 4A and B) was significantly reduced to a similar degree in both control and ethanol-exposed populations (n = 8 for both groups). The most intense effect on modulation of expiratory time was observed in ethanol-exposed animals with a dose-dependent decrease starting at 0.05 μm, whereas expiratory time in control animals was not significantly decreased by strychnine (Fig. 4A and B). Expiratory time before addition of strychnine was much longer in ethanol-exposed animals and the difference between the two populations was abolished in the presence of 0.1 μm strychnine. Post-inspiratory time in both populations (Fig. 4C) was reduced to a similar degree by strychnine. In contrast, the second phase of expiration (or stage 2) was reduced only in ethanol-exposed animals (Fig. 4C). In summary, strychnine affected inspiratory, post-inspiratory and stage 2 of expiratory times in ethanol-exposed animals, but only affected inspiratory and post-inspiratory times in the control population. These effects on phase durations induced an increase in Phr nerve frequency (Fig. 4D) with a higher sensitivity of ethanol-exposed animals, as 0.1 μm strychnine was effective compared to 0.2 μm in control animals. However, the difference in burst frequency was abolished between the two populations only with 0.2 μm strychnine. The effects on burst amplitude were variable in each group but no significant change was observed until 0.5 μm strychnine (Fig. 4D).
Figure 4. Strychnine and phrenic nerve activity in situ.

A, recording of Phr nerve activity in the presence of 0.2 μm strychnine. At this concentration, phase durations and frequency were similar in the two populations. B, strychnine decreased the duration of inspiration (left) to a similar degree in the two populations, but decreased expiratory duration only in ethanol-exposed animals (right). C, post-inspiratory time was reduced to a similar degree in the two populations (left), whereas the second expiratory phase was reduced only in ethanol-exposed animals (right). D, Phr frequency was increased in both populations, but at a lower dose for the control group (left) and burst amplitude was not affected up to a concentration of 0.5 μm strychnine in both populations. Data for B–D are expressed as means ±s.e.m.iP = 0.05, *P < 0.05, **P < 0.01, ***P < 0.001 as compared to baseline values before strychnine. #P < 0.05 and ##P < 0.01, between populations.
Response to ischaemia in the presence of strychnine
The dose–response to strychnine revealed a higher sensitivity of the respiratory cycle duration in ethanol-exposed animals. We therefore tested whether the response to ischaemia in ethanol-exposed animals was also sensitive to strychnine. We tested 0.2 μm strychnine since at that concentration the lower basal respiratory frequency was compensated (Fig. 4D).
Under these conditions, Phr burst frequency in the ethanol group (22.9 ± 4.7 cycles min−1; n = 4) was higher than without strychnine (12.5 ± 3.1, P < 0.05; +83%) but similar to that obtained in control animals without strychnine (19.8 ± 2.6, P > 0.05). The response to ischaemia showed a significant increase in respiratory frequency reaching 47.1 ± 9.8 cycles min−1 (+105%, P < 0.05, Fig. 5A–C) compared to ethanol-exposed animals with strychnine-free perfusate (+37%, P > 0.05; see Fig. 3B). Burst amplitude was also more markedly increased than without strychnine (+30%versus+15%Figs 5A–C and 3B), but was still not significant due to the marked variability of response and the presence of a tonic phase. Gasping activity appeared even earlier (i.e. apnoea duration was even shorter) than without strychnine (Table 1‘0.2 μm strychnine’ part). Total number of gasps was unchanged, while gasp frequency was increased (Tables 1 and 2). Time to recovery of the first incrementing Phr burst after ischaemia was significantly reduced to a value similar to that of control animals without strychnine (84.5 s versus 76.1 s in control animals, P > 0.05, Table 1).
Figure 5. Phrenic nerve response to ischaemia in situ in the presence of 0.2 μm strychnine or 8 mm[K+]o.

A, recordings of Phr nerve response. Tonic activity was observed during the first minute of ischaemia in most cases. Note the same gasping latency and recovery time to the first incrementing burst (single and double arrows, respectively). B, Phr response to ischaemia of an ethanol-exposed animal in the presence of 8 mm potassium instead of 6.25 mm. Note the absence of increase in frequency and amplitude, the time to first gasp and the very slow recovery. C, increase in Phr nerve frequency (left) was restored in ethanol-exposed animals and the difference between populations was abolished. The increase in burst amplitude (right) was similar between the two populations.
Table 2.
