

Abstract
Upregulation of CCR2 chemokine receptor expression by dorsal root ganglion (DRG) neurons is an important process in the development and maintenance of neuropathic pain. CCR2 is not expressed by DRG neurons under normal conditions but is upregulated in several animal models of neuropathic pain where its signaling is excitatory. However, the molecular mechanisms underlying neuronal upregulation of CCR2 have not been investigated. We examined the promoter region of the CCR2 gene and found that a binding site for the nuclear factor of activated T-cells (NFAT) was conserved among species. The NFAT element was functional since the CCR2 promoter was activated by a constitutively active form of calcineurin A, whereas a point mutation in the NFAT binding site abrogated it. Activation of the NFAT pathway in the DRG neuronal cell line F11 increased CCR2 promoter activity and induced CCR2 transcription. Moreover, depolarization of cultured DRG neurons induced de novo synthesis of CCR2 mRNA, which was blocked by the calcineurin inhibitors cyclosporin A and FK506. These data indicate that CCR2 is a target of the NFAT pathway and suggest that tonic excitation of DRG neurons in association with chronic pain may lead to neuronal CCR2 upregulation via activation of the NFAT pathway.
Keywords: Chemokine receptors, dorsal root ganglion, neuropathic pain, NFAT, calcineurin, FK506, cyclosporin A
Introduction
Long-term changes in the profile of gene transcription in dorsal root ganglion (DRG) cells and in the central nervous system (CNS) are believed to underlie the heightened pain sensitivity observed in neuropathic pain states. Many genes are differentially regulated by DRG neurons and glia in different animal models of chronic pain, suggesting multiple signaling pathways may contribute to this process (Basbaum and Woolf, 1999; Ji and Strichartz, 2004; Mogil et al., 2000). However, the mechanism by which such changes occur is poorly understood. Expression of the CCR2 chemokine receptor has been found to be upregulated by DRG neurons in association with several animal models of neuropathic pain (Bhangoo et al., 2006; White et al., 2005b). Under normal circumstances neither CCR2 nor its ligand MCP-1/CCL2 are expressed by DRG neurons, whereas their expression is upregulated under pathological conditions (Bhangoo et al., 2006; Sun et al., 2006; White et al., 2005b). Importantly, DRG neurons from animals exhibiting neuropathic pain are strongly excited by the application of MCP-1, whereas neurons from naïve animals are not (Sun et al., 2006; White et al., 2005b). Moreover, CCR2 knockout mice do not develop neuropathic pain following spared nerve injury (Abbadie et al., 2003), suggesting that upregulation of CCR2 is a crucial step in the generation and/or maintenance of neuropathic pain. However, the transcriptional regulation of CCR2 receptors in DRG neurons has not been examined. Consequently, we wished to define the signaling pathway responsible for the upregulation of these receptors in DRG neurons. In addition to its diverse roles in the development and function of the immune system, it has recently been shown that the nuclear factor of activated T-cells (NFAT) family of transcription factors can play an important role in mediating long-term changes in neuronal excitability (Graef et al., 1999). NFAT cannot normally enter the nucleus until it is dephosphorylated, but can be activated by a Ca2+-dependent phophatase calcineurin (CN). Thus, an increase in [Ca2+]i turns on the transcription of NFAT-dependent genes and this is believed to link transient neuronal excitation to a long-lasting transcriptional activation (Graef et al., 1999; Zanzouri et al., 2006).
Here we report that CCR2 is a target gene of the NFAT pathway in DRG neurons. We demonstrate that there is a functional and conserved NFAT binding element in the promoter region of the CCR2 gene. Activation of the NFAT pathway in DRG neurons and the DRG neuronal cell line F11 increased the CCR2 promoter activity and initiated CCR2 transcription. These data suggest that repetitive excitation of DRG neurons under pathological circumstances might turn on activity-dependent transcription of CCR2 via NFAT signaling. Hence, antagonism of the NFAT pathway may be a novel point for therapeutic intervention in treating neuropathic pain.
Results
1. The structure of the mouse CCR2 gene
We examined the genomic organization of the mouse CCR2 gene. It is composed of three exons with the entire protein coding region residing in the third exon (Fig 1A). The length of the 5’-untranslated region (5’-UTR) varies among the reported mRNA sequences (Fig 1A). The expected length of the first exon deduced from the longest reported sequence (AK046579) was 258 nt (Fig 1A). In order to see if this represents the transcription initiation site, we performed a primer extension experiment with a primer selected from 61 nt downstream of the putative initiation site (Fig 1B). RNA isolated from a mouse monocytic cell line WEHI265.1, from which the first mouse CCR2 cDNA was cloned, was used (Kurihara and Bravo, 1996). A specific band of the expected size was generated indicating that the transcription initiation site lies around the expected region. To confirm this, RT-PCR was performed using a combination of a reverse primer located in exon 3 and a forward primer located in exon 1 or in exon 2. As expected, specific bands were only generated from the cDNA of WEHI265.1 cells but not from F11 cells, a DRG neuronal cell line, using both combinations of primer sets (Kurihara and Bravo, 1996) (Fig 1C). However, a distinct band that was around 100 bp shorter than the expected size was also generated when the forward primer in exon 1 was used (Fig 1C: left diagrams). The two bands were cloned and their sequence identities were verified. It was found that there is a complete set of splice donor and acceptor sites within the exon 1, and the both mRNA species with the complete exon 1 and deleted exon 1 co-exist in WEHI265.1 cells (Fig 1D).
