

Abstract
Background and Aims
Signal transducer and activator of transcription 3 (STAT3) is known to be activated in human alcoholic liver disease, but its role in the pathogenesis of alcoholic liver injury remains obscure.
Methods
The role of STAT3 in alcoholic liver injury was investigated in hepatocyte-specific STAT3 knockout (H-STAT3KO) mice and macrophage/neutrophil-specific STAT3 KO (M/N-STAT3KO) mice. Alcoholic liver injury was achieved by feeding mice with a liquid diet containing 5% ethanol for up to 8 weeks.
Results
Compared to wild type mice, feeding H-STAT3KO mice with an ethanol containing diet induced greater hepatic steatosis, hypertriglyceridemia, and hepatic expression of lipogenic genes (SREBP1, FAS, ACC1, and SCD1), but less inflammation and lower expression of hepatic proinflammatory cytokines. In contrast, ethanol-fed M/N STAT3KO mice showed more hepatic inflammation, worse injury, and elevated hepatic expression of proinflammatory cytokines compared with wild-type mice. Kupffer cells isolated from ethanol-fed H-STAT3KO mice produced similar amounts of ROS and TNF-α, while Kupffer cells from M/N-STAT3KO mice produced more ROS and TNF-α compared with wild-type controls.
Conclusions
These findings suggest that STAT3 regulates hepatic inflammation in a cell type-dependent manner during alcoholic liver injury: STAT3 in hepatocytes promotes whereas STAT3 in macrophages/Kupffer cells suppresses inflammation. In addition, activation of hepatocellular STAT3 ameliorates alcoholic fatty liver via inhibition of SREBP1c expression.
Keywords: STAT3, alcohol, macrophage, inflammation, fatty liver
Introduction
Alcohol drinking is a major etiologic factor in chronic liver disease worldwide, causing fatty liver, alcoholic hepatitis, cirrhosis, and/or hepatocellular carcinoma.1, 2 In the last few decades, major progress has been made in our understanding of the molecular mechanisms underlying alcoholic liver injury. These mechanisms include alcohol-induced hepatotoxicity and oxidative stress, as well as complex interactions between alcohol metabolism, various hepatic cells, multiple cytokines, and the immune system.3–8 Studies using animal models suggest that alcohol intake permeabilizes the gut resulting in elevated hepatic endotoxin levels, which is followed by activation of hepatic macrophages/Kupffer cells. Activated macrophages/Kupffer cells generate oxidative stress and produce a variety of soluble factors and cytokines.9 Among them, tumor necrosis factor α (TNF-α) is considered one of the most important cytokines involved in the pathogenesis of alcoholic liver injury, with evidence showing that disruption of the TNF-αR1 gene or treatment with a TNF-α neutralizing antibody reduces alcoholic liver injury in mice.10, 11 In contrast, adiponectin and interleukin-6 (IL-6) have been shown to protect against alcoholic liver injury.12–16 For example, genetic ablation of the IL-6 gene enhanced alcohol-induced liver injury, whereas IL-6 treatment ameliorated alcoholic liver injury and alcoholic fatty liver transplant.14–16 The important role of IL-6 in hepatoprotection has also been demonstrated in experimental models of liver injury induced by partial hepatectomy, concanavalin A, Fas agonist (Jo2), high fat diet, hepatotoxins, etc. (see review17), as well as in chronic human liver disease.18.
The hepatoprotective action of IL-6 is mainly mediated via STAT3 activation. In addition to IL-6, many other factors and cytokines activate STAT3 in the liver, including IL-22, IL-10, EGF, and hepatitis viral proteins.17 Activation of STAT3 in hepatocytes has been reported in virtually all models of liver injury17 and has been shown to contribute to acute phase responses,19 liver regeneration,20, 21 amelioration of liver injury,22–24 regulation of gluconeogenesis and carbohydrate metabolism.25 Activation of STAT3 has also been reported in chronic human alcoholic hepatitis and alcoholic cirrhosis.26, 27 However, the role of STAT3 in alcoholic liver disease remains elusive. In this study, we explored the functions of STAT3 in alcoholic liver injury using hepatocyte-specific STAT3 knockout (H-STAT3KO) mice and macrophage/neutrophil-specific STAT3 knockout (M/N-STAT3KO) mice. Our findings suggest that in hepatocytes, STAT3 plays an important role in inhibiting steatosis but promoting inflammation, while in macrophages STAT3 activation inhibits liver inflammation during chronic alcoholic liver injury.
Methods and Materials
Mice
STAT3flox/flox mice were developed as described previously.28 Hepatocyte-specific STAT3KO (H-STAT3KO)(STAT3flox/floxAlbCre+/−) mice were generated by crossing STAT3flox/flox mice with albumin-promoter Cre transgenic mice (the Jackson Laboratory, Bar Harbor, Maine). STAT3flox/floxAlbCre−/− mice were used as littermate wild-type (WT) controls.
Macrophage/neutrophil-specific STAT3KO (STAT3flox/floxLysCre+/−)(M/N-STAT3KO) mice were generated by crossing STAT3flox/flox mice with lysozyme M-promoter Cre transgenic mice (the Jackson Laboratory). M/N-STAT3KO mice were fed a regular chow. A small percentage of M/N-STAT3KO mice developed colitis after 12 weeks. Mice with obvious symptoms of colitis were removed from the study. STAT3flox/floxLysMCre−/− mice were used as littermate WT controls. M/N-STAT3KO mice have been proven to be a valuable tool for conditional knockout of the STAT3 gene in monocytes/macrophages and neutrophils.29.
Other Materials and Methods
Other materials and methods are described in the supporting document. These include: mouse model for chronic ethanol consumption, blood chemistry, hepatic lipid contents, histology, immunohistochemistry, Oil red O staining lipid content, serum cytokine levels, Western blotting, isolation of hepatocytes and Kupffer cells, measurement of reactive oxidative species (ROS), semiquantitive RT-PCR, real-time PCR, lipopolysaccharide (LPS) stimulation of Kupffer cells and TNF-α production. Flow cytometric analysis. Primers used in RT-PCR and real-time PCR are listed in the supporting document.
Statistical Analysis
Data are expressed as means ± SD. To compare values obtained from 2 groups, the Student t test was performed. To compare values obtained from three or more groups, 1-factor analysis of variance (ANOVA) was used, followed by Tukey's post hoc test. Statistical significance was taken at the P <0.05 level. The statistical comparisons between pair-fed knockout mice and pair-fed WT mice or between ethanol-fed knockout and ethanol-fed WT mice are shown in the Fig. 1–Fig. 7. The statistical comparisons between pair-fed and ethanol-fed WT mice or between pair-fed and ethanol-fed knockout mice are shown in sFig. 1–sFig. 7 in supporting materials.
Figure 1.