Effect of early ethanol exposure on blood oxygen status in normoxia
| Control group | Ethanol-exposed group | |
|---|---|---|
| (n = 12) | (n = 12) | |
| Total O2 content (vol percentage) | 8.7 ± 0.7 | 11.7 ± 0.4 (**) |
| Hematocrit (%) | 24.6 ± 1.4 | 30.6 ± 0.7 (**) |
| Total Hb content (g dl−1) | 7.9 ± 0.5 | 9.9 ± 0.2 (**) |
Early chronic ethanol exposure induced a polycythemia that significantly raised the total blood oxygen content. Data are expressed as mean ±s.e.m. (**) P < 0.01, significant difference between the control group ant the ethanol-exposed group.
In control preparations exposed to ischaemia in the presence of 0.2 μm strychnine (n = 4), Phr nerve frequency increased significantly to 30.7 ± 2.4 cycles min−1 (P < 0.05; Fig. 5A–C). During ischaemia, these animals showed a 75% increase in respiratory frequency (P < 0.05) and a 32.7% increase in burst amplitude (P = 0.05). These increases were lower than those obtained without strychnine (see Fig. 2B). The time to first gasping burst was decreased compared to control animals without strychnine (Fig. 5A, Table 1‘0.2 μm strychnine’ part). In contrast to ethanol-exposed animals, the time to recovery of the first incrementing burst was not affected by strychnine in the control population.
Effects of ethanol exposure on [3H]strychnine binding parameters
Ethanol exposure increased the Bmax value for [3H]strychnine in the medulla from juvenile rats (controls, 564.9 ± 40 fmol (mg protein)−1; ethanol, 712 ± 35.5; P < 0.05) and the dissociation constant (Kd) was significantly lower in ethanol-exposed animals (39.4 ± 3.2 versus 52.2 ± 3.5 nm in control group, P < 0.05).
Increasing extracellular K+ concentration
To confirm that compensation of basal respiratory frequency by blockade of glycinergic inhibitions was specifically involved in the response to ischaemia, respiratory frequency was increased by elevating extracellular potassium concentration in the perfusate to 8 mm[K+]o in four ethanol-exposed animals (Fig. 5B). Prior to ischaemia and in the presence of 6.25 mm[K+]o, spontaneous Phr nerve frequency was 13.5 ± 1.4 cycles min−1 and reached 23.0 ± 2.7 cycles min−1 in the presence of 8 mm[K+]o. This value was similar to that observed in the control group in the presence of 6.25 mm[K+]o (19.8 ± 2.6 cycles min−1, P > 0.05) and in the ethanol-exposed group in the presence of strychnine (22.9 ± 4.7 cycles min−1, P > 0.05). During ischaemia and in the presence of 8 mm[K+]o, neither Phr nerve respiratory frequency nor amplitude was increased (frequency, +7%± 9%, P > 0.05; amplitude, +8 ± 6%, P > 0.05; Fig. 5C). Apnoea duration was not modified (19.0 ± 3.4 s, P > 0.05) compared to ethanol-exposed animals in the presence of 6.25 mm[K+]o. [K+]o at 8 mm did not modify the latency to first gasp (47.8 ± 4.0 s, P > 0.05) and the total number of gasps (8 ± 1.5, P > 0.05) compared to 6.25 mm[K+]o. However, gasp frequency was increased to 21.3 ± 2 cycles min−1 (P < 0.05). After reoxygenation, ethanol-exposed animals exhibited a longer time to recover the first incrementing Phr burst (359.1 ± 49.4 s, P < 0.05), compared to both 6.25 mm[K+]o ethanol-exposed and control animals.
Increasing extracellular potassium concentration therefore did not restore the response to ischaemia of ethanol-exposed animals, but increased gasp frequency and further delayed recovery of the first incrementing Phr burst.
Arterial blood sample analysis
Measurements (Table 2) revealed that total blood oxygen content was significantly higher (P < 0.01) in ethanol-exposed animals than in control animals. Haematocrit was significantly increased (P < 0.01) after ethanol exposure and accordingly, total haemoglobin content was also significantly increased (P < 0.01). In summary, early chronic ethanol exposure induced polycythaemia, responsible for higher total arterial blood oxygen content.
Heart and lung weights
Body weight was not significantly different between the two groups of animals (controls, 56 ± 1.1 g; ethanol, 58 ± 0.7 g, P > 0.05). After correction for body weight, no significant difference was observed between ethanol-exposed rats and control rats in terms of lung and heart weights, neither wet nor dry (P > 0.05, Table 3). In addition, the ratio lung weight/body weight was also similar.
