

Abstract
Arteriolar myogenic vasoconstriction occurs when stretch or increased membrane tension leads to smooth muscle cell (SMC) depolarization and opening of voltage-gated Ca2+ channels. While the mechanism underlying the depolarization is uncertain a role for non-selective cation channels has been demonstrated. As such channels may be expected to pass Na+, we hypothesized that reverse mode Na+/Ca2+ exchange (NCX) may act to remove Na+ and in addition play a role in myogenic signalling through coupled Ca2+ entry. Further, reverse (Ca2+ entry) mode function of the NCX is favoured by the membrane potential found in myogenically active arterioles. All experiments were performed on isolated rat cremaster muscle first order arterioles (passive diameter ∼150 μm) which were pressurized in the absence of intraluminal flow. Reduction of extracellular Na+ to promote reverse-mode NCX activity caused significant, concentration-dependent vasoconstriction and increased intracellular Ca2+. This vasoconstriction was attenuated by the NCX inhibitors KB-R7943 and SEA 04000. Western blotting confirmed the existence of NCX protein while real-time PCR studies demonstrated that the major isoform expressed in the arteriolar wall was NCX1. Oligonucleotide knockdown (24 and 36 h) of NCX inhibited the vasoconstrictor response to reduced extracellular Na+ while also impairing both steady-state myogenic responses (as shown by pressure–diameter relationships) and acute reactivity to a 50 to 120 mmHg pressure step. The data are consistent with reverse mode activity of the NCX in arterioles and a contribution of this exchanger to myogenic vasoconstriction.
Arterioles typically exist in a state of partial constriction, or tone, which results from mechanical forces exerted by intraluminal pressure. This constrictor response is inherent to the vascular smooth muscle layer and is referred to as myogenic tone or myogenic responsiveness. While gaps exist in our knowledge of the underlying signal transduction mechanisms it is believed that this pressure-induced response plays an important physiological role by regulating local vascular resistance, contributing to autoregulation of microvascular blood flow and pressure, and providing a level of tone which can be acted upon by various vasodilator stimuli (Johnson, 1980; Davis & Hill, 1999).
At a cellular level myogenic constriction has been shown to occur as a result of the mechanical stimulus causing the opening of smooth muscle non-selective cation channels (NSCC), membrane depolarization, Ca2+ entry through voltage-gated Ca2+ channels and subsequent activation of acto-myosin interactions (Davis et al. 1992; Zou et al. 1995; Davis & Hill, 1999; Kotecha & Hill, 2005). This basic pathway may also be supported by events including intracellular Ca2+ release, Ca2+ sensitization and cytoskeletal rearrangement (Gokina & Osol, 2002; Lagaud et al. 2002; Flavahan et al. 2005). A number of the non-selective cation channels proposed to play a role in pressure/stretch-induced membrane depolarization would be expected to pass Na+ in physiological solutions. For example, stretch activated cation currents (Davis et al. 1992; Wu & Davis, 2001) and members of the TRP family of channels (Slish et al. 2002; Welsh et al. 2002; Earley et al. 2004; Earley et al. 2007) have both been implicated in pressure-induced constriction and pass Na+ as their principal ion. On the basis of these observations we hypothesized that a Na+ removal mechanism, in particular reverse mode Na+/Ca2+ exchange (NCX), may act to remove Na+ and hence play a role in myogenic signalling. Support for this hypothesis is also found in myocardium where stretch induced Na+ influx (via the Na+/H+exchanger) is coupled to reverse (Ca2+ entry) mode NCX activation (von Lewinski et al. 2003; Luers et al. 2005). Further, smooth muscle cells stimulated with ATP show a functional coupling between TRPC6-mediated Na+ entry and Ca2+ entry by reverse mode action of the NCX (Lemos et al. 2007). Linking these observations is the demonstration by Welsh et al. (2002) that TRPC6 is critical to arteriolar myogenic reactivity.
While the NCX was functionally identified in vascular smooth muscle some 34 years ago (Reuter et al. 1973), its exact role in ion handling in arterial vessels remains uncertain. This in part relates to its ability to operate in both the forward (allowing Na+ entry and Ca2+ removal) and reverse modes, considerable molecular diversity, and a relative lack of specific inhibitors (Blaustein & Lederer, 1999). A factor which may increase the functional importance of the NCX in resistance arteries and arterioles, particularly as relates to reverse mode operation, is the resting membrane potential (Em). A number of studies have shown cannulated and pressurized arterioles to be relatively depolarized at physiological intraluminal pressures with Em in the range of −45 to −30 mV (Knot & Nelson, 1998; Kotecha & Hill, 2005). At these levels of Em, Lederer and colleagues (Eisner & Lederer, 1985; Blaustein & Lederer, 1999) have suggested that the NCX is likely to operate in the ‘reverse mode’ removing Na+ from the cell in exchange for Ca2+. Further, the NCX has been suggested to colocate with a number of signalling molecules/structures involved in mechanotransduction including caveolae and closely apposed plasma membrane-sarcoplasmic reticulum junctions (Moore et al. 1993; Juhaszova et al. 1994). Thus the NCX would make an attractive mechanism for removing stretch induced increases in cellular Na+ while perhaps providing Ca2+ to locally modulate intracellular Ca2+ dynamics (Poburko et al. 2004a,b).
On the basis of the above, a series of studies were undertaken to identify functionality of the NCX in arterioles, demonstrate its presence at the cellular level and examine its role in myogenic vasoconstriction by manipulation of expression using antisense oligonucleotides. The data confirm the presence of the NCX and suggest that its reverse mode activity plays an important role in the arteriolar myogenic response.
Methods
Studies used male Sprague–Dawley rats of approximately 200–300 g body weight. Prior to experiments rats were housed in an animal facility with controlled temperature and lighting and allowed free access to water and a commercial rodent chow. All procedures were approved by the Animal Ethics and Experimentation Committee of RMIT University, Melbourne, Australia.
Rats were anaesthetized with sodium thiopental (Pentothal; 10 mg (100 g body weight)−1, intraperitoneal) after which the cremaster muscles were surgically removed and placed in a cooled (4°C) dissection chamber. Following removal of tissue, and while remaining under anaesthesia, rats were killed by cervical dislocation. Segments of the first order arteriole (passive diameter approximately 150 μm) were isolated and cannulated on glass pipettes as previously described (Meininger et al. 1991). The cannulation chamber was then placed on the stage of an inverted microscope and the vessel segment superfused (4 ml min−1) with a modified Krebs buffer solution (mm: NaCl 111, NaHCO3 25.7, KCl 4.9, CaCl2 2, MgSO4 1.2, KH2PO4 1.2 glucose 11, Hepes 10). During an equilibration period vessels were pressurized to 70 mmHg (in the absence of intraluminal flow), temperature gradually raised to 34°C, tested for pressure leaks and adjusted to optimal length (Potocnik et al. 2000). Vessels with leaks were discarded. Changes in arteriole diameter were monitored by video microscopy using an electronic calliper or a vessel diameter measuring program (Diamtrak, version 2.1c, South Australia, Australia).