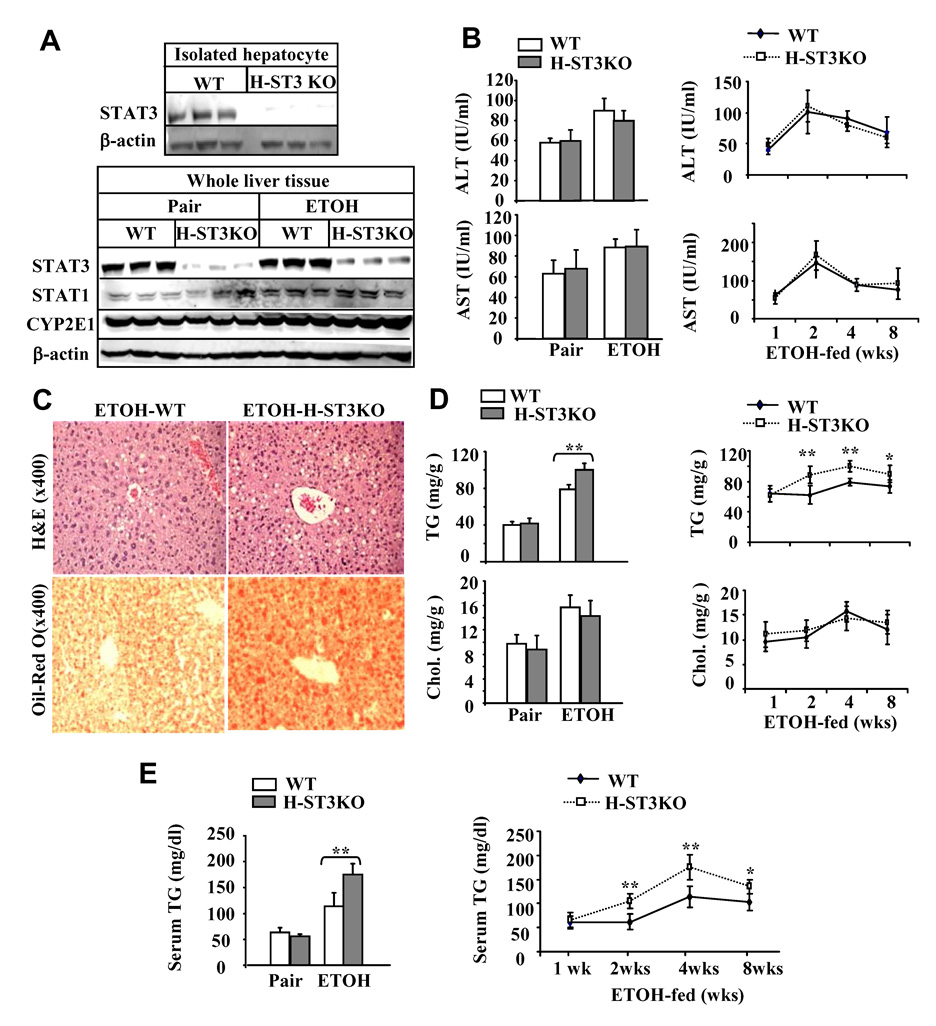
H-STAT3KO mice are more susceptible to ethanol-induced fatty liver. (A) Western blot analyses of STAT3 protein from the isolated hepatocytes or from the whole liver tissues. (B–E) WT and H-STAT3KO mice were pair-fed or fed ethanol diet for 4 weeks or various weeks. Sera were collected for measurement of ALT/AST (B); Liver tissues collected for H&E and Oil-red O staining (C) and for measurement of hepatic lipid contents (D); (E) Serum triglyceride levels. Values shown in the left panels B, D, and E, represent 4 week-feeding, in the right panels represent 1 to 8 week-feeding. The values represent means ± SD (n=6 mice per group in 1 or 8 week groups; n=8 mice per group in 2 week groups; In 4 week groups, n= 8 mice per group in pair-fed groups, n=15 mice per group in ethanol-fed groups). *P<0.05, **P<0.01 denotes significant difference from corresponding ethanol-fed WT groups. H-ST3KO: H-STAT3KO; TG: triglyceride; Chol.: cholesterol.
Figure 7.
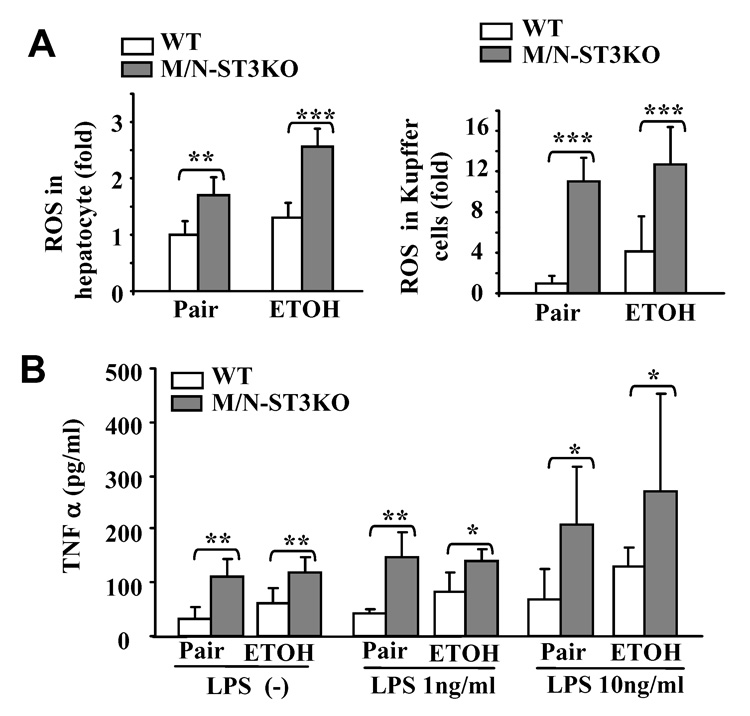
Increased production of ROS and TNF-α by Kupffer cells from M/N-STAT3KO mice. Feeding of mice as in Fig. 5. (A) Hepatocytes and Kupffer cells were isolated and ROS production measured. (B) Kupffer cells WT and M/NSTAT3KO mice were stimulated with LPS for 4 h. TNF-α production was measured. Values represent means ± SD (n=5–6). *P<0.05, **P<0.01, ***P<0.001 denotes significant differences in comparison with WT groups.
Results
H-STAT3KO mice are more susceptible to ethanol-induced fatty liver
Fig. 1A documents the complete loss of STAT3 protein expression in hepatocytes from H-STAT3KO mice, confirming the deletion of STAT3 in these cells. Weak expression of the STAT3 protein in whole liver tissue of H-STAT3KO mice in Fig. 1A is probably due to the expression of STAT3 in nonparenchymal cells. Expression of STAT1 protein was slightly higher in H-STAT3KO mice compared with WT mice, and was further increased after ethanol feeding. Expression of CYP2E1 was similar in H-STAT3KO and WT mice and, as expected, was elevated after ethanol feeding (Fig. 1A). Next, the effects of alcohol feeding on liver injury were compared in H-STAT3KO mice and WT controls. As shown in Fig. 1B, serum ALT and AST levels were similar in H-STAT3KO and WT mice, and higher in the ethanol-fed groups than in the respective pair-fed groups. H&E and oil-red-O staining revealed greater steatosis (fat deposition) in the livers of H-STAT3KO mice compared with WT mice after ethanol feeding (Fig. 1C), which was also confirmed via biochemical analyses (Fig. 1D). Furthermore, ethanol-fed H-STAT3KO mice showed significantly higher triglyceride levels in the serum than corresponding WT controls (Fig. 1E). In contrast, no significant difference was found in cholesterol levels in the liver and serum between ethanol-fed H-STAT3KO and WT mice, although alcohol feeding increased the amount of cholesterol (Fig. 1D).
To examine the time course of liver injury, we also investigated the effects of ethanol feeding for various weeks in H-STAT3KO and WT mice. There was no difference in serum ALT and AST levels between these 2 groups (Fig. 1B), while the hepatic and serum triglyceride levels were significantly higher in H-STAT3KO mice at 2, 4, and 8 weeks post ethanol feeding (Fig. 1D–E). Levels of hepatic cholesterol and serum cholesterol, glucose, and insulin were similar in these 2 groups at all time points post ethanol feeding (Fig. 1D and sFig. 8).
Ethanol upregulates the expression of lipogenic genes in H-STAT3KO mice
To identify the mechanism by which ethanol induces more steatosis in H-STAT3KO mice than in WT mice, we profiled the expression of a group of fat metabolism-related genes. As shown in Figs. 2A–B, mRNA expression of SREBP1c and the SREBP1c-regulated genes, FAS, ACC1, and SCD1, were higher in ethanol-fed H-STAT3KO mice than in WT mice. Expression of CPT1 and PPARα mRNA, which accelerate fatty acid lipid oxidation, was also upregulated in ethanol-fed H-STAT3KO mice compared to WT groups. Upregulation of SREBP1c protein was further confirmed by Western blotting. As shown in Figs. 2C–D, consistent with previous reports,30 ethanol feeding increased mature form of nuclear SREBP1c protein expression about 2 fold in WT (white bars). Interestingly, the same feeding increased expression of nuclear SREBP1 protein about 10 fold in H-STAT3KO mice (grey bars). As expected, nuclear pSTAT3 protein was much lower in H-STAT3 KO mice than in WT mice (Fig. 2C).