Table 3.
effect of early ethanol exposure on lungs and heart weights
| Control group | Ethanol-exposed | |
|---|---|---|
| (n = 5) | group (n = 5) | |
| Lungs (g) | ||
| Wet weight/100 g body weight | 0.842 ± 0.020 | 0.859 ± 0.017 |
| Dry weight/100 g body weight | 0.157 ± 0.004 | 0.160 ± 0.003 |
| Dry weight/wet weight | 0.187 ± 0.001 | 0.186 ± 0.002 |
| Heart (g) | ||
| Wet weight/100 g body weight | 0.662 ± 0.031 | 0.658 ± 0.024 |
| Dry weight/100 g body weight | 0.126 ± 0.005 | 0.122 ± 0.005 |
| Dry weight/wet weight | 0.191 ± 0.002 | 0.185 ± 0.004 |
Discussion
In this investigation, a marked effect of ethanol exposure was observed on glycinergic inhibitions in the respiratory network of juvenile rats that affected both eupnoeic activity and response to low oxygen. The in vivo ventilatory response and the time course of phrenic nerve response to ischaemia were both reduced after ethanol exposure. A shorter latency to gasping activity and a longer time to recovery of eupnoeic activity after reoxygenation were also observed in situ. The strychnine dose–response curve revealed an increase in glycinergic inhibitions during normoxia. A low concentration of strychnine in ethanol-exposed animals restored basal respiratory frequency, the fictive hyperventilatory response to ischaemia and the recovery time. Moreover, binding experiments revealed a higher Bmax and a higher affinity for [3H]strychnine in the medulla of ethanol-exposed animals. Finally, ethanol-exposed animals developed polycythaemia with no alteration of lung and heart weights.
Pre- and postnatal ethanol exposure alters baseline respiratory parameters
Baseline values under normoxic conditions for the in vivo control group were within the same range as those reported previously for juvenile rats (Mortola & Saiki, 1996; Peyronnet et al. 2000; Dubois et al. 2006).We have previously reported similar baseline respiratory values for the ethanol-exposed group (Dubois et al. 2006). Thus, the breathing pattern consisted of a lower respiratory frequency due to an increase in expiratory time with no alteration of inspiratory time. Moreover, in ethanol-exposed animals, the low VT combined with low frequency resulted in lower minute ventilation under normoxic conditions. Additionally, the VT/Ti ratio, an index of inspiratory inflow, was significantly lower in ethanol-exposed animals, suggesting less efficient pulmonary gas exchange during normoxia. However, ethanol exposure induced a polycythaemia in juvenile rats that possibly compensated for the lower baseline respiratory activity at rest. Similar alterations of baseline ventilation measured in intact animals were observed for phrenic nerve activity recorded in situ, equivalent to the results reported by Dubois et al. (2006). Importantly, our model of ethanol exposure induced significant blood ethanol levels in pregnant rats (Naassila & Daoust, 2002) comparable to those observed in pregnant women with chronic alcohol consumption (Halmesmaki, 1988) and which can be considered to be moderate (Eckardt et al. 1998).
Respiratory response to low oxygen is blunted in ethanol-exposed animals
The main change during hypoxia in vivo was a failure to increase minute ventilation 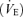 in ethanol-exposed animals, suggesting either a reduced central inspiratory drive or increased inhibition. Studies on in situ preparations revealed that ethanol exposure induced certain modifications of the central respiratory network. During ischaemia in situ, the respiratory network showed the classical response with acceleration of respiratory activity (fictive hyperventilation) at onset of stimulation, followed by a decline resulting in apnoea (Guntheroth & Kawabori, 1975; St-John & Paton, 2000; St-John & Leiter, 2002), and ending with gasping activity corresponding to an ‘autoresuscitation’ mechanism (St-John & Paton, 2000; St-John & Leiter, 2002; Fewell, 2005). The phrenic nerve response in ethanol-exposed animals was abolished in terms of both amplitude (a surrogate of VT) and frequency. The lack of increase in phrenic nerve amplitude, in accordance with the in vivo findings, supported the hypothesis that increased central inspiratory drive in response to low oxygen was not efficient after ethanol exposure. Moreover, the lack of increase in frequency, only seen in situ, suggests that a central component was impaired by ethanol, but was probably compensated in the intact animal. However, in this preparation, the lungs are not functional, the chest wall has been removed, and the Hering–Breuer reflex and sensory mechanical afferent feedback are absent. This might partly explain the lack of control of respiratory frequency during ischaemia (Harris & St-John, 2003, 2005). Altogether, these results indicate that animals exposed to chronic ethanol early in life have a blunted response to low oxygen environment after 3 weeks of life.