For measurement of changes in intracellular Ca2+, smooth cells of the vessel wall were loaded with the acetomethoxyester form of the Ca2+ sensitive indicator fura-2 (2 μm). To restrict loading to the smooth muscle layer fura-2 was applied abluminally (60 min, room temperature) followed by extensive washout of excess indicator (Meininger et al. 1991; Hill et al. 2000). Changes in fluorescence emission (510 nm) following alternating excitation at 340 and 380 nm, provided by a spinning filter wheel, were monitored by photometry (Texas A & M University, College Station, TX, USA). Changes in [Ca2+]i were expressed as changes in the 340/380 nm fluorescence ratio after background subtraction. Under the conditions of the described studies autofluorescence of unloaded arterioles was found to be negligible.
Western blotting
Vessel segments were dissected as above, cannulated at one end to allow clearance of blood cells, and stored at −80°C prior to use. Segments were homogenized in cold (< 4°C) buffer (25 mm Tris-HCl (ph 6.8); 1% SDS; 5 mm EGTA; 50 mm sodium fluoride; 1 mm sodium vanadate; 10% glycerol and protease cocktail inhibitor). Samples were then diluted in sample buffer (250 mm Tris-HCl (pH 6.8); 2% SDS; 10% glycerol; 10 mm dithiothreitol; 2% mercaptoethanol and 0.01% bromophenol blue) and separated on 8% acrylamide gels. Samples were transferred to nitrocellulose using a semidry blotting system. Membranes were then probed with an anti-NCX antibody (1: 1000) followed by secondary antibody conjugated to StrepTactin-HRP. Bands were then visualized using enhanced chemiluminescence (Perkin Elmer, MA, USA). After probing for NCX membranes were stripped (Restore™, Pierce Biotechnology, Inc., Rockford, IL, USA) and re-probed using an anti-α-actin antibody. NCX data were normalized to the density of the actin band to account for variation in protein loading. An internal standard of a pooled rat aorta homogenate was included in each electrophoretic separation.
Real time PCR
Total RNA was extracted using an RNeasy kit (Qiagen, Victoria, Australia) according to the manufacturer's instructions. In addition to arterioles, RNA was extracted from heart, aorta and skeletal muscle for comparison to the literature and, in the case of skeletal muscle, as a control for possible contamination. RNA content and purity were established by measurement of absorbance at 260 and 280 nm. Samples were reverse transcribed (Taqman, Applied Biosystems, Foster City, CA, USA) at a final concentration of 10 ng μl−1 and stored at −20°C.
Forward and reverse primers were designed to mammalian NCX1, 2 and 3 using Primer Express software (Applied Biosystems): NCX1 (forward 5′-AGCAAGGCGGCTTCTCTTTT and reverse 5′- GCTGGTCTGTCTCCTTCATGT); NCX2 (CACTACGAG-GATGCTTGTGG and CCTTCTTCTCATACTCTT-CGT); and NCX3 (CCTGTGGCTCCTCTACGTACTCTT and GAGGTCTTGTTCTGGTGGTTCA). 18S mRNA was used as a constitutively expressed housekeeping gene. Real time PCR was performed with the fluorescent interchelating dye, Sybr Green, using a Bio-Rad iCycler. Thermal cycling conditions were 2 min at 50°C and 10 min at 95°C, followed by 50 cycles of denaturing (30 s at 95°C), annealing (30 s at 60°C), and extending (60 s at 72°C) (Seiler et al. 2004).
Relative differences in mRNA were standardized by normalizing the threshold fluorescence levels (Ct) of the sample to the internal standard (18S). Results were calculated and reported as fold changes, using the comparative 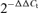 method (Livak & Schmittgen, 2001). Melting analysis was performed at the end of each PCR run to exclude the presence of non-specific products and formation of primer-dimers.
method (Livak & Schmittgen, 2001). Melting analysis was performed at the end of each PCR run to exclude the presence of non-specific products and formation of primer-dimers.
Reverse permeabilization and administration of oligonucleotides
Reverse permeabilization was based on methods published by Morgan & Morgan (1982) and Rembold & Murphy (1986). In brief, arterioles were dissected and transferred to a plastic slide in which narrow channels had been machined, kept on ice and exposed to a series of four solutions. Composition of the solutions was as follows. Solution 1 (mm): EGTA 10; KCl 120; ATP 5; MgCl2 2; and N-Tris[hydroxymethyl]methyl-2-amino-ethane sulphonic acid (Tes) 20, pH 6.8. Solution 2: EGTA 0.1; KCl 120; ATP 5; MgCl2 2; Tes 20, pH 6.8. Solution 3: EGTA 0.1; KCl 120; ATP 5; MgCl2 10; Tes 20, pH 6.8. Solution 4: NaCl 140; KCl 5; MgCl2 10; Mops 2, pH 7.1 (Rembold & Murphy, 1986). Solution 1 was added to the channels of the plastic chamber, making sure that the arteriole was submerged in the solution for 60 min. During this procedure the permeability of vascular cells was increased (Morgan & Morgan, 1982). This initial step was followed by solutions 2, 3 and 4 sequentially. Solution 2 (loading solution) contained either antisense/sense (1 or 10 μm; antisense (5′–3′) AGGAACACGTTCACGGCGTT or sense (5′–3′) AACGGCGTGAACGTGTTCCT) or no oligonucleotides (permeabilization control group). Solution 3 restores permeability towards the baseline state (Morgan & Morgan, 1982) while Solution 4 returns the vessel to physiological conditions by being gradually warmed to 22°C and with CaCl2 being added at 10 minute intervals to achieve Ca2+ concentrations of 0.001, 0.01, 0.1 and 1.6 mm. Arterioles were incubated for 24–36 h in fresh Krebs buffer solution with antisense/sense (1 or 10 μm) or no ODNs containing 1% dilution of penicillin–streptomycin solution (Sigma, St Louis, MO, USA).
As in previous studies (Richards et al. 2003) efficacy of the reverse permeabilization process was confirmed by uptake of a cell-impermeant form of protein kinase A inhibitor (PKI, 0.1 mm; Sigma) and subsequent significant attenuation of forskolin-mediated vasodilatation. Vessels remained viable as assessed by ability to develop myogenic tone, reactivity to exogenous agonists and a lack of nuclear staining by propridium iodide (Richards et al. 2003) (data not shown).
Protocols
To demonstrate reverse mode NCX activity cannulated arterioles were exposed to decreasing extracellular Na+ concentrations (137–25 mm). Na+ in the physiological salt solution was replaced with choline chloride to maintain iso-osmotic conditions. Diameter measurements were taken after achieving a steady-state vasomotor response. Diameter responses were examined in intact arterioles and vessels in which the endothelium had been removed by controlled passage of an air bubble through the lumen. In a subset of experiments the effect of extracellular Na+ reduction on intracellular Ca2+ levels was examined following loading of vascular smooth muscle with Fura-2. Concentration–response curves were established for NCX inhibitors, KB-R4793 (0.1–30 μm) and SEA-0700 (0.001–3 μm) and optimal concentrations (KB-R4793, 10 μm; SEA-0700, 1 μm) used for demonstrating that extracellular Na+ reduction affected reverse mode NCX activity.