Figure 2.
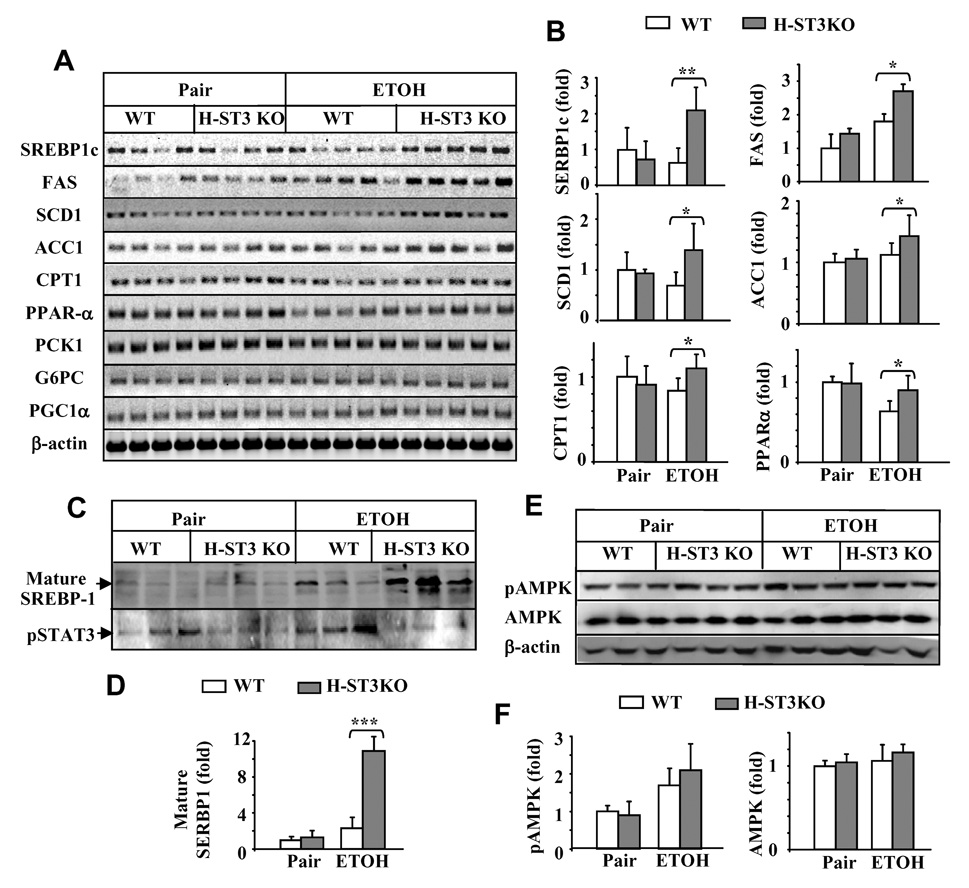
Upregulation of lipogenic gene expression in ethanol-fed H-STAT3KO mice. Liver samples from 4 week-fed WT and H-STAT3KO mice in Fig. 1 were used to analyze the expression of lipid metabolism- and gluconeogenesis-related genes (mRNAs) by semi-quantitative RT-PCR (A). The densities in panel A were quantified by densitometry (B). (C–D) Expression of nuclear SREBP1c and pSTAT3 proteins was examined (C) and quantified (D). (E–F) pAMPK and AMPK proteins in whole liver tissues were examined by Western blotting (E) and quantified (F). Values in panels B, D, and F represent means ± SD (n= 3–5). *P<0.05, **P<0.01 denotes significant differences in comparison with WT groups.
In contrast, expression of gluconeogenesis-related genes such as PCK1, G6PC, and PGC-1α was similar in pair- and ethanol-fed WT and H-STAT3KO mice (Fig. 2A). Finally, there was no significant difference in phospho-AMPKα expression between H-STAT3KO and WT mice; however, ethanol feeding increased phospho-AMPKα levels relative to the control diet (Figs. 2E–F).
H-STAT3KO mice are resistant to ethanol-induced hepatic inflammation, expression of pro-inflammatory cytokines and chemokines
Ethanol feeding increased the number of inflammatory foci and neutrophils (myeloperoxidase [MPO]+ cells) in both H-STAT3KO and WT mice, but such increase was less pronounced in H-STAT3 KO mice (Fig. 3A). This suggests that H-STAT3KO mice are not similarly prone to inflammation as WT mice after ethanol feeding.
Figure 3.
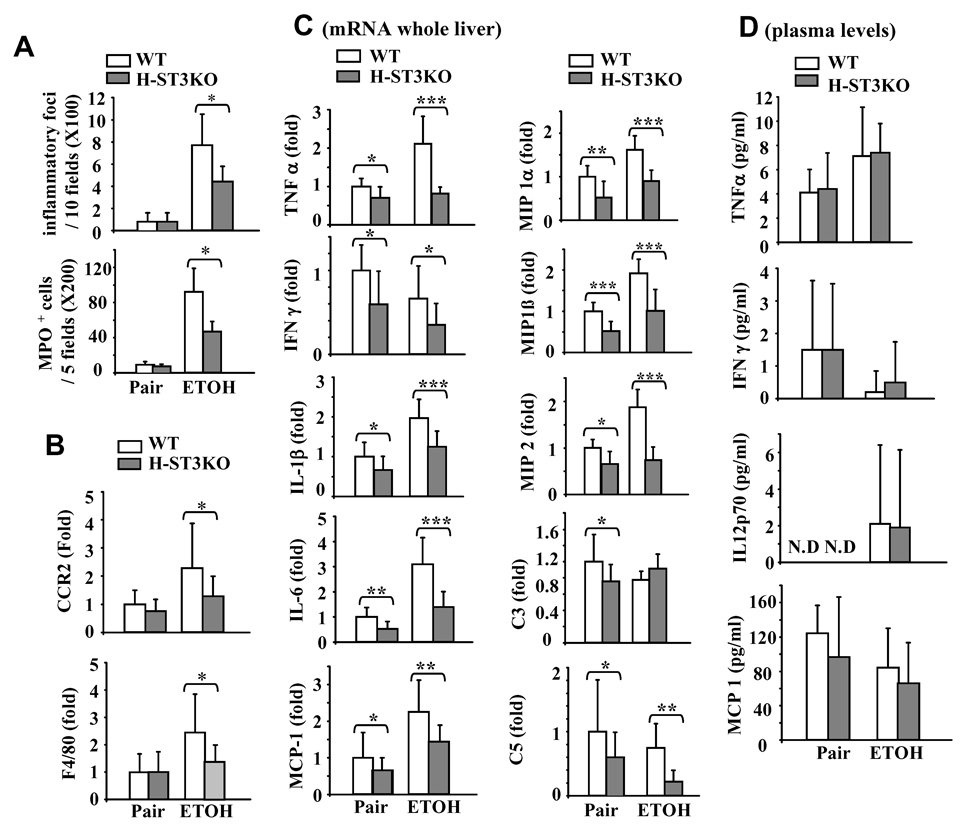
H-STAT3KO mice are resistant to ethanol-induced hepatic inflammation. Feeding of mice for 4 weeks as in Fig. 1. Liver tissues were collected for H&E staining and MPO staining. (A) The number of inflammatory foci and the number of MPO positive cells were counted. (B) Real-time PCR analyses for CCR2 and F4/80 expression in liver. (C) Real-time PCR analyses of cytokine and chemokine expression in the liver. (D) Serum cytokine and chemokine levels. Values represent means ± SD (n=8–10 mice). *P<0.05, **P<0.01, ***P<0.001 denotes significant differences in comparison with corresponding WT groups.