Figure 1. The genomic organization of the mouse CCR2 gene.

A) Reported mRNA species and their coverage on genomic DNA sequences (numbered boxes: exons; lines between exons: introns; a black box: protein coding region). B) Primer extension experiment. The location of the primer is denoted in the diagram on the left (W: WEHI265.1 monocytic cell line). C) RT-PCR experiments. Relative location of primers is denoted on the upper diagram (arrow heads: primers). Two sets of primers were used: E1up/E3dn and E2up/E3dn (left two and right two lanes, respectively). The structure of the PCR product is denoted in the left and right diagrams (W: WEHI265.1 monocytic cell line; F: F11 DRG neuronal cell line). D) The sequence of the exon 1 (E1). Exonic sequence is in upper cases and non-exonic sequence in lower cases. An additional intronic sequence within the exon 1 is underlined. AG and GT consensus splice dinucleotides are in bold letters. An mRNA with this sequence spliced out corresponds to the shorter band in the leftmost lane in C.
2. Cell type-specific activity of the CCR2 promoter
CCR2 is normally expressed by several cell types of the immune system including monocytes, B-cells, activated T-cells, NK cells, and immature dendritic cells. On the other hand, under normal conditions, DRG neurons do not express CCR2 receptors. However, CCR2 expression can be upregulated in DRG neurons under pathological conditions. Accordingly, the DRG neuronal cell line F11 did not express detectable levels of CCR2 mRNA, although this mRNA was highly expressed by WEHI265.1 cells (Fig 1C and Fig 2A: left panels). The normal expression of CCR2 mRNA in WEHI265.1 was due to active ongoing transcription since the half-life of CCR2 mRNA in these cells was very short (Fig 2A: right panels). In order to define the minimal promoter region responsible for cell type-specific CCR2 expression, a 1 kb fragment upstream of the transcription initiation site was cloned into a luciferase reporter vector, and the relative promoter activity was examined in these two cell lines. The activity of the CCR2 promoter recapitulated the transcriptional activity and it was maintained even with a 0.5 kb proximal fragment, suggesting that important regulatory cis-elements lie in the relatively short fragment of the promoter (Fig 2B).
Figure 2. Cloning of a functional CCR2 promoter.

A) Cell type-specific CCR2 transcriptional activity. CCR2 was expressed by the WEHI265.1 (W) monocytic cell line but not by the F11 DRG neuronal cell line (F) under basal condition (left panels). The half-life of CCR2 mRNA was examined in WEHI265.1 (right panels). After the addition of a transcriptional inhibitor actinomycin D (ActD: 1 µg/ml), the CCR2 mRNA level rapidly decreased. 28S rRNA was used as an internal control. B) Cell type specific CCR2 promoter activity. 1.0 kb and 0.5 kb fragments upstream of exon 1 (E1) were cloned into a luciferase reporter vector (Luc). Promoter activity was high in WEHI265.1 cells but very low in F11 cells (**p<0.01 and *p<0.05 between WEHI265.1 and F11, unpaired t-test).
3. A functional NFAT binding element in the CCR2 promoter
In order to define signaling pathways whose activation might lead to the upregulation of neuronal CCR2 receptors, putative transcription binding elements in the shorter fragment of the CCR2 promoter were sought using standard programs (MatInspector v6.0, Genomatix Software GmbH; MacVector, Accelrys). Among several sites, we located an NFAT binding element conserved between human and mouse in the proximity of the transcription initiation site (Fig 3A). To examine if this NFAT site is functional, we first performed an electrophoretic mobility shift assay (EMSA) using nuclear extracts from WEHI265.1 cells. A specific binding activity was observed (Fig 3B: filled arrowheads), which could be competed for by unlabeled self-competitors but not by mutated competitors (GGAAAA → GGTTTT). The presence of NFAT in these binding complexes was confirmed by ‘supershift’ assays. The formation of specific binding complexes (Fig 3B: filled arrowheads) but not nonspecific ones (Fig 3B: empty arrowheads) was inhibited by the addition of a pan-specific antibody to NFATc (which recognizes NFATc1 to c4) but not by an isotype-matched control antibody. Next, we co-transfected the CCR2 promoter with a constitutively active form of calcineurin A (a catalytic subunit) (caCnA) to examine if the CCR2 promoter could be activated by the NFAT pathway. CCR2 promoter activity was increased by caCnA, and this was blocked by a point mutation in the NFAT binding sequence, indicating that the NFAT site in the CCR2 promoter is functional (Fig 3C).
Figure 3. An NFAT element in the promoter.