in ethanol-exposed animals, suggesting either a reduced central inspiratory drive or increased inhibition. Studies on in situ preparations revealed that ethanol exposure induced certain modifications of the central respiratory network. During ischaemia in situ, the respiratory network showed the classical response with acceleration of respiratory activity (fictive hyperventilation) at onset of stimulation, followed by a decline resulting in apnoea (Guntheroth & Kawabori, 1975; St-John & Paton, 2000; St-John & Leiter, 2002), and ending with gasping activity corresponding to an ‘autoresuscitation’ mechanism (St-John & Paton, 2000; St-John & Leiter, 2002; Fewell, 2005). The phrenic nerve response in ethanol-exposed animals was abolished in terms of both amplitude (a surrogate of VT) and frequency. The lack of increase in phrenic nerve amplitude, in accordance with the in vivo findings, supported the hypothesis that increased central inspiratory drive in response to low oxygen was not efficient after ethanol exposure. Moreover, the lack of increase in frequency, only seen in situ, suggests that a central component was impaired by ethanol, but was probably compensated in the intact animal. However, in this preparation, the lungs are not functional, the chest wall has been removed, and the Hering–Breuer reflex and sensory mechanical afferent feedback are absent. This might partly explain the lack of control of respiratory frequency during ischaemia (Harris & St-John, 2003, 2005). Altogether, these results indicate that animals exposed to chronic ethanol early in life have a blunted response to low oxygen environment after 3 weeks of life.
Chemosensory failure may have occurred at different sites along the chemoreflex pathway including peripheral chemoreceptor sensitivity or structure, central nervous system (i.e. central cardiorespiratory neuronal network), chest/lung compliance or the cardiovascular system. However, it is unlikely that such failure was due to chest/lung mechanical properties or cardiovascular defects since ethanol-exposed animals did not present right ventricular hypertrophy or pulmonary oedema, for example. These measurements therefore suggest that the lungs and heart developed normally after birth in ethanol-exposed animals. Nevertheless, it cannot be excluded that peripheral chemoreceptors were affected, as the respiratory response at the onset of hypoxia is due to stimulation of the carotid bodies.
Compared to control animals, gasping activity during hypoxia occurred earlier in ethanol-exposed animals but, once elicited, the frequency of this activity was not affected. Our results suggest that (1) microenvironmental changes necessary to trigger gasping activity were induced more rapidly after early ethanol exposure and (2) neuronal and cellular mechanisms underlying gasping activity were not impaired. Finally, recovery of the first incrementing phrenic burst after ischaemia was delayed in ethanol-exposed animals, revealing difficulties in the reorganization of breathing function after episodes of hypoxia.
Pre- and postnatal ethanol exposure increases glycinergic inhibitions
Glycinergic inhibition, together with GABAergic inhibition, participates in the rhythmic discharge of respiratory neurons, and this is supported by computer models of central respiratory rhythmogenesis in mature mammals (Pierrefiche et al. 1998; Smith et al. 2000; Richter & Spyer, 2001). Recent studies have demonstrated the importance of glycine inhibition for the formation of normal respiratory rhythm in mature rodents. Glycine receptor blockade impairs coordination of cranial and spinal respiratory motor outflow in juvenile rats (Dutschmann & Paton, 2002; Paton & Dutschmann, 2002) and transforms the three phases of normal respiratory rhythm (inspiration, post-inspiration, stage 2 expiration) into a faster two-phase rhythm (inspiration, stage 2 expiration) in mature mice (Büsselberg et al. 2001; Dutschmann & Paton, 2002; Paton & Dutschmann, 2002). A faster rhythm was also observed in both animal populations with strychnine, but this was due to a reduction in inspiratory and post-inspiratory phase durations in control animals whereas inspiration, post-inspiration and stage 2 expiration were reduced in ethanol-exposed animals. This demonstrates that ethanol exposure increased glycinergic inhibitions at least throughout stage 2 of expiration during eupnoea. Further, strychnine binding density and glycine receptor affinity for strychnine in the medulla were both increased after ethanol exposure. This suggests that the stronger effect of strychnine throughout the respiratory cycle in ethanol-exposed animals might be due to ethanol-induced increased expression of glycine receptors and increased affinity for glycine. In this context, it has been shown that acute application of ethanol on neonatal rat brainstem slices preferentially modulates glycine receptors in hypoglossal motoneurons of juvenile rats (Sebe et al. 2003) by increasing their affinity for glycine (Eggers & Berger, 2004).