Functional experiments were repeated in arterioles treated with antisense oligonucleotides to determine the effects of NCX suppression on myogenic reactivity. Vessels receiving a sense oligonucleotide sequence were used as controls. In addition to the functional tests above, pressure (30–150 mmHg)–diameter relationships were established in oligonucleotide treated vessels
Data handling and statistical methods
In all protocols with exception of Western blotting, a given n represents both the number of vessels and number of animals studied. For Western blotting three to four vessel segments (based on that used in cannulated preparations) were pooled to ensure adequate amounts of protein. Diameters of blood vessels have been normalized relative to the passive diameter at an intraluminal pressure of 70 mmHg. Myogenic index (MI) was calculated as a measure of strength of an acute myogenic response according to Halpern et al. (1984):
where Di is initial diameter before the change in intraluminal pressure; ΔD is the diameter change in response to the pressure change; and Δp is the magnitude of the applied change in pressure. Diameter measurements were taken after development of a stable vasomotor response (approximately 5 min following application of the pressure step).
Results are presented as means ± s.e.m. Comparison of means was performed using ANOVA for multiple groups or Student's t test where means of two groups were compared. Significance was assumed at P < 0.05.
Chemicals and reagents
Unless stated all general chemicals were obtained from Sigma Chemical Company (Sydney, Australia). KB-R7943 was purchased from Tocris (MO, USA) and SEA-0400 was a kind gift from Taisho Pharmaceutical Company Limited (Saito, Japan). Monoclonal NCX antibody (R3F1) was obtained from Swant (Switzerland) and monoclonal actin antibody was obtained from Dako Cytomation (Denmark). Reagents for Western blotting and real time PCR were obtained from Bio-Rad (New South Wales, Australia). Oligonucleotides were synthesized by Sigma Genosys (New South Wales, Australia).
Results
Arterioles used in the functional studies described below had a diameter of 81.9 ± 1.5 μm under active conditions and 151.8 ± 3.0 μm in the passive state (following superfusion with 0 mm Ca2+ Krebs containing 2 mm EGTA) (n = 72).
Pharmacological studies
Functional significance of reverse-mode NCX was initially demonstrated by examining vasomotor responses to reduced extracellular [Na+] (137, 100, 75 and 25 mm). Arterioles (70 mmHg intralumenal pressure) showed concentration-dependent vasoconstriction together with an increase in [Ca2+]i (Fig. 1A and B). At 25 mm extracellular Na+ arterioles had constricted from 50.2 ± 1.7 to 36.8 ± 1.3% of maximal passive diameter while intracellular Ca2+ had increased by 9% from 0.59 ± 0.02 to 0.65 ± 0.03 (relative to levels at 137 mm extracellular Na+) (Fig. 1A and B). Decreased extracellular Na+ was associated with impairment of acute myogenic vasoconstriction to a 50–120 mmHg pressure step as shown by a significant (P < 0.001; ANOVA) increase (i.e. less reactive) in calculated myogenic index as extracellular Na+ was reduced (Fig. 1E). Responses to decreased extracellular Na+ were not affected by removal of the endothelium (Fig. 1D and E; no significant difference in calculated myogenic index, at a given extracellular [Na+], between endothelium intact and endothelium removed vessels) indicating that the observed responses were predominately an effect at the level of the vascular smooth muscle. As a control experiment, arterioles were preconstricted with either 30 or 60 mm KCl to determine whether constriction from baseline in itself altered myogenic responsiveness. Despite constriction from 48.5 ± 3 (diameter/diameter passive × 100; n = 5) to 32.8 ± 1.5 in the presence of 30 mm KCl and to 24.4 ± 0.8 following 60 mm KCl, calculated myogenic index for the 50–120 mmHg pressure step was not significantly different from that in the absence of preconstriction (Fig. 1F). The baseline response was repeated after washout of the raised K+ concentrations to show a lack of time-dependent effects (Fig. 1F).
Figure 1. Effect of extracellular Na+ reduction on arteriolar myogenic responsiveness.

A (n = 13) illustrates that reduction of Na+ causes a concentration-dependent vasoconstriction (diameter expressed relative to passive diameter at 70 mmHg), which is associated with an increase in global intracellular Ca2+ (B; n = 13). Changes in Ca2+ are expressed as the fold change in 340/380 nm fluorescence ratio relative to that at 137 mm extracellular Na+. C and D, the effect of Na+ reduction on the steady-state responses to a 50–120 mmHg pressure step. C represents intact arterioles (n = 6) while D shows data for de-endothelialized vessels (n = 6) illustrating that responses are at the level of the vascular smooth muscle. E, the pressure step data expressed in terms of the myogenic index (see text). Myogenic index declines with extracellular Na+ concentration in both intact (n = 6) and de-endothelialized (n = 6) vessels. F, that vasoconstriction with KCl (30 or 60 mm) does not significantly alter myogenic index (n = 4) suggesting the data in E are not simply a result of an alteration in starting diameter. Myogenic responsiveness was determined before and after washout of KCl as a control for time. Results are expressed as means ± s.e.m.; ***P < 0.001.
Inhibitors of NCX, KB-R7943 (10−7–3 × 10−5m) and SEA-0400 (10−9–3 × 10−6m) caused dose-dependent vasodilatation of arterioles pressurized at an intralumenal pressure of 70 mmHg and exhibiting spontaneous myogenic tone (Fig. 2A and B). Calculated −log IC50 values were 5.4 ± 0.07 for KB-R7943 and 6.7 ± 0.01 for SEA-0400. Maximal dilatation to the inhibitors was 96 ± 1.0 (diameter/passive diameter at 70 mmHg × 100%) for KB-R7943 and 76 ± 5.0 for SEA-0400.
Figure 2. Effect of NCX inhibitors KB-R7943 and SEA 0400 on arteriolar diameter (A and B) and responses to extracellular Na+ reduction (C and D).

KB-R7943 (A; n = 9) and SEA 0400 (B; n = 9) caused concentration-dependent vasodilatation with the former causing near maximal relaxation. Both inhibitors blocked the vasoconstrictor response to reduced extracellular Na+ concentrations (C and D) consistent with an effect on reverse mode (Ca2+ influx) NCX activity. Data in C (n = 9) and D (n = 9) have been normalized such that D/Dmax at an extracellular Na+ concentration of 137 mm is represented as 100%. As the NCX inhibitors caused vasodilatation control studies were performed with nifedipine (10−6m). Nifedipine, while causing vasodilatation, did not inhibit subsequent constriction in response to extracellular Na+ reduction (E; n = 4). Results are expressed as means ± s.e.m.; ***P < 0.001.
In de-endothelialized arterioles, vasoconstriction to decreasing extracellular Na+ was inhibited by both KB-R7943 (10−5m) and SEA-0400 (10−6m) (Fig. 2C and D). As both inhibitors caused vasodilatation at physiological Na+ concentrations additional control studies (for the effects of dilatation per se) were performed in the presence of either the voltage-gated Ca2+ channel inhibitor, nifedipine (10−6m) or the cAMP-mediated vasodilator, adenosine (10−5m). Nifedipine (n = 4) caused a significant (P < 0.01) loss of myogenic tone (52.4 ± 1.4% at baseline to 76.8 ± 4.3% (D/Dmax× 100)) but, in contrast to the NCX inhibitors, did not prevent the vasoconstrictor response to decreased extracellular Na+ (Fig. 2E). Adenosine (n = 3) significantly (P < 0.01) dilated arterioles from a baseline diameter of 51.9 ± 0.9% (D/Dmax) to 76.5 ± 2.0%. Similarly to the case of nifedipine, dilatation did not impair the vasoconstrictor response to reduction of extracellular Na+.