Expression of CCR2 and F4/80, markers for monocytes and macrophages, respectively, were examined to detect the infiltration of monocytes/macrophages. As shown in Fig. 3B, expression of CCR2 and F4/80 increased after ethanol feeding in WT mice, but such increase was less evident in H-STAT3KO mice. Taken together, this implies that ethanol-induced infiltration of inflammatory cells is attenuated in H-STAT3KO mice.
Expression of a variety of proinflammatiory cytokines in the liver was examined by real-time PCR. As shown in Fig. 3C, ethanol feeding increased the hepatic expression of several proinflammatory cytokines and chemokines in WT mice (white bars); however, such increase was not observed, or was less pronounced, in H-STAT3KO mice (grey bars). In contrast, ethanol decreased hepatic IFN-γ mRNA expression in both WT and H-STAT3KO mice. Moreover, ethanol feeding decreased slightly the expression of hepatic complement 3 (C3) and C5 mRNA expression in WT mice, and the decrease in C5 was more pronounced in H-STAT3KO mice after ethanol feeding (Fig. 3C).
Serum levels of several proinflammatory cytokines were also examined. As shown in Fig. 3D, ethanol increased serum levels of TNF-α and IL-12p70, but decreased IFN-γ and MCP-1 in WT mice. No significant differences were found in TNF-α, IFN-γ, IL12p70 and MCP1 levels between WT and H-STAT3KO mice. Serum levels of IL-6 and IL-10 were under the assay detection limits in each group (data not shown).
Kupffer cells from ethanol-fed H-STAT3KO mice and WT mice respond similarly to LPS stimulation
Fig. 4A shows that ethanol feeding induces ROS production by hepatocytes and Kupffer cells in WT mice. In hepatocytes from H-STAT3KO mice, ethanol-induced ROS was reduced, while Kupffer cells from WT and H-STAT3KO mice produced similar levels of ROS after ethanol feeding. Moreover, Kupffer cells from ethanol-fed mice produced higher basal levels of TNF-α compared with pair-fed mice (Fig. 4B). Production of TNF-α by Kupffer cells from ethanol-fed mice further increased with LPS treatment. This increase was less evident in Kupffer cells from pair-fed mice, in agreement with previous findings that ethanol increases Kupffer cell sensitivity to LPS stimulation due to increased expression of the LPS receptor, CD14, after ethanol feeding.31 There was no difference in TNF-α production by Kupffer cells from either WT or H-STAT3KO mice (Fig. 4B).
Figure 4.
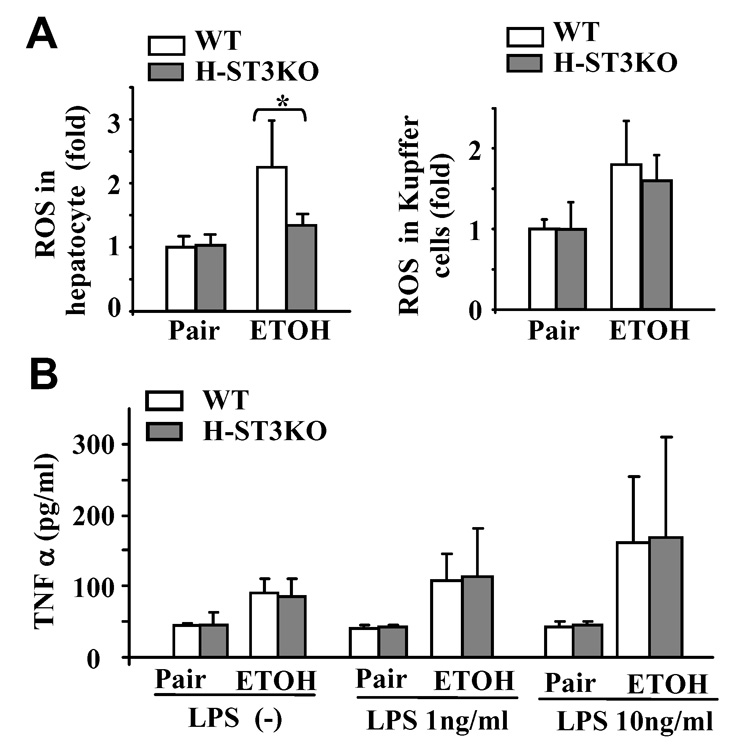
Kupffer cells from H-STAT3KO and WT mice produce similar levels of ROS and respond similarly to LPS stimulation. Feeding of mice for 4 weeks as in Fig. 1. (A) ROS production in hepatocytes and Kupffer from WT or H-STAT3KO mice. (B) Kupffer cells from WT or H-STAT3KO mice were stimulated with LPS for 4 h. TNF-α production was measured. Values represent means ± SD (n=5–6 mice per group.) *P<0.05 denotes significant differences in comparison with WT groups.
M/N-STAT3 KO mice are more susceptible to ethanol-induced hepatic injury and inflammation
Fig. 5A shows STAT3 protein expression was markedly reduced in peritoneal macrophages and hepatic macrophages/Kupffer cells from M/N-STAT3KO mice. The significant reduction of STAT3 activation (pSTAT3) in hepatic macrophages/Kupffer cells was also confirmed by flow cytometric analyses (sFig. 10). Fig. 5B shows that ethanol feeding elevated serum ALT/AST levels in both M/N-STAT3KO and WT groups, such increase was more evident in the M/N-STAT3KO group (grey bars). Although liver histology showed similar levels of fat deposition, greater inflammation was observed from livers of ethanol-fed M/N-STAT3KO versus WT mice (Fig. 5C). The similar levels of hepatic fat deposition in these 2 groups were further confirmed by measurement of lipid contents (Fig. 5D). Serum levels of triglyceride, cholesterol, glucose, and insulin were also similar in M/N-STAT3KO and WT mice (sFig. 9). Finally, more hepatic inflammation in M/N-STAT3KO versus WT mice was further demonstrated by increased numbers of inflammatory foci and neutrophils, and increased expression of CCR2 and F4/80 mRNA (Figs. 5E–F).
Figure 5.
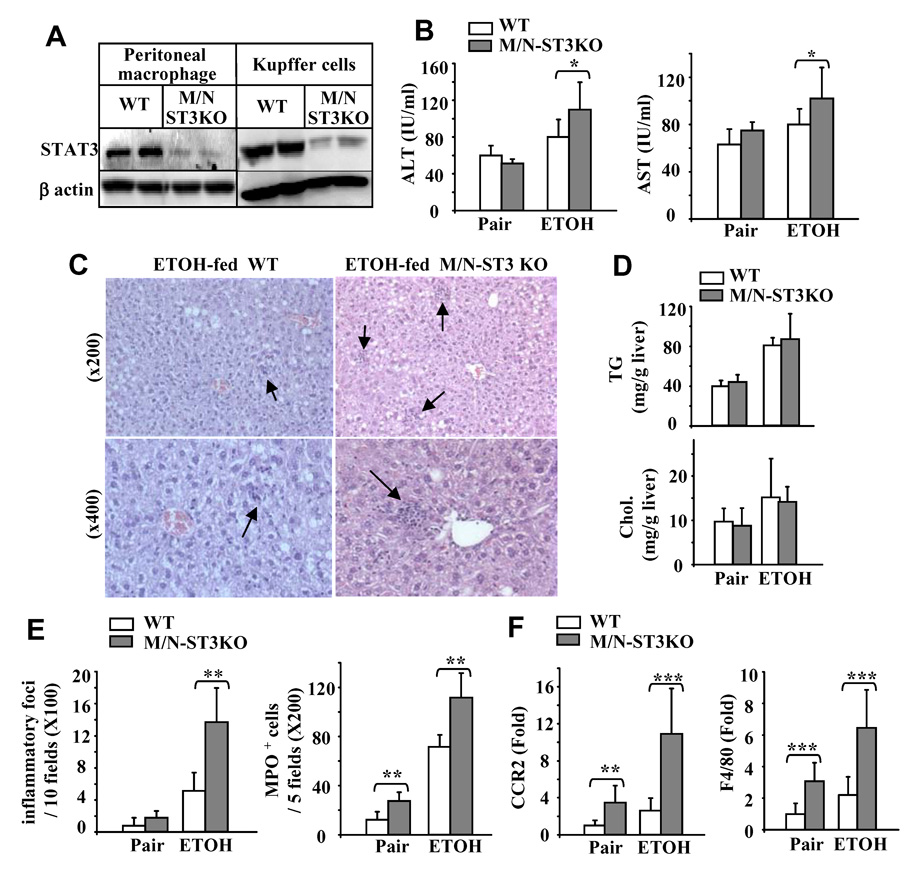
M/N-STAT3KO mice are more susceptible to ethanol-induced liver inflammation. (A) Western blot analyses of STAT3 protein expression in peritoneal macrophages and Kupffer cells. (B–F) WT and M/N-STAT3KO mice were given an ethanol-diet or pair-fed with a control diet for 4 weeks. Sera were collected for measurement of ALT/AST (B); Liver tissues collected for H&E staining (C) or for measurement of lipid contents (D). (E) The number of inflammatory foci and MPO positive cells in the liver. (F) R real-time PCR analyses for CCR2 and F4/80 expression. Values shown in panels B, D, E, and F represent means ± SD (n=6 in pair-fed group [one M/N-STAT3KO mouse was removed from the study due to enterocolitis. n=10 in ethanol-fed group [two M/N-STAT3KO mice were removed due to enterocolitis]). *P<0.05, **P<0.01, **P<0.001 denotes significant differences in comparison with WT groups. M/N-ST3KO: M/N-STAT3KO.