During the first minutes of hypoxia, glutamate and GABA both accumulate in the ventral respiratory group of the adult cat medulla (Richter et al. 1999). High levels of glycine are also released in other parts of the central nervous system during hypoxia (Phillis & O'Regan, 2003; Kawasaki et al. 2004). Other neurotransmitters might have also participated in the reported results. Serotonin and adenosine are two possible candidates since they are released during hypoxia (Richter et al. 1999) and greatly disturbed by chronic ethanol exposure (Druse et al. 1991; Watson et al. 1999). Nevertheless, the reduced frequency during low oxygen episodes suggests that ethanol exposure impairs excitatory processes or, conversely, increases inhibitory processes in the respiratory network. In this context, we have previously shown that, after similar ethanol exposure, the respiratory network became tolerant to muscimol, a GABAA agonist, suggesting that GABA release during hypoxia probably does not induce any additional inhibition of the respiratory network (Dubois et al. 2006). It therefore seems more plausible that the respiratory network was either unable to respond to low oxygen-induced release of glutamate or alternatively that an inhibitory neurotransmitter other than GABA was strongly released and/or became more effective after ethanol exposure. The reduced frequency response observed in situ could also have been due to a slower spontaneous respiratory frequency during the normoxia. However, increasing cycle frequency by elevating [K+]o to 8 mm did not restore fictive hyperventilation. We interpret these results as evidence for an ethanol-induced increase in glycinergic neurotransmission in the network at least partly responsible for both the low baseline respiratory frequency and the blunted hyperventilatory response. However, another possible explanation is that respiratory neurons became less sensitive to glutamate after ethanol exposure.
Gasping activity per se is independent of respiratory network inhibitory transmissions involving GABA and glycine (St-John & Paton, 2002). Our results on the effects of strychnine during both eupnoea and ischaemia in ethanol-exposed animals are in accordance with those reported by St-John & Paton (2002) and St-John & Leiter (2002). In our study, strychnine in control animals mimicked the shorter latency to first gasping burst (St-John & Leiter, 2002) as observed in ethanol-exposed animals without strychnine. However, in ethanol-exposed animals strychnine was still effective in reducing this delay. The apparent ambiguity between an increase in glycinergic inhibitions after ethanol during ‘normoxia’ and a shorter delay to gasping activity during ischaemia suggests that the properties of glycinergic inhibitions during apnoea were altered and/or that another neurotransmitter released or functioning during ischaemia was also modified after ethanol exposure. This might be the case for adenosine for example as cited above.
Another result was that the time to recovery of the first incrementing phrenic burst was restored to control values in the presence of strychnine, whereas there was no effect on this time in control animals. This reveals a new role for glycinergic inhibitions after chronic ethanol exposure during the recovery period following hypoxic episodes. This recovery time was further prolonged in the presence of high extracellular potassium concentrations in ethanol-exposed animals and this effect may be due to depolarization of glycinergic neurons resulting in a high level of activity during the reoxygenation period.
Overall, our results support the proposal of an increase in glycinergic inhibitions in the respiratory network after prenatal and postnatal ethanol exposure, as strychnine (i) restored respiratory frequency during eupnoea, (ii) restored abolition of increased phrenic nerve frequency and amplitude during hypoxia, and (iii) restored the recovery time, and these effects were associated with an increase in binding site density and affinity for strychnine in the medulla.
Conclusion
Moderate chronic ethanol exposure during brain development in rats induced polycythaemia in the offspring, which was probably responsible for breathing adaptation at rest. Furthermore, early ethanol exposure increased glycinergic inhibition in the central respiratory network and this participates, at least in part, in the lower breathing frequency at rest, the blunted hyperventilation during low oxygen episodes and the slower recovery after reoxygenation. These effects are possibly due to an interaction between chronic ethanol exposure and expression of glycine receptors and changes in receptor affinity for glycine. Moreover, autoresuscitation mechanisms underlying gasping activity were induced earlier suggesting a higher sensitivity of glycine receptor blockade during hypoxia. We speculate that ethanol exposure during gestation may induce several disturbances of glycine-dependent defensive reflexes of the upper airways in the offspring that may put neonates at risk during their first days of life.
Acknowledgments
This work was funded by the Council of Picardie. C. Dubois is supported by the University of Picardie Jules Verne and Houchi H. is supported by the Council of Picardie. The authors would like to thank Dr A. Saul for editing the manuscript.