In addition to determining the effects of the NCX inhibitors on myogenic responsiveness studies were conducted to examine their inhibitory actions on phenylephrine (10−9–10−5m)-induced vasoconstriction. While both KB-R7943 (10−5m) and SEA-0400 (10−6m) caused vasodilatation, as described above, KB-R7943 did not significantly alter the calculated −log EC50 in response to phenylephrine (7.69 ± 0.25 compared to 7.50 ± 0.32; n.s., n = 3) while SEA-0400 caused a significant decrease in the −log EC50 (7.13 ± 0.22 compared to 7.96 ± 0.25; P < 0.01, n = 4) as indicated by a rightward shift in the concentration–response curve.
Identification of arteriolar NCX by Western transfer and real-time PCR
Western transfer using a monoclonal anti-NCX antibody (Swant R3F1) showed the presence of two principal bands, at approximately 120 and 70 kDa (Fig. 3; typical of n = 6), consistent with the intact exchanger protein and a proteolytic fragment as previously described (Juhaszova et al. 1994). Results were confirmed using a commercially available polyclonal rabbit anti-NCX antibody (Swant π 11–13; rabbit anti-NCX) with bands at 120 and 70 kDa similarly detected (n = 3; data not shown). Real-time PCR identified the presence of mRNA for the three isoforms, NCX1, 2 and 3. Relative expression of NCX1 was significantly (P < 0.01) greater than either NCX2 or NCX3. Skeletal muscle samples from cremaster showed higher expression of NCX 2 and 3 indicating that the arteriole samples were unlikely to have been significantly contaminated by surrounding skeletal muscle tissue.
Figure 3. Demonstration of NCX protein by Western blotting (A; typical of n = 6) and mRNA by real-time PCR (B; n = 6) in cremaster muscle arteriolar wall.

Loading controls were actin for protein (A) and 18S RNA for NCX mRNA (B). The additional protein band between the 70 and 120 kDa markers (A, left two lanes) is presumed to be a degradation production recognized by the antibody. Results are expressed as mean ± s.e.m.
Oligonucleotide suppression of NCX expression
Control studies (reactivity to agonists and development of myogenic tone) were performed to test if reactivity of arterioles was compromised by the reverse permeabilization conditions or by the time taken to undertake these procedures (data not shown; see also Richards et al. 2003). Further, as a positive control for the effectiveness of the loading procedure during reverse permeabilization, inhibition of forskolin-mediated vasodilatation was tested by uptake of cell-impermeant PKI (forskolin −log EC50 8.2 ± 0.4 under control conditions and 6.8 ± 0.04 in the presence of PKI, P < 0.05; see also Richards et al. 2003).
In an initial series of experiments, arterioles were treated with either sense or antisense oligonucleotides (10−6m) for 24 h. Western transfer showed a decrease of approximately 25–30% in NCX expression (relative to α-smooth muscle actin) in vessels treated with the antisense oligonucleotide compared to those treated with the sense sequence (Fig. 4A and B). Steady-state pressure–diameter relationships were unaffected by oligonucleotide treatment (Fig. 4C); however, myogenic reactivity to an acute 50–120 mmHg pressure step (as shown by the calculated myogenic index) was significantly (P < 0.05) decreased in vessels exposed to the antisense oligonucleotide (Fig. 4D and E).
Figure 4. Effect of 24 h NCX oligonucleotide exposure on NCX expression (A and B) and myogenic responsiveness (C–E).

Western blotting showed a significant (P < 0.05) reduction in NCX expression of approximately 30% after 24 h treatment with the antisense oligonucleotide compared to the sense sequence (A and B). A standard aorta homogenate was included as a between-gel control (A). Twenty-four hour exposure to the antisense oligonucleotide did not alter the steady-state pressure–diameter relationship (C) but decreased myogenic responsiveness to an acute pressure step (50–120 mmHg; D) as indicated by a significant (P < 0.05) reduction in myogenic index (E). Results are expressed as means ± s.e.m.; *P < 0.05, n = 5 for antisense and sense treatments.
In a second series of experiments vessels underwent a more stringent exposure to olignucleotides with concentration being increased to 10−5m for a duration of 36 h. Antisense oligonucleotide-treated arterioles showed a significant shift in their steady-state pressure–diameter relationship showing vasodilatation (relative to sense treated vessels) and attenuation of pressure-dependent contractile responsiveness as well as decreased reactivity to an acute pressure step (Fig. 5A and B). Antisense treated vessels further showed a decreased vasoconstrictor response to reduction in extracellular Na+ (Fig. 5C) consistent with the oligonucleotide treatment reducing reverse mode NCX.
Figure 5. Effect of 36 h NCX oligonucleotide exposure on arteriolar myogenic reactivity (A and B) and responsiveness to extracellular Na+ reduction (C) and phenylephrine (D).

Antisense treatment caused a significant impairment in both the steady-state pressure–diameter relationship (A) and responsiveness to an acute pressure step (50–120 mmHg) as indicated by a significant (P < 0.05) reduction the myogenic index (B). Similarly, treatment with the antisense oligonucleotide significantly impaired contraction to extracellular Na+ reduction and topically applied phenylephrine (as determined by calculated EC50 values; see text) compared to treatment with the sense sequence (C and D, respectively). Results are expressed as means ± s.e.m.; *P < 0.05, ***P < 0.001; n = 5 for antisense and sense treatments.
In addition to determining the effect of oligonucleotide treatment on myogenic responsiveness its effect on phenylephrine (10−9–10−4m)-induced constriction was examined. Antisense oligonucleotide treated vessels showed a significant (P < 0.01) decrease in sensitivity to phenylephrine compared to sense treated arterioles as assessed by −log EC50 values (6.25 ± 0.13 compared to 7.02 ± 0.16, n = 6) (Fig. 5D).
Discussion
The results of the current studies support the presence of a functional NCX mechanism in arterioles from skeletal muscle and suggest that reverse mode activity (Na+ extrusion/Ca2+ entry) contributes to myogenic responsiveness. The data add to a growing literature supporting the role of reverse mode NCX activity in the control of vascular tone (Slodzinski et al. 1995; Arnon et al. 2000; Iwamoto et al. 2004b; Dong et al. 2006; Poburko et al. 2006). As myogenic, or pressure-induced, vasoconstriction is believed to involve an initial opening of non-selective cation channels (for example stretch-activated (Davis et al. 1992; Wu & Davis, 2001) and TRP channels, including TRPC6 (Welsh et al. 2002)), it is conceivable that Na+ entry through such mechanisms provides the necessary drive for reverse mode activity of the NCX. Consistent with this, several recent studies in smooth muscle have suggested that a localized increase in intracellular Na+ can drive the NCX (Wu & Fry, 2001; Lemos et al. 2007). A further consequence of this coupling would be Ca2+ entry, which may then contribute to local Ca2+ handling due to the exchanger being located in regions of the plasma membrane that are closely associated with the sarcoplasmic reticulum (Arnon et al. 2000).