Upregulation of proinflammatory cytokines and chemokines in the liver and serum of ethanol-fed M/N-STAT3KO versus WT mice
Fig. 6A shows that in pair-fed groups, basal levels of various hepatic pro-inflammatory cytokines and chemokines were higher in M/N-STAT3KO than in WT mice. Ethanol feeding slightly increased the expression of all of those cytokines and chemokines in WT mice, but dramatically increased their expression levels in M/N-STAT3KO mice. Interestingly, ethanol feeding decreased hepatic expression of IFN-γ in M/N-STAT3KO mice. Similarly, in pair-fed mice, serum levels of TNF-α, IFN-γ, and IL-6 were higher in M/N-STAT3KO than in WT mice (Fig. 6B). Ethanol feeding further increased TNF-α and IL-6 but decreased IFN-γ serum levels in M/N-STAT3KO mice (Fig. 6B).
Figure 6.
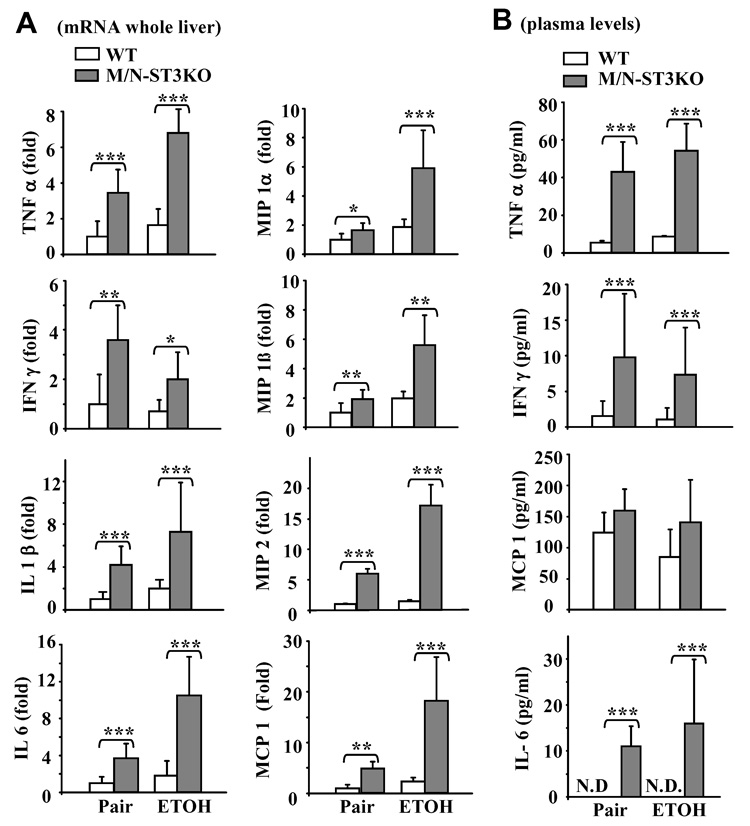
Upregulation of proinflammatory cytokines and chemokines in M/N-STAT3 KO mice. Feeding of mice as in Fig. 5. (A) Real-time PCR analyses of gene expression in liver. (B) Serum cytokine and chemokine levels. Values represent means ± SD (N=6–10). **P<0.01, ***P<0.001 denotes significant differences in comparison with WT groups.
Ethanol feeding induces greater production of ROS and TNF-α in Kupffer cells from M/N-STAT3KO compared to WT mice
Ethanol feeding enhanced ROS production by hepatocytes and Kupffer cells in WT mice (white bars in Fig. 7A). Basal levels of ROS production in pair-fed groups were higher in hepatocytes and Kupffer cells from M/N-STAT3KO compared with WT mice. The high basal levels of ROS found in M/N-STAT3KO mice were further increased after ethanol feeding. Similarly, as shown in Fig. 7B, ethanol feeding increased basal and LPS-stimulated TNF-α production in Kupffer cells from WT mice (white bars), whereas in M/N-STAT3KO mice it only enhanced TNF-α production stimulated by 10 but not by 1 ng/mL of LPS, probably due to the very high basal levels of TNF-α (grey bars). Finally, basal and LPS-stimulated TNF-α production by Kupffer cells was greater in M/N-STAT3KO than in WT mice in pair-fed or ethanol-fed groups (grey bars vs. white bars).
Discussion
It is widely accepted that chronic alcohol consumption permeabilizes the gut, leading to the accumulation of endotoxins in the liver. These endotoxins activate macrophages/Kupffer cells produce ROS and a variety of soluble factors and cytokines (eg, TNF-α), precipitating liver injury.9 It is also known that endotoxin-activated macrophages/Kupffer cells produce IL-6 and IL-10, which in turn activate STAT3 in hepatocytes and macrophages/Kupffer cells. Indeed, STAT3 activation was detected in human alcoholic hepatitis and alcoholic cirrhosis.26,27 The present findings suggest that during alcoholic liver injury, activation of STAT3 in hepatocytes serves a compensatory role in preventing fatty liver, but also has a detrimental effect in promoting inflammation, whereas in macrophages/Kupffer cells the same STAT3 activation has antinflammatory effects. We have integrated these findings in a new model (summarized in Fig. 8) that depicts the role of STAT3 in alcoholic liver injury.
Figure 8.