References
- Aguayo LG, Pancetti FC. Ethanol modulation of the gamma-aminobutyric acidA- and glycine-activated Cl− current in cultured mouse neurons. J Pharmacol Exp Ther. 1994;270:61–69. [PubMed] [Google Scholar]
- Aguayo LG, Tapia JC, Pancetti FC. Potentiation of the glycine-activated Cl− current by ethanol in cultured mouse spinal neurons. J Pharmacol Exp Ther. 1996;279:1116–1122. [PubMed] [Google Scholar]
- Barker DJ. The developmental origins of adult disease. J Am Coll Nutr. 2004;6(Suppl.):588S–595S. doi: 10.1080/07315724.2004.10719428. [DOI] [PubMed] [Google Scholar]
- Bartlett D, Jr, Tenney SM. Control of breathing in experimental anemia. Respir Physiol. 1970;10:384–395. doi: 10.1016/0034-5687(70)90056-3. [DOI] [PubMed] [Google Scholar]
- Burd L, Klug M, Martsolf J. Increased sibling mortality in children with fetal alcohol syndrome. Addict Biol. 2004;9:179–188. doi: 10.1080/13556210410001717088. [DOI] [PubMed] [Google Scholar]
- Burd L, Wilson H. Fetal, infant, and child mortality in a context of alcohol use. Am J Med Genet C Semin Med Genet. 2004;127:51–58. doi: 10.1002/ajmg.c.30016. [DOI] [PubMed] [Google Scholar]
- Busselberg D, Bischoff AM, Paton JF, Richter DW. Reorganisation of respiratory network activity after loss of glycinergic inhibition. Pflugers Arch. 2001;441:444–449. doi: 10.1007/s004240000453. [DOI] [PubMed] [Google Scholar]
- Cohen G, Roux JC, Grailhe R, Malcolm G, Changeux JP, Lagercrantz H. Perinatal exposure to nicotine causes deficits associated with a loss of nicotinic receptor function. Proc Natl Acad Sci U S A. 2005;102:3817–3821. doi: 10.1073/pnas.0409782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa ET, Savage DD, Valenzuela CF. A review of the effects of prenatal or early postnatal ethanol exposure on brain ligand-gated ion channels. Alcohol Clin Exp Res. 2000;24:706–715. [PubMed] [Google Scholar]
- Druse MJ, Kuo A, Tajuddin N. Effects of in utero ethanol exposure on the developing serotoninergic system. Alcohol Clin Exp Res. 1991;15:678–684. doi: 10.1111/j.1530-0277.1991.tb00578.x. [DOI] [PubMed] [Google Scholar]
- Dubois C, Naassila M, Daoust M, Pierrefiche O. Early chronic ethanol exposure in rats disturbs respiratory network activity and increases sensitivity to ethanol. J Physiol. 2006;576:297–307. doi: 10.1113/jphysiol.2006.111138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutschmann M, Paton JF. Glycinergic inhibition is essential for co-ordinating cranial and spinal respiratory motor outputs in the neonatal rat. J Physiol. 2002;543:643–653. doi: 10.1113/jphysiol.2001.013466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckardt MJ, File SE, Gessa GL, Grant KA, Guerri C, Hoffman PL, Kalant H, Koob GF, Li TK, Tabakoff B. Effects of moderate alcohol consumption on the central nervous system. Alcohol Clin Exp Res. 1998;22:998–1040. doi: 10.1111/j.1530-0277.1998.tb03695.x. [DOI] [PubMed] [Google Scholar]
- Eggers ED, Berger AJ. Mechanisms for the modulation of native glycine receptor channels by ethanol. J Neurophysiol. 2004;91:2685–2695. doi: 10.1152/jn.00907.2003. [DOI] [PubMed] [Google Scholar]
- Fewell JE. Protective responses of the newborn to hypoxia. Respir Physiol Neurobiol. 2005;149:243–255. doi: 10.1016/j.resp.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Fewell JE, Smith FG, Ng VK. Prenatal exposure to nicotine impairs protective response of rat pups to hypoxia in an age-dependent manner. Respir Physiol Neurobiol. 2001;127:61–73. doi: 10.1016/s0034-5687(01)00232-8. [DOI] [PubMed] [Google Scholar]
- Fujii M, Arata A, Kanbara-Kume N, Saito K, Yanagawa Y, Obata K. Respiratory activity in brainstem of fetal mice lacking glutamate decarboxylase 65/67 and vesicular GABA transporter. Neuroscience. 2007;146:1044–1052. doi: 10.1016/j.neuroscience.2007.02.050. [DOI] [PubMed] [Google Scholar]
- Gleed RD, Mortola JP. Ventilation in newborn rats after gestation at simulated high altitude. J Appl Physiol. 1991;70:1146–1151. doi: 10.1152/jappl.1991.70.3.1146. [DOI] [PubMed] [Google Scholar]
- Gozal D, Reeves SR, Row BW, Neville JJ, Guo SZ, Lipton AJ. Respiratory effects of gestational intermittent hypoxia in the developing rat. Am J Respir Crit Care Med. 2003;167:1540–1547. doi: 10.1164/rccm.200208-963OC. [DOI] [PubMed] [Google Scholar]