Using Western blotting the present studies showed the arteriolar wall to contain NCX immunoreactivity of approximately 120 and 70 kDa. The latter has been reported to be an N-terminal degradation fragment (Juhaszova et al. 1994). In addition, real-time PCR indicated that the predominant form of NCX mRNA was NCX1 with NCX2 and NCX3 mRNA being present at much lower levels. This overall expression pattern is consistent with a number of studies performed in larger diameter arterial vessels and other tissues (Nakasaki et al. 1993; Quednau et al. 1997). Although not examined in the present study, additional molecular diversity may exist as NCX1 is known to exist in many splice variant forms (Nakasaki et al. 1993; Lee & Lytton, 1994).
The approach taken in our studies to confirm a functional role of the NCX in arteriolar myogenic responsiveness was to first demonstrate its role pharmacologically and subsequently decrease its expression through the use of oligonucleotides administered by reverse permeabilization. The initial experiments showed that both inhibitors of NCX, KB-R7943 and SEA-0400, caused a loss of myogenic tone at concentrations comparable to that previously reported for an action on NCX (Iwamoto et al. 1996, 2004a; Iwamoto, 2004). Thus for an inhibitory effect on myogenic tone approximate EC50 values were found to be 4 μm for KB-R7943 and 0.2 μm for SEA-0400.
Decreasing extracellular Na+ (135–25 mm) caused an increase in intracellular Ca2+ and vasoconstriction, an effect blocked by both KB-R7943 and SEA-0400. In contrast, nifedipine did not block the constrictor response to extracellular Na+ reduction suggesting that the effect of the NCX inhibitors could not be explained by a non-specific effect relating to vasodilatation and loss of basal myogenic tone, nor only an action at the level of voltage-gated Ca2+ channels as has been suggested for KB-R7943 (Ouardouz et al. 2005). Similarly, reducing basal tone with adenosine did not prevent vasoconstriction to extracellular Na+ reduction. These control data would similarly argue against solely a role for inhibition of events leading to myogenic depolarization and constriction (Kotecha & Hill, 2005), although KB-R7943 has recently been proposed to both inhibit the putative mechanosensory TRPC6 (Welsh et al. 2002) at concentrations below that required to block NCX (Kraft, 2007) and cause hyperpolarization (−34.2 ± 0.9 mV before and −42.8 ± 1.9 mV following 3 × 10−5m KB-R7943; P < 0.05, n = 4, unpublished data) of our arteriolar preparations.
Both KB-R7943 and SEA-0400 have been reported to preferentially inhibit reverse mode NCX activity and further, the latter has been shown to effectively inhibit the predominant vascular isoforms of NCX1 (1.3 and 1.7) when overexpressed in fibroblasts (Iwamoto et al. 2004b). Decreasing extracellular Na+ concentrations, in addition to causing vasoconstriction, impaired myogenic responsiveness as shown by a decrease in the calculated myogenic index. While reduction in extracellular Na+ stimulates reverse mode NCX activity with a resultant Ca2+ entry, the decreased Na+ availability may impair upstream myogenic signalling events, namely the NSCC-mediated membrane depolarization (Davis & Hill, 1999; Kotecha & Hill, 2005). Alternatively, the extent of vasoconstriction resulting from stimulating reverse mode NCX activity limits the ability to vasoconstrict to an added pressure stimulus. The latter explanation appears unlikely as control experiments showed that constriction with KCl (30 or 60 mm) did not significantly decrease the calculated myogenic index.
In part, because of concerns relating to the specificity of the pharmacological approaches, additional studies were performed using an oligonucleotide knockdown approach. Treatment of isolated arterioles with antisense oligonucleotides (24 and 36 h) was observed to impair myogenic reactivity and vasoconstriction to lowering of extracellular Na+. The effects on myogenic reactivity were more evident after a 36 h exposure to the nucleotide sequence, perhaps consistent with the endogenous protein having a half-life of approximately 33 h as reported for neonatal cardiomyocytes (Slodzinski & Blaustein, 1998). After 24 h treatment with the antisense oligonucleotide, a significant inhibitory effect was seen on the response to the acute 50–120 mmHg pressure step, but not the steady-state pressure–diameter relationship. Rather than this being an apparent contradiction it is suggested this reflects a threshold effect for the antisense treatment conditions used. Slodzinski and Blaustein, however, required longer time periods (approximately 5 days) to see a reduction in protein content (Slodzinski et al. 1995; Slodzinski & Blaustein, 1998). In contrast, in studies of primary neurons it was reported that a decrease in NCX activity followed 12–24 h exposure to an antisense oligonucleotide (Ranciat-McComb et al. 2000). While such differences may reflect differing methodologies for nucleotide delivery, or cell types, it is unlikely that the results can be explained by non-specific effects as sense sequences did not alter myogenic responsiveness or vasoconstrictor responses to reduced extracellular Na+.
Exposure of arterioles to the NCX antisense oligonucleotide (36 h) was observed to result in inhibition of phenylephrine-induced constriction as well as inhibiting myogenic responsiveness. Rather than an indication of non-specificity of the oligonucleotide approach it is likely that NCX may be involved in both contractile events. Consistent with this both mechanisms have been reported to involve non-selective cation channels, such as TRP channels, which would be expected to pass Na+ (Davis & Hill, 1999; Welsh et al. 2002; Earley et al. 2004; Spassova et al. 2006; Hill & Davis, 2007). However, as the vessels exposed to the sense oligonucleotide showed significantly greater sensitivity to phenylephrine compared to the antisense treated arterioles, it appears reasonable to assume that the differential effects of the oligonucleotides relate to a specific effect on NCX rather than a non-specific action relating to reverse permeabilization or introduction of nucleotide sequences.
While the exact physiological role of NCX in smooth muscle has remained uncertain, interest in the role of reverse mode NCX activity in smooth muscle has, in particular, stemmed from the work of Blaustein and colleagues (Slodzinski et al. 1995; Arnon et al. 2000). Thus, Arnon et al. (2000) suggested that Na+ entry through receptor-mediated activation of non-selective cation channels stimulated reverse mode NCX with subsequent Ca2+ entry and interaction with the SR. This was facilitated by NCX being concentrated in areas of the plasma membrane which closely oppose the superficial regions of the SR (Moore et al. 1993). Supporting such a mechanism it is evident from a number of recent studies that colocalization of signalling molecules within restricted membrane domains allows localized changes in ion concentrations to regulate signalling mechanisms. For example, release of Ca2+ from the superficial SR into the restricted space formed by the close apposition of SR and plasma membranes acts to modulate Ca2+-activated ion channels (Nelson et al. 1995; Zhuge et al. 2002).
An alternate role for NCX has been suggested to be re-filling of intracellular Ca2+ stores following store depletion-mediated opening of a non-selective plasma membrane cation channel (Hirota et al. 2006), most likely involving members of the TRP family. Such a relationship is not limited to vascular smooth muscle nor contraction. For example, acetylcholine-induced contraction in airway smooth muscle activates reverse mode NCX activity as a source of Ca2+ for store refilling (Hirota et al. 2006). In studies of secretory activity of colonic epithelial cells Ca2+ store depletion leads to opening of non-selective cation channels and inhibition of forward mode NCX, which raises [Ca2+]i (Seip et al. 2001).