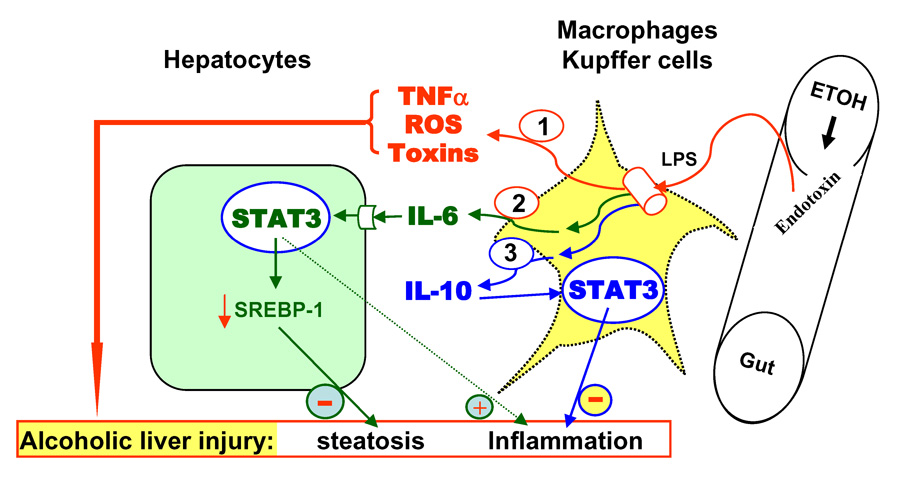
Schematic representation of roles of hepatocyte- and macrophage-STAT3 in alcoholic liver injury. Alcohol leads to the accumulation of endotoxin in the liver, followed by activation of macrophages/Kupffer cells and subsequent production of a variety of toxic mediators, leading to hepatic steatosis and inflammation. Macrophages/Kupffer cells also produce IL-6 and IL-10. IL-6 activates STAT3 in hepatocytes, which attenuates steatosis via inhibition of SREBP1c but promotes inflammation. IL-10 activates STAT3 in macrophages/Kupffer cells, which acts as an anti-inflammatory signal to inhibit inflammation during alcoholic liver injury.
Hepatic STAT3 promotes lipolysis in ethanol-induced fatty liver
Changes in liver histology and serum biochemistry shown in Fig. 1 clearly illustrate that H-STAT3KO mice are more prone to develop ethanol-induced fatty liver. Several lines of evidence suggest that the greater steatosis in H-STAT3KO mice is due to increased triglyceride synthesis. First, both serum and hepatic levels of triglyceride were higher in H-STAT3KO than in WT mice after ethanol feeding, while levels of cholesterol were similar (Fig. 1 and sFig. 8). Second, expression of SREBP1c, a key regulator of fatty acid synthesis,32 was higher in the livers of H-STAT3KO than WT mice after ethanol feeding. Consistently, SREBP1-controlled lipogenic genes (eg, FAS, SCD1, ACC1) were also more highly expressed in the livers of ethanol-fed H-STAT3KO versus WT mice. The important role of SREBPs in the development of fatty liver has been well documented.33, 34 Thus, activation of SREBP-1 in H-STAT3KO mice likely contributes to the increased steatosis in these mice after ethanol feeding. Furthermore, it has been reported that insulin and glucose induce expression of SREBPs; however, serum levels of insulin and glucose were similar in H-STAT3KO and WT mice after being fed an ethanol diet, suggesting that higher levels of SREBP1 in H-STAT3KO occur independently of insulin and glucose. Moreover, transfection with STAT3 has been shown to inhibit SRBEP1 promoter activity in hepatic cells,35 suggesting that STAT3 directly attenuates SREBP1c gene transcription, consequently inhibiting fatty acid synthesis and the development of fatty liver during ethanol feeding. The protective role of STAT3 in fatty livers has also been suggested in several models of nonalcoholic fatty liver diseases.15, 25 Collectively, hepatic STAT3 activation ameliorates alcoholic and nonalcoholic fatty livers. Interestingly, hepatic STAT3 signaling is impaired in ethanol-36 or high fructose diet-induced steatosis,37 suggesting that impaired STAT3 signaling could accelerate the development of hepatic steatosis in ethanol or high fructose-induced liver injury.
STAT3 in hepatocytes acts as a pro-inflammatory signal during alcoholic liver injury
Although H-STAT3KO mice had greater steatosis than WT mice after ethanol feeding, they had less inflammation in the liver compared to the WT control group. Compared with ethanol-fed WT mice, the number of inflammatory foci and neutrophils, and expression of proinflammatory cytokines and chemokines in the liver were lower in ethanol-fed H-STAT3KO mice (Fig. 3). In addition, expression of C3 and C5, two complement components that contribute to inflammation and steatosis in alcoholic liver injury,38 was also lower in H-STAT3KO mice compared to WT mice after ethanol feeding. These findings clearly suggest that STAT3 activation in hepatocytes is a proinflammatory signal during ethanol-induced liver injury. However, the mechanism by which STAT3 functions as a proinflammatory signal in hepatocytes is not clear. Previous studies suggest that IL-6/STAT3 stimulates inflammation via induction of MCP-1, ICAM-1, and VEGF, and subsequent inhibition of macrophage infiltration in murine models of spinal injury39 and in laser-induced choroidal neovascularization.40 Here we have shown that expression of MCP-1, MIP-1, and MIP-2 is lower in ethanol-fed H-STAT3KO compared with WT mice (Fig. 3), suggesting that STAT3 in hepatocytes is involved in the induction of proinflammatory cytokines and chemokines, followed by increased monocyte/macrophage infiltration into the liver, leading to more inflammation.
It is generally believed that accumulation of triglycerides in the hepatocyte is the first step, which leads to inflammation and acts as “lipotoxicity”, in alcoholic liver injury as well as in non-alcoholic liver injury.41 However, in our model, ethanol-fed H-STAT3KO mice had more steatosis than WT mice, but the degree of liver injury was unexpectedly similar in these 2 groups, suggesting that steatosis and injury may be independent. This notion was also supported by several other recent studies. For example, Pritchard et al. reported that C3 KO mice had less steatosis but more liver injury, while C5 KO mice had more steatosis but less liver injury in a model of alcohol feeding.38 Moreover, Yamaguchi et al. reported that inhibition of triglyceride synthesis improved steatosis but exacerbated liver damage and fibrosis.42 These findings suggest that steatosis may not be necessarily correlated with the liver injury and other factors may also contribute to liver injury. Indeed, the less severe hepatic inflammation we observed in ethanol-fed H-STAT3KO compared to wild-type mice may explain why liver injury was similar in the two strains despite of the greater degree of steatosis in the knockouts.
STAT3 in macrophage acts as an anti-inflammatory signal during alcoholic liver injury
In contrast to hepatocytes, where STAT3 functions as a proinflammatory signal, in macrophages and neutrophils it acts as an anti-inflammatory signal during alcoholic liver injury, as suggested by greater inflammation and expression of pro-inflammatory cytokines/chemokines in the livers of M/N-STAT3KO compared to WT mice (Fig. 5–Fig. 6). M/N-STAT3KO mice were generated by crossbreeding lysozyme M-Cre mice with STAT3flox/flox mice. This strategy has been documented to result in the selective deletion of the STAT3 gene in bone-marrow-derived macrophages and neutrophils,29 which was also confirmed here (Fig. 5A). However, the efficiency of lysozyme M-Cre recombination-mediated gene deletion in Kupffer cells is not clear. Maeda et al reported that lysozyme M-Cre recombination-mediated deletion of IKKβ is efficient in circulating macrophage but not useful in Kupffer cells.43 However, the Western blot data in Fig. S6 from Dr. Maeda’s paper43 showed that expression of IKKβ protein was significantly lower in Kupffer cells from Lyz-Cre IKKβFlox/Flox mice, suggesting IKKβ protein was partially deleted in Kupffer cells. Here we showed that STAT3 expression and activation were markedly reduced in Kupffer cells from M/N-STAT3KO mice (Fig. 5A and sFig. 10), suggesting that STAT3 is efficiently but not completely deleted in these cells. The weak STAT3 protein expression detected in these cells may be due to contamination of some non-macrophages or the heterogeneity of macrophages44. Taken together, STAT3 likely acts as an anti-inflammatory signal in macrophages/Kupffer cells in alcoholic liver injury; however, further studies are required to confirm the absolute role of STAT3 in Kupffer cells using a promoter more specific to Kupffer cells to achieve cell-specific deletion of the STAT3 gene.