- Guntheroth WG, Kawabori I. Hypoxic apnea and gasping. J Clin Invest. 1975;56:1371–1377. doi: 10.1172/JCI108217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habbick BF, Nanson JL, Snyder RE, Casey RE. Mortality in foetal alcohol syndrome. Can J Public Health. 1997;88:181–183. doi: 10.1007/BF03403884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halmesmaki E. Alcohol counselling of 85 pregnant problem drinkers: effect on drinking and fetal outcome. Br J Obstet Gynaecol. 1988;95:243–247. [PubMed] [Google Scholar]
- Harris MB, St-John WM. Tonic pulmonary stretch receptor feedback modulates both eupnea and gasping in an in situ rat preparation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R215–R221. doi: 10.1152/ajpregu.00112.2003. [DOI] [PubMed] [Google Scholar]
- Harris MB, St-John WM. Phasic pulmonary stretch receptor feedback modulates both eupnea and gasping in an in situ rat preparation. Am J Physiol Regul Integr Comp Physiol. 2005;289:R450–R455. doi: 10.1152/ajpregu.00750.2004. [DOI] [PubMed] [Google Scholar]
- Inselman LS, Fisher SE, Spencer H, Atkinson M. Effect of intrauterine ethanol exposure on fetal lung growth. Pediatr Res. 1985;19:12–14. doi: 10.1203/00006450-198501000-00004. [DOI] [PubMed] [Google Scholar]
- Iqbal U, Dringenberg HC, Brien JF, Reynolds JN. Chronic prenatal ethanol exposure alters hippocampal GABAA receptors and impairs spatial learning in the guinea pig. Behav Brain Res. 2004;150:117–125. doi: 10.1016/S0166-4328(03)00246-8. [DOI] [PubMed] [Google Scholar]
- Iyasu S, Randall LL, Welty TK, Hsia J, Kinney HC, Mandell F, McClain M, Randall B, Habbe D, Wilson H, Willinger M. Risk factors for sudden infant death syndrome among northern plains Indians. JAMA. 2002;288:2717–2723. doi: 10.1001/jama.288.21.2717. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Fujita T, Kumamoto E. Enhancement of the releases of GABA and glycine during ischemia in rat spinal dorsal horn. Biochem Biophys Res Commun. 2004;316:553–558. doi: 10.1016/j.bbrc.2004.02.078. [DOI] [PubMed] [Google Scholar]
- Kinkead R, Genest SE, Gulemetova R, Lajeunesse Y, Laforest S, Drolet G, Bairam A. Neonatal maternal separation and early life programming of the hypoxic ventilatory response in rats. Respir Physiol Neurobiol. 2005;149:313–324. doi: 10.1016/j.resp.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Filiano JJ, White WF. Medullary serotonergic network deficiency in the sudden infant death syndrome: review of a 15-year study of a single dataset. J Neuropathol Exp Neurol. 2001;60:228–247. doi: 10.1093/jnen/60.3.228. [DOI] [PubMed] [Google Scholar]
- Leiter JC, St-John WM. Phrenic, vagal and hypoglossal activities in rat: pre-inspiratory, inspiratory, expiratory components. Respir Physiol Neurobiol. 2004;142:115–126. doi: 10.1016/j.resp.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Mortola JP, Saiki C. Ventilatory response to hypoxia in rats: gender differences. Respir Physiol. 1996;106:21–34. doi: 10.1016/0034-5687(96)00064-3. [DOI] [PubMed] [Google Scholar]
- Naassila M, Daoust M. Effect of prenatal and postnatal ethanol exposure on the developmental profile of mRNAs encoding NMDA receptor subunits in rat hippocampus. J Neurochem. 2002;80:850–860. doi: 10.1046/j.0022-3042.2002.00755.x. [DOI] [PubMed] [Google Scholar]
- Paton JF. The ventral medullary respiratory network of the mature mouse studied in a working heart-brainstem preparation. J Physiol. 1996;493:819–831. doi: 10.1113/jphysiol.1996.sp021425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton JF, Dutschmann M. Central control of upper airway resistance regulating respiratory airflow in mammals. J Anat. 2002;201:319–323. doi: 10.1046/j.1469-7580.2002.00101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyronnet J, Roux JC, Geloen A, Tang LQ, Pequignot JM, Lagercrantz HC, Dalmaz Y. Prenatal hypoxia impairs the postnatal development of neural and functional chemoafferent pathway in rat. J Physiol. 2000;524:525–537. doi: 10.1111/j.1469-7793.2000.00525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillis JW, O'Regan MH. Characterization of modes of release of amino acids in the ischemic/reperfused rat cerebral cortex. Neurochem Int. 2003;43:461–467. doi: 10.1016/s0197-0186(03)00035-4. [DOI] [PubMed] [Google Scholar]