An additional consideration in postulating a role for reverse mode NCX in myogenically active arterioles is the resting Em of vascular smooth muscle. Cannulated arterioles exhibiting spontaneous myogenic tone have been shown to be considerably depolarized across a physiological range of intraluminal pressures. For example in skeletal muscle arterioles we have reported Em values of between −45 and −30 mV over a pressure range of 30–120 mmHg (Kotecha & Hill, 2005). The relevance of this relates to the electrotonic behaviour of the NCX with depolarization favouring reverse mode action of the exchanger (Eisner & Lederer, 1985; Blaustein & Lederer, 1999). Specifically the exchanger is suggested to have a greater probability of functioning in reverse mode at an Em of approximately −30 mV (see Fig. 15 in Blaustein & Lederer, 1999). Thus acute and steady-state pressure-induced depolarization would itself favour the reverse mode function, perhaps enhancing the effect of localized Na+ gradients. With respect to the latter point it should be emphasized that under pressurized conditions arteriolar smooth muscle is an active state that favours reverse mode NCX (Poburko et al. 2006).
Although the exact mechanism underlying pressure-induced membrane depolarization is uncertain, antisense oligonucleotide administration to isolated cerebral arterioles has suggested a role for TRPC6 (Welsh et al. 2002). TRPC6, perhaps in conjunction with TRPM4, form NSCCs, which act to cause depolarization presumably via Na+ entry (Welsh et al. 2002; Earley et al. 2004; Spassova et al. 2006; Earley et al. 2007; Hill & Davis, 2007). TRPC6 has also been shown to be a necessary component in α-adrenergic-mediated vasoconstriction (Albert & Large, 2003; Hill et al. 2006). In isolated smooth muscle cells ATP stimulation similarly leads to TRPC6-mediated Na+ entry which has further been shown to be coupled to reverse mode NCX activity (Lemos et al. 2007) Analgous to these studies the related TRPC3 has been shown to associate with NCX in a HEK-293 cell expression system (Rosker et al. 2004). Collectively these studies suggest a common sequence of events, including NSCC and NCX, may exist to promote mechanical activation and the actions of, at least some, contractile agonists.
Given the bidirectional nature of the NCX it is difficult to exclude that some of the results of the present studies (for example for the low extracellular Na+ protocols) could be affected by a contribution of forward mode exchange inhibition rather than stimulation of the reverse mode. Using the isolated arteriole preparation, however, it is not possible to directly measure the directionality of ion fluxes. However, given the action of the reverse mode selective inhibitors, the depolarized resting smooth muscle membrane potential and analogous findings in other vascular preparations (Arnon et al. 2000; Lee et al. 2001) the available evidence is consistent with a role for reverse mode NCX in arterioles. Supporting this, Dong et al. (2006) have recently suggested an increasing contribution of reverse mode NCX with decreasing vessel diameter.
In regard to the physiological significance of the NCX in the myogenic regulation of vascular resistance, it is of interest to note that mice over-expressing NCX1.3 exhibit a slight hypertension (Iwamoto et al. 2004b). Systolic blood pressure in the N1.3Tg/Tg mouse is reported to be approximately 10 mmHg greater than the wild-type control and further is exacerbated by high salt feeding (Iwamoto et al. 2004b). Collectively the data from Iwamoto and those of the present pharmacological and oligonucleotide suppression studies support an important role for the NCX in the modulation of physiological myogenic tone and further suggest that abnormal NCX function may contribute to pathophysiological states such as hypertension.
In summary, the results of the present study provide evidence for functional reverse mode activity of the NCX in myogenically active arterioles. Presumably the driving force for reverse mode function is provided by NSCC, activated as a result of an intraluminal pressure stimulus and the consequent influx of Na+. In addition, Ca2+ entry via the NCX appears to contribute to contractile activation but whether this occurs through an effect on bulk cytosolic Ca2+ or through events such as an action on Ca2+-modulated ion channels or the filling state of intracellular stores remains to be determined.
Acknowledgments
Studies described in this manuscript were supported by grants from the National Health and Research Council, Australia and the National Heart Foundation of Australia to M.A.H. Thanks are extended to Simon Potocnik and Timothy Murphy for constructive input into the studies and to Michael Davis for critically reviewing this paper prior to submission. Taisho Pharmaceutical Company Limited (Saito, Japan) is thanked for supplying the SEA-0400.
References
- Albert AP, Large WA. Synergism between inositol phosphates and diacylglycerol on native TRPC6-like channels in rabbit portal vein myocytes. J Physiol. 2003;552:789–795. doi: 10.1113/jphysiol.2003.052977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnon A, Hamlyn JM, Blaustein MP. Na+ entry via store-operated channels modulates Ca2+ signaling in arterial myocytes. Am J Physiol Cell Physiol. 2000;278:C163–C173. doi: 10.1152/ajpcell.2000.278.1.C163. [DOI] [PubMed] [Google Scholar]
- Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- Davis MJ, Donovitz JA, Hood JD. Stretch-activated single-channel and whole cell currents in vascular smooth muscle cells. Am J Physiol Cell Physiol. 1992;262:C1083–C1088. doi: 10.1152/ajpcell.1992.262.4.C1083. [DOI] [PubMed] [Google Scholar]
- Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- Dong H, Jiang Y, Triggle CR, Li X, Lytton J. Novel role for K+-dependent Na+/Ca2+ exchangers in regulation of cytoplasmic free Ca2+ and contractility in arterial smooth muscle. Am J Physiol Heart Circ Physiol. 2006;291:H1226–H1235. doi: 10.1152/ajpheart.00196.2006. [DOI] [PubMed] [Google Scholar]
- Earley S, Straub SV, Brayden J. Protein kinase C regulates vascular myogenic tone through activation of TRPM4. Am J Physiol Heart Circ Physiol. 2007;292:H2613–2622. doi: 10.1152/ajpheart.01286.2006. [DOI] [PubMed] [Google Scholar]