Collectively, STAT3 plays a dual role in regulating inflammation: a proinflammatory effect in hepatocytes and an anti-inflammatory effect in macrophages in alcoholic liver injury, suggesting a complex role of STAT3 in regulating inflammation, which has been well documented in many other models.45,46 These findings lead to the question of what is the net effect of STAT3 on the hepatic inflammatory response during alcoholic liver injury. Since macrophages/Kupffer cells play a major role in inducing inflammation in the liver, the anti-inflammatory effect of STAT3 in macrophages/Kupffer cells likely dominates over the proinflammatory effect in hepatoctyes. Indeed, hepatic inflammation and expression of proinflammatory cytokines were reduced by only half in ethanol-fed H-STAT3KO versus WT mice, but increased by up to 15-fold in ethanol-fed M/N-STAT3KO versus WT controls. In addition, STAT3 in endothelial cells has been shown to act as an anti-inflammatory signal.47 Thus, it is likely that the overall effect of STAT3 is anti-inflammatory during alcoholic liver injury. The next obvious question is how STAT3 exerts opposite effects on inflammation in hepatocytes and macrophages/Kupffer cells. Both IL-6 and IL-10 activate STAT3, but they play differing roles, with IL-6 as a proinflammatory cytokine and IL-10 as an anti-inflammatory cytokine.48, 49 Hepatocytes only express functional receptors for IL-6, but not for IL-10,50 while macrophages/Kupffer cells express functional receptors for both cytokines, which may partly explain why STAT3 promotes inflammation in hepatocytes, but inhibits it in macrophages/Kupffer cells during chronic alcoholic liver injury (Fig.8).
Clinical data indicate that STAT3 is activated in alcoholic hepatitis and alcoholic cirrhosis.26, 27 Compared to viral hepatitis, alcoholic hepatitis presents with greater STAT3 activation,26 whereas STAT3 activation is less pronounced in alcoholic cirrhosis.27, 51 This suggests that STAT3 may be more active in the early stages of alcoholic liver disease, but then becomes suppressed in end-stage disease. The stronger activation of STAT3 in early alcoholic liver injury (alcoholic hepatitis) may be compensatory to ameliorate fatty liver and inhibit inflammation, while decreased STAT3 activation in the later stages of alcoholic liver injury (cirrhosis) could contribute to the progression of chronic alcoholic liver disease.
Supplementary Material
Acknowledgement
This work was supported by the intramural program of NIAAA, NIH. Dr. Norio Horiguchi was supported partially by the Japan Society for the Promotion of Science (JSPS) award at NIH.
This work was supported by the intramural program of NIAAA, NIH. No conflicts of interest exist for all authors.
Abbreviations
- STAT3
signal transducer and activator of transcription 3
- H-STAT3KO mice
hepatocyte-specific STAT3 knock out
- M/N-STAT3KO
macrophage/neutrophil-specific STAT3 KO
- SREBP
sterol regulatory element-binding protein
- AMPK
AMP-activated protein kinase
- ALT
alanine transaminase
- AST
aspartate aminotransferase
- FAS
fatty acid synthase
- ACC1
acetyl-CoA carboxylase-1
- SCD1
stearoyl-CoA desaturase 1
- CPT1
carnitine palmitoyltransferase I
- PPAR-α
peroxysome proliferators-activated receptor-α
- G6PC
glucose-6 phosphatase
- PCK-1
phosphoenolpyruvate carboxylase-1
- PGC1 α
peroxisome proliferator-activated receptor-γ coactivator-1α
- ROS
reactive oxygen species
- MCP-1
monocyte chemoattractant protein 1
- MIP
macrophage inflammatory protein
- C3
complement 3
- MPO
myeloperoxidase
- CCR2
CC chemokine receptor 2
- LPS
lipopolysaccharide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zakhari S, Li TK. Determinants of alcohol use and abuse: Impact of quantity and frequency patterns on liver disease. Hepatology. 2007;46:2032–2039. doi: 10.1002/hep.22010. [DOI] [PubMed] [Google Scholar]
- 2.Williams R. Global challenges in liver disease. Hepatology. 2006;44:521–526. doi: 10.1002/hep.21347. [DOI] [PubMed] [Google Scholar]
- 3.Tsukamoto H, Lu SC. Current concepts in the pathogenesis of alcoholic liver injury. Faseb J. 2001;15:1335–1349. doi: 10.1096/fj.00-0650rev. [DOI] [PubMed] [Google Scholar]
- 4.Dey A, Cederbaum AI. Alcohol and oxidative liver injury. Hepatology. 2006;43:S63–S74. doi: 10.1002/hep.20957. [DOI] [PubMed] [Google Scholar]
- 5.Gramenzi A, Caputo F, Biselli M, Kuria F, Loggi E, Andreone P, et al. Review article: alcoholic liver disease--pathophysiological aspects and risk factors. Aliment Pharmacol Ther. 2006;24:1151–1161. doi: 10.1111/j.1365-2036.2006.03110.x. [DOI] [PubMed] [Google Scholar]
- 6.Arteel GE. Oxidants and antioxidants in alcohol-induced liver disease. Gastroenterology. 2003;124:778–790. doi: 10.1053/gast.2003.50087. [DOI] [PubMed] [Google Scholar]
- 7.Hoek JB, Pastorino JG. Ethanol, oxidative stress, and cytokine-induced liver cell injury. Alcohol. 2002;27:63–68. doi: 10.1016/s0741-8329(02)00215-x. [DOI] [PubMed] [Google Scholar]
- 8.Nagy LE. Recent insights into the role of the innate immune system in the development of alcoholic liver disease. Exp Biol Med (Maywood) 2003;228:882–890. doi: 10.1177/153537020322800803. [DOI] [PubMed] [Google Scholar]
- 9.Uesugi T, Froh M, Arteel GE, Bradford BU, Wheeler MD, Gabele E, et al. Role of lipopolysaccharide-binding protein in early alcohol-induced liver injury in mice. J Immunol. 2002;168:2963–2969. doi: 10.4049/jimmunol.168.6.2963. [DOI] [PubMed] [Google Scholar]
- 10.Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, et al. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117:942–952. doi: 10.1016/s0016-5085(99)70354-9. [DOI] [PubMed] [Google Scholar]
- 11.Ji C, Deng Q, Kaplowitz N. Role of TNF-alpha in ethanol-induced hyperhomocysteinemia and murine alcoholic liver injury. Hepatology. 2004;40:442–451. doi: 10.1002/hep.20309. [DOI] [PubMed] [Google Scholar]
- 12.You M, Considine RV, Leone TC, Kelly DP, Crabb DW. Role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice. Hepatology. 2005;42:568–577. doi: 10.1002/hep.20821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong F, Kim WH, Tian Z, Jaruga B, Ishac E, Shen X, et al. Elevated interleukin-6 during ethanol consumption acts as a potential endogenous protective cytokine against ethanol-induced apoptosis in the liver: involvement of induction of Bcl-2 and Bcl-x(L) proteins. Oncogene. 2002;21:32–43. doi: 10.1038/sj.onc.1205016. [DOI] [PubMed] [Google Scholar]
- 15.Hong F, Radaeva S, Pan HN, Tian Z, Veech R, Gao B. Interleukin 6 alleviates hepatic steatosis and ischemia/reperfusion injury in mice with fatty liver disease. Hepatology. 2004;40:933–941. doi: 10.1002/hep.20400. [DOI] [PubMed] [Google Scholar]
- 16.Sun Z, Klein AS, Radaeva S, Hong F, El-Assal O, Pan HN, et al. In vitro interleukin-6 treatment prevents mortality associated with fatty liver transplants in rats. Gastroenterology. 2003;125:202–215. doi: 10.1016/s0016-5085(03)00696-6. [DOI] [PubMed] [Google Scholar]
- 17.Gao B. Cytokines, STATs and liver disease. Cell Mol Immunol. 2005;2:92–100. [PubMed] [Google Scholar]
- 18.Streetz KL, Tacke F, Leifeld L, Wustefeld T, Graw A, Klein C, et al. Interleukin 6/gp130-dependent pathways are protective during chronic liver diseases. Hepatology. 2003;38:218–229. doi: 10.1053/jhep.2003.50268. [DOI] [PubMed] [Google Scholar]