- Pierrefiche O, Schwarzacher SW, Bischoff AM, Richter DW. Blockade of synaptic inhibition within the pre-Botzinger complex in the cat suppresses respiratory rhythm generation in vivo. J Physiol. 1998;509:245–254. doi: 10.1111/j.1469-7793.1998.245bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasch V. Cigarette, alcohol, and caffeine consumption: risk factors for spontaneous abortion. Acta Obstet Gynecol Scand. 2003;82:182–188. doi: 10.1034/j.1600-0412.2003.00078.x. [DOI] [PubMed] [Google Scholar]
- Richardson DP, Byrnes ML, Brien JF, Reynolds JN, Dringenberg HC. Impaired acquisition in the water maze and hippocampal long-term potentiation after chronic prenatal ethanol exposure in the guinea-pig. Eur J Neurosci. 2002;16:1593–1598. doi: 10.1046/j.1460-9568.2002.02214.x. [DOI] [PubMed] [Google Scholar]
- Richter DW, Schmidt-Garcon P, Pierrefiche O, Bischoff AM, Lalley PM. Neurotransmitters and neuromodulators controlling the hypoxic respiratory response in anaesthetized cats. J Physiol. 1999;514:567–578. doi: 10.1111/j.1469-7793.1999.567ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter DW, Spyer KM. Studying rhythmogenesis of breathing: comparison of in vivo and in vitro models. Trends Neurosci. 2001;24:464–472. doi: 10.1016/s0166-2236(00)01867-1. [DOI] [PubMed] [Google Scholar]
- Sebe JY, Eggers ED, Berger AJ. Differential effects of ethanol on GABAA and glycine receptor-mediated synaptic currents in brain stem motoneurons. J Neurophysiol. 2003;90:870–875. doi: 10.1152/jn.00119.2003. [DOI] [PubMed] [Google Scholar]
- Smith JC, Butera RJ, Koshiya N, Del Negro C, Wilson CG, Johnson SM. Respiratory rhythm generation in neonatal and adult mammals: the hybrid pacemaker-network model. Respir Physiol Neurobiol. 2000;122:131–147. doi: 10.1016/s0034-5687(00)00155-9. [DOI] [PubMed] [Google Scholar]
- St-John WM. Neurogenesis of patterns of automatic ventilatory activity. Prog Neurobiol. 1998a;56:97–117. doi: 10.1016/s0301-0082(98)00031-8. [DOI] [PubMed] [Google Scholar]
- St-John WM. Maternal cocaine alters eupneic ventilation but not gasping of neonatal rats. Neurosci Lett. 1998b;246:137–140. doi: 10.1016/s0304-3940(98)00225-0. [DOI] [PubMed] [Google Scholar]
- St-John WM, Leiter JC. Gasping is elicited by briefer hypoxia or ischemia following blockade of glycinergic transmission. Respir Physiol Neurobiol. 2002;133:167–171. doi: 10.1016/s1569-9048(02)00164-7. [DOI] [PubMed] [Google Scholar]
- St-John WM, Paton JF. Characterizations of eupnea, apneusis and gasping in a perfused rat preparation. Respir Physiol. 2000;123:201–213. doi: 10.1016/s0034-5687(00)00177-8. [DOI] [PubMed] [Google Scholar]
- St-John WM, Paton JF. Neurogenesis of gasping does not require inhibitory transmission using GABAA or glycine receptors. Respir Physiol Neurobiol. 2002;132:265–277. doi: 10.1016/s1569-9048(02)00079-4. [DOI] [PubMed] [Google Scholar]
- St-John WM, Rudkin AH, Harris MR, Leiter JC, Paton JF. Maintenance of eupnea and gasping following alterations in potassium ion concentration of perfusates of in situ rat preparation. J Neurosci Methods. 2005;142:125–129. doi: 10.1016/j.jneumeth.2004.08.006. [DOI] [PubMed] [Google Scholar]
- van Zundert B, Albarran FA, Aguayo LG. Effects of chronic ethanol treatment on gamma-aminobutyric acidA and glycine receptors in mouse glycinergic spinal neurons. J Pharmacol Exp Ther. 2000;295:423–429. [PubMed] [Google Scholar]
- Watson CS, White SE, Homan JH, Fraher L, Brien JF, Bocking AD. The adenosine A1-receptor antagonist 8-CPT reverses ethanol-induced inhibition of fetal breathing movements. J Appl Physiol. 1999;87:1333–1338. doi: 10.1152/jappl.1999.87.4.1333. [DOI] [PubMed] [Google Scholar]
- Ye JH, Tao L, Ren J, Schaefer R, Krnjevic K, Liu PL, Schiller DA, McArdle JJ. Ethanol potentiation of glycine-induced responses in dissociated neurons of rat ventral tegmental area. J Pharmacol Exp Ther. 2001;296:77–83. [PubMed] [Google Scholar]