- Earley S, Waldron BJ, Brayden JE. Critical role for transient receptor potential channel TRPM4 in myogenic constriction of cerebral arteries. Circ Res. 2004;95:922–929. doi: 10.1161/01.RES.0000147311.54833.03. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Lederer WJ. Na-Ca exchange: stoichiometry and electrogenicity. Am J Physiol Circ Physiol. 1985;248:C189–C202. doi: 10.1152/ajpcell.1985.248.3.C189. [DOI] [PubMed] [Google Scholar]
- Flavahan NA, Bailey SR, Flavahan WA, Mitra S, Flavahan S. Imaging remodeling of the actin cytoskeleton in vascular smooth muscle cells after mechanosensitive arteriolar constriction. Am J Physiol Heart Circ Physiol. 2005;288:H660–H669. doi: 10.1152/ajpheart.00608.2004. [DOI] [PubMed] [Google Scholar]
- Gokina NI, Osol G. Actin cytoskeletal modulation of pressure-induced depolarization and Ca2+ influx in cerebral arteries. Am J Physiol Heart Circ Physiol. 2002;282:H1410–H1420. doi: 10.1152/ajpheart.00441.2001. [DOI] [PubMed] [Google Scholar]
- Halpern W, Osol G, Coy GS. Mechanical behavior of pressurized in vitro prearteriolar vessels determined with a video system. Ann Biomed Eng. 1984;12:463–479. doi: 10.1007/BF02363917. [DOI] [PubMed] [Google Scholar]
- Hill MA, Davis MJ. Coupling a change in intraluminal pressure to vascular smooth muscle depolarization: still stretching for an explanation. Am J Physiol Heart Circ Physiol. 2007;292:H2570–H2572. doi: 10.1152/ajpheart.00331.2007. [DOI] [PubMed] [Google Scholar]
- Hill AJ, Hinton JM, Cheng H, Gao Z, Bates DO, Hancox JC, Langton PD, James AF. A TRPC-like non-selective cation current activated by α1-adrenoceptors in rat mesenteric artery smooth muscle cells. Cell Calcium. 2006;40:29–40. doi: 10.1016/j.ceca.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Hill MA, Zou H, Davis MJ, Potocnik SJ, Price S. Transient increases in diameter and [Ca2+]i are not obligatory for myogenic constriction. Am J Physiol Heart Circ Physiol. 2000;278:H345–H352. doi: 10.1152/ajpheart.2000.278.2.H345. [DOI] [PubMed] [Google Scholar]
- Hirota SA, Pertens E, Janssen LJ. The reverse-mode of the sodium-calcium exchanger provides a source of calcium for store-refilling following agonist-induced calcium mobilization. Am J Physiol Lung Cell Mol Physiol. 2006;292:L438–L447. doi: 10.1152/ajplung.00222.2006. [DOI] [PubMed] [Google Scholar]
- Iwamoto T. Forefront of Na+/Ca2+ exchanger studies: molecular pharmacology of Na+/Ca2+ exchange inhibitors. J Pharmacol Sci. 2004;96:27–32. doi: 10.1254/jphs.fmj04002x6. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Kita S, Uehara A, Imanaga I, Matsuda T, Baba A, Katsuragi T. Molecular determinants of Na+/Ca2+ exchange (NCX1) inhibition by SEA0400. J Biol Chem. 2004a;279:7544–7553. doi: 10.1074/jbc.M310491200. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Kita S, Zhang J, Blaustein MP, Arai Y, Yoshida S, Wakimoto K, Komuro I, Katsuragi T. Salt-sensitive hypertension is triggered by Ca2+ entry via Na+/Ca2+ exchanger type-1 in vascular smooth muscle. Nat Med. 2004b;10:1193–1199. doi: 10.1038/nm1118. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Watano T, Shigekawa M. A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J Biol Chem. 1996;271:22391–22397. doi: 10.1074/jbc.271.37.22391. [DOI] [PubMed] [Google Scholar]
- Johnson PC. The myogenic response. In: Berne RM, Sperelakis N, editors. Handbook of Physiology, section 2, The Cardiovascular System, vol. II, Vascular Smooth Muscle. Bethesda: American Physiological Society; 1980. pp. 409–442. [Google Scholar]
- Juhaszova M, Ambesi A, Lindenmayer GE, Bloch RJ, Blaustein MP. Na+-Ca2+ exchanger in arteries: identification by immunoblotting and immunofluorescence microscopy. Am J Physiol Cell Physiol. 1994;266:C234–C242. doi: 10.1152/ajpcell.1994.266.1.C234. [DOI] [PubMed] [Google Scholar]
- Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol. 1998;508:199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha N, Hill MA. Myogenic contraction in rat skeletal muscle arterioles: smooth muscle membrane potential and Ca2+ signaling. Am J Physiol Heart Circ Physiol. 2005;289:H1326–H1334. doi: 10.1152/ajpheart.00323.2005. [DOI] [PubMed] [Google Scholar]
- Kraft R. The Na+/Ca2+ exchange inhibitor KB-R7943 potently blocks TRPC channels. Biochem Biophys Res Commun. 2007;361:230–236. doi: 10.1016/j.bbrc.2007.07.019. [DOI] [PubMed] [Google Scholar]
- Lagaud G, Gaudreault N, Moore ED, Van Breemen C, Laher I. Pressure-dependent myogenic constriction of cerebral arteries occurs independently of voltage-dependent activation. Am J Physiol Heart Circ Physiol. 2002;283:H2187–H2195. doi: 10.1152/ajpheart.00554.2002. [DOI] [PubMed] [Google Scholar]
- Lee SL, Yu AS, Lytton J. Tissue-specific expression of Na+-Ca2+ exchanger isoforms. J Biol Chem. 1994;269:14849–14852. [PubMed] [Google Scholar]
- Lee CH, Poburko D, Sahota P, Sandhu J, Ruehlmann DO, van Breemen C. The mechanism of phenylephrine-mediated [Ca2+]i oscillations underlying tonic contraction in the rabbit inferior vena cava. J Physiol. 2001;534:641–650. doi: 10.1111/j.1469-7793.2001.t01-1-00641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos VS, Poburko D, Liao CH, Cole WC, van Breemen C. Na+ entry via TRPC6 causes Ca2+ entry via NCX reversal in ATP stimulated smooth muscle cells. Biochem Biophys Res Commun. 2007;352:130–134. doi: 10.1016/j.bbrc.2006.10.160. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Luers C, Fialka F, Elgner A, Zhu D, Kockskamper J, von Lewinski D, Pieske B. Stretch-dependent modulation of [Na+]i, [Ca2+]i, and pHi in rabbit myocardium – a mechanism for the slow force response. Cardiovasc Res. 2005;68:454–463. doi: 10.1016/j.cardiores.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Meininger GA, Zawieja DC, Falcone JC, Hill MA, Davey JP. Calcium measurement in isolated arterioles during myogenic and agonist stimulation. Am J Physiol Heart Circ Physiol. 1991;261:H950–H959. doi: 10.1152/ajpheart.1991.261.3.H950. [DOI] [PubMed] [Google Scholar]
- Moore ED, Etter EF, Philipson KD, Carrington WA, Fogarty KE, Lifshitz LM, Fay FS. Coupling of the Na+/Ca2+ exchanger, Na+/K+ pump and sarcoplasmic reticulum in smooth muscle. Nature. 1993;365:657–660. doi: 10.1038/365657a0. [DOI] [PubMed] [Google Scholar]
- Morgan JP, Morgan KG. Vascular smooth muscle: the first recorded Ca2+ transients. Pflugers Arch. 1982;395:75–77. doi: 10.1007/BF00584972. [DOI] [PubMed] [Google Scholar]