- 19.Alonzi T, Maritano D, Gorgoni B, Rizzuto G, Libert C, Poli V. Essential role of STAT3 in the control of the acute-phase response as revealed by inducible gene inactivation [correction of activation] in the liver. Mol Cell Biol. 2001;21:1621–1632. doi: 10.1128/MCB.21.5.1621-1632.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li W, Liang X, Kellendonk C, Poli V, Taub R. STAT3 contributes to the mitogenic response of hepatocytes during liver regeneration. J Biol Chem. 2002;277:28411–28417. doi: 10.1074/jbc.M202807200. [DOI] [PubMed] [Google Scholar]
- 21.Yeoh GC, Ernst M, Rose-John S, Akhurst B, Payne C, Long S, et al. Opposing roles of gp130-mediated STAT-3 and ERK-1/ 2 signaling in liver progenitor cell migration and proliferation. Hepatology. 2007;45:486–494. doi: 10.1002/hep.21535. [DOI] [PubMed] [Google Scholar]
- 22.Haga S, Terui K, Zhang HQ, Enosawa S, Ogawa W, Inoue H, et al. Stat3 protects against Fas-induced liver injury by redox-dependent and -independent mechanisms. J Clin Invest. 2003;112:989–998. doi: 10.1172/JCI17970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein C, Wustefeld T, Assmus U, Roskams T, Rose-John S, Muller M, et al. The IL-6-gp130-STAT3 pathway in hepatocytes triggers liver protection in T cell-mediated liver injury. J Clin Invest. 2005;115:860–869. doi: 10.1172/JCI200523640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Streetz KL, Wustefeld T, Klein C, Kallen KJ, Tronche F, Betz UA, et al. Lack of gp130 expression in hepatocytes promotes liver injury. Gastroenterology. 2003;125:532–543. doi: 10.1016/s0016-5085(03)00901-6. [DOI] [PubMed] [Google Scholar]
- 25.Inoue H, Ogawa W, Ozaki M, Haga S, Matsumoto M, Furukawa K, et al. Role of STAT-3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism in vivo. Nat Med. 2004;10:168–174. doi: 10.1038/nm980. [DOI] [PubMed] [Google Scholar]
- 26.Larrea E, Aldabe R, Molano E, Fernandez-Rodriguez CM, Ametzazurra A, Civeira MP, et al. Altered expression and activation of signal transducers and activators of transcription (STATs) in hepatitis C virus infection: in vivo and in vitro studies. Gut. 2006;55:1188–1196. doi: 10.1136/gut.2005.070060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horiguchi N, Ishac EJ, Gao B. Liver regeneration is suppressed in alcoholic cirrhosis: correlation with decreased STAT3 activation. Alcohol. 2007;41:271–280. doi: 10.1016/j.alcohol.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Welte T, Zhang SS, Wang T, Zhang Z, Hesslein DG, Yin Z, et al. STAT3 deletion during hematopoiesis causes Crohn's disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc Natl Acad Sci U S A. 2003;100:1879–1884. doi: 10.1073/pnas.0237137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 30.You M, Fischer M, Deeg MA, Crabb DW. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP) J Biol Chem. 2002;277:29342–29347. doi: 10.1074/jbc.M202411200. [DOI] [PubMed] [Google Scholar]
- 31.Enomoto N, Schemmer P, Ikejima K, Takei Y, Sato N, Brenner DA, et al. Long-term alcohol exposure changes sensitivity of rat Kupffer cells to lipopolysaccharide. Alcohol Clin Exp Res. 2001;25:1360–1367. [PubMed] [Google Scholar]
- 32.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ji C, Chan C, Kaplowitz N. Predominant role of sterol response element binding proteins (SREBP) lipogenic pathways in hepatic steatosis in the murine intragastric ethanol feeding model. J Hepatol. 2006;45:717–724. doi: 10.1016/j.jhep.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 34.Shimano H, Horton JD, Hammer RE, Shimomura I, Brown MS, Goldstein JL. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J Clin Invest. 1996;98:1575–1584. doi: 10.1172/JCI118951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ueki K, Kondo T, Tseng YH, Kahn CR. Central role of suppressors of cytokine signaling proteins in hepatic steatosis, insulin resistance, and the metabolic syndrome in the mouse. Proc Natl Acad Sci U S A. 2004;101:10422–10427. doi: 10.1073/pnas.0402511101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao B. Therapeutic potential of interleukin-6 in preventing obesity- and alcohol-associated fatty liver transplant failure. Alcohol. 2004;34:59–65. doi: 10.1016/j.alcohol.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 37.Roglans N, Vila L, Farre M, Alegret M, Sanchez RM, Vazquez-Carrera M, et al. Impairment of hepatic Stat-3 activation and reduction of PPARalpha activity in fructose-fed rats. Hepatology. 2007;45:778–788. doi: 10.1002/hep.21499. [DOI] [PubMed] [Google Scholar]
- 38.Pritchard MT, McMullen MR, Stavitsky AB, Cohen JI, Lin F, Medof ME, et al. Differential contributions of C3, C5, and decay-accelerating factor to ethanol-induced fatty liver in mice. Gastroenterology. 2007;132:1117–1126. doi: 10.1053/j.gastro.2007.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okada S, Nakamura M, Mikami Y, Shimazaki T, Mihara M, Ohsugi Y, et al. Blockade of interleukin-6 receptor suppresses reactive astrogliosis and ameliorates functional recovery in experimental spinal cord injury. J Neurosci Res. 2004;76:265–276. doi: 10.1002/jnr.20044. [DOI] [PubMed] [Google Scholar]
- 40.Izumi-Nagai K, Nagai N, Ozawa Y, Mihara M, Ohsugi Y, Kurihara T, et al. Interleukin-6 Receptor-Mediated Activation of Signal Transducer and Activator of Transcription-3 (STAT3) Promotes Choroidal Neovascularization. Am J Pathol. 2007;170:2149–2158. doi: 10.2353/ajpath.2007.061018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology. 2004;40:185–194. doi: 10.1002/hep.20283. [DOI] [PubMed] [Google Scholar]
- 42.Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–1374. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 43.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 44.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 45.Pfitzner E, Kliem S, Baus D, Litterst CM. The role of STATs in inflammation and inflammatory diseases. Curr Pharm Des. 2004;10:2839–2850. doi: 10.2174/1381612043383638. [DOI] [PubMed] [Google Scholar]
- 46.Sakamori R, Takehara T, Ohnishi C, Tatsumi T, Ohkawa K, Takeda K, et al. Signal transducer and activator of transcription 3 signaling within hepatocytes attenuates systemic inflammatory response and lethality in septic mice. Hepatology. 2007;46:1564–1573. doi: 10.1002/hep.21837. [DOI] [PubMed] [Google Scholar]
- 47.Kano A, Wolfgang MJ, Gao Q, Jacoby J, Chai GX, Hansen W, et al. Endothelial cells require STAT3 for protection against endotoxin-induced inflammation. J Exp Med. 2003;198:1517–1525. doi: 10.1084/jem.20030077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ishihara K, Hirano T. IL-6 in autoimmune disease and chronic inflammatory proliferative disease. Cytokine Growth Factor Rev. 2002;13:357–368. doi: 10.1016/s1359-6101(02)00027-8. [DOI] [PubMed] [Google Scholar]
- 49.Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 50.Shen X, Hong F, Nguyen VA, Gao B. IL-10 attenuates IFN-alpha-activated STAT1 in the liver: involvement of SOCS2 and SOCS3. FEBS Lett. 2000;480:132–136. doi: 10.1016/s0014-5793(00)01905-0. [DOI] [PubMed] [Google Scholar]
- 51.Starkel P, De Saeger C, Leclercq I, Strain A, Horsmans Y. Deficient Stat3 DNA-binding is associated with high Pias3 expression and a positive anti-apoptotic balance in human end-stage alcoholic and hepatitis C cirrhosis. J Hepatol. 2005;43:687–695. doi: 10.1016/j.jhep.2005.03.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.