- Nakasaki Y, Iwamoto T, Hanada H, Imagawa T, Shigekawa M. Cloning of the rat aortic smooth muscle Na+/Ca2+ exchanger and tissue-specific expression of isoforms. J Biochem (Tokyo) 1993;114:528–534. doi: 10.1093/oxfordjournals.jbchem.a124211. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- Ouardouz M, Zamponi GW, Barr W, Kiedrowski L, Stys PK. Protection of ischemic rat spinal cord white matter: Dual action of KB-R7943 on Na+/Ca2+ exchange and L-type Ca2+ channels. Neuropharmacology. 2005;48:566–575. doi: 10.1016/j.neuropharm.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Poburko D, Kuo KH, Dai J, Lee CH, van Breemen C. Organellar junctions promote targeted Ca2+ signaling in smooth muscle: why two membranes are better than one. Trends Pharmacol Sci. 2004a;25:8–15. doi: 10.1016/j.tips.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Poburko D, Lee CH, van Breemen C. Vascular smooth muscle mitochondria at the cross roads of Ca2+ regulation. Cell Calcium. 2004b;35:509–521. doi: 10.1016/j.ceca.2004.01.020. [DOI] [PubMed] [Google Scholar]
- Poburko D, Potter K, van Breemen E, Fameli N, Liao CH, Basset O, Ruegg UT, van Breemen C. Mitochondria buffer NCX-mediated Ca2+-entry and limit its diffusion into vascular smooth muscle cells. Cell Calcium. 2006;40:359–371. doi: 10.1016/j.ceca.2006.04.031. [DOI] [PubMed] [Google Scholar]
- Potocnik SJ, Murphy TV, Kotecha N, Hill MA. Effects of mibefradil and nifedipine on arteriolar myogenic responsiveness and intracellular Ca2+ Br J Pharmacol. 2000;131:1065–1072. doi: 10.1038/sj.bjp.0703650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quednau BD, Nicoll DA, Philipson KD. Tissue specificity and alternative splicing of the Na+/Ca2+ exchanger isoforms NCX1, NCX2, and NCX3 in rat. Am J Physiol Cell Physiol. 1997;272:C1250–C1261. doi: 10.1152/ajpcell.1997.272.4.C1250. [DOI] [PubMed] [Google Scholar]
- Ranciat-McComb NS, Bland KS, Huschenbett J, Ramonda L, Bechtel M, Zaidi A, Michaelis ML. Antisense oligonucleotide suppression of Na+/Ca2+ exchanger activity in primary neurons from rat brain. Neurosci Lett. 2000;294:13–16. doi: 10.1016/s0304-3940(00)01524-x. [DOI] [PubMed] [Google Scholar]
- Rembold CM, Murphy RA. Myoplasmic calcium, myosin phosphorylation, and regulation of the crossbridge cycle in swine arterial smooth muscle. Circ Res. 1986;58:803–815. doi: 10.1161/01.res.58.6.803. [DOI] [PubMed] [Google Scholar]
- Reuter H, Blaustein MP, Haeusler G. Na-Ca exchange and tension development in arterial smooth muscle. Philos Trans R Soc Lond B Biol Sci. 1973;265:87–94. doi: 10.1098/rstb.1973.0011. [DOI] [PubMed] [Google Scholar]
- Richards K, Davis MJ, Potocnik SJ, Murphy TV, Bishara NB, Rajanayagam MA, Darby IA, Hill MA. Approaches for introducing peptides into intact and functional arteriolar smooth muscle: manipulation of protein kinase-based signalling. Clin Exp Pharmacol Physiol. 2003;30:653–658. doi: 10.1046/j.1440-1681.2003.03892.x. [DOI] [PubMed] [Google Scholar]
- Rosker C, Graziani A, Lukas M, Eder P, Zhu MX, Romanin C, Groschner K. Ca2+ signaling by TRPC3 involves Na+ entry and local coupling to the Na+/Ca2+ exchanger. J Biol Chem. 2004;279:13696–13704. doi: 10.1074/jbc.M308108200. [DOI] [PubMed] [Google Scholar]
- Seiler PU, Stypmann J, Breithardt G, Schulze-Bahr E. Real-time RT-PCR for gene expression profiling in blood of heart failure patients-a pilot study: gene expression in blood of heart failure patients. Basic Res Cardiol. 2004;99:230–238. doi: 10.1007/s00395-004-0467-6. [DOI] [PubMed] [Google Scholar]
- Seip G, Schultheiss G, Kocks SL, Diener M. Interaction between store-operated non-selective cation channels and the Na+ -Ca2+ exchanger during secretion in the rat colon. Exp Physiol. 2001;86:461–468. doi: 10.1113/eph8602243. [DOI] [PubMed] [Google Scholar]
- Slish DF, Welsh DG, Brayden JE. Diacylglycerol and protein kinase C activate cation channels involved in myogenic tone. Am J Physiol Heart Circ Physiol. 2002;283:H2196–H2201. doi: 10.1152/ajpheart.00605.2002. [DOI] [PubMed] [Google Scholar]
- Slodzinski MK, Blaustein MP. Na+/Ca2+ exchange in neonatal rat heart cells: antisense inhibition and protein half-life. Am J Physiol Cell Physiol. 1998;275:C459–C467. doi: 10.1152/ajpcell.1998.275.2.C459. [DOI] [PubMed] [Google Scholar]
- Slodzinski MK, Juhaszova M, Blaustein MP. Antisense inhibition of Na+/Ca2+ exchange in primary cultured arterial myocytes. Am J Physiol Cell Physiol. 1995;269:C1340–C1345. doi: 10.1152/ajpcell.1995.269.5.C1340. [DOI] [PubMed] [Google Scholar]
- Spassova MA, Hewavitharana T, Xu W, Soboloff J, Gill DL. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc Natl Acad Sci U S A. 2006;103:16586–16591. doi: 10.1073/pnas.0606894103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh DG, Morielli AD, Nelson MT, Brayden JE. Transient receptor potential channels regulate myogenic tone of resistance arteries. Circ Res. 2002;90:248–250. doi: 10.1161/hh0302.105662. [DOI] [PubMed] [Google Scholar]
- von Lewinski D, Stumme B, Maier LS, Luers C, Bers DM, Pieske B. Stretch-dependent slow force response in isolated rabbit myocardium is Na+ dependent. Cardiovasc Res. 2003;57:1052–1061. doi: 10.1016/s0008-6363(02)00830-1. [DOI] [PubMed] [Google Scholar]
- Wu X, Davis MJ. Characterization of stretch-activated cation current in coronary smooth muscle cells. Am J Physiol Heart Circ Physiol. 2001;280:H1751–H1761. doi: 10.1152/ajpheart.2001.280.4.H1751. [DOI] [PubMed] [Google Scholar]
- Wu C, Fry CH. Na+/Ca2+ exchange and its role in intracellular Ca2+ regulation in guinea pig detrusor smooth muscle. Am J Physiol Cell Physiol. 2001;280:C1090–C1096. doi: 10.1152/ajpcell.2001.280.5.C1090. [DOI] [PubMed] [Google Scholar]
- Zhuge R, Fogarty KE, Tuft RA, Walsh JV., Jr Spontaneous transient outward currents arise from microdomains where BK channels are exposed to a mean Ca2+ concentration on the order of 10 μM during a Ca2+ spark. J Gen Physiol. 2002;120:15–27. doi: 10.1085/jgp.20028571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Ratz PH, Hill MA. Role of myosin phosphorylation and [Ca2+]i in myogenic reactivity and arteriolar tone. Am J Physiol Heart Circ Physiol. 1995;269:H1590–H1596. doi: 10.1152/ajpheart.1995.269.5.H1590. [DOI] [PubMed] [Google Scholar]