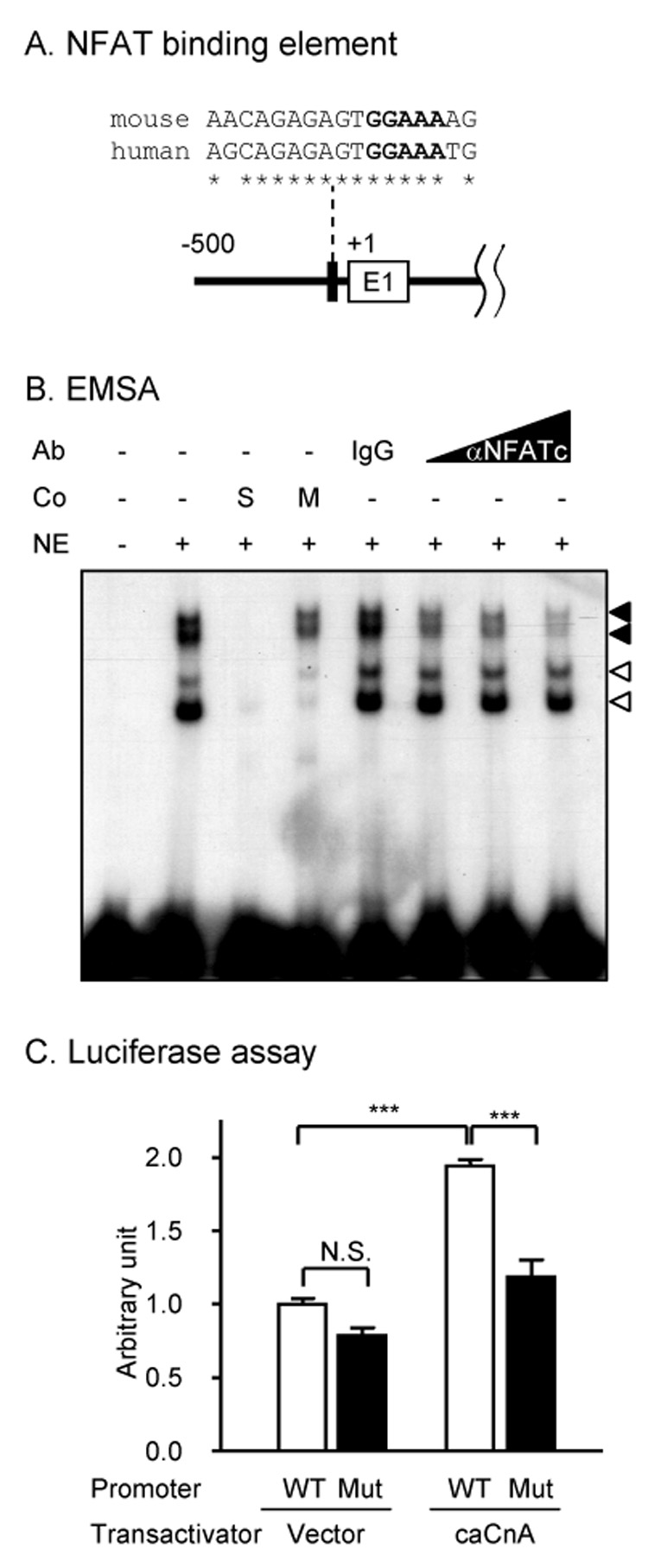
A) The NFAT binding element near the transcription initiation site is conserved (*) between mouse and human (E1: exon 1). The core binding sequences are in bold. B) A specific NFAT binding activity in the nuclear extract of WEHI265.1 cells (Co: cold competitors; S: self-competitors; M: GGAAA → GGTTT mutated competitors; NE: nuclear extract from WEH265.1 cells; a filled arrowhead: specific NFAT binding activity; an empty arrowhead: binding activity that is not specific to the core NFAT sequence; Ab: antibody; IgG: an isotype-matched control antibody; αNFATc: a pan-specific antibody to NFATc). C) The NFAT binding element in the CCR2 promoter is functional. The CCR2 promoter (WT) or the promoter with a point mutation in the NFAT binding site (Mut: GGAAA → GGTTT) was co-transfected with a constitutively active form of calcineurin A (caCnA) or an empty vector (***p<0.001; N.S.: not significant, a post hoc Bonferroni’s multiple comparison test following one-way ANOVA).
4. Involvement of the NFAT pathway in excitation-transcription coupling in DRG neurons
We used the DRG neuronal cell line F11 as a model for DRG neurons. F11 cells retain several features of mature sensory neurons including the expression of voltage-sensitive Ca2+ channels (VSCCs) and Substance P-like peptides (Francel et al., 1987), opioid receptors (Fan et al., 1992), and transient receptor potential (TRP) channels (Jahnel et al., 2003). They also express mRNA for calcitonin gene-related peptide (CGRP) and preprotachykinin (PPT; a precursor for Substance P) (supplementary figure 1). We sought to examine if NFAT activation in DRG neurons or F11 cells leads to upregulation of CCR2. First, we examined whether DRG neurons and F11 cells express components of the NFAT pathway by RT-PCR. CnA and all isoforms of NFAT were expressed in both DRG and F11 neurons (Fig 4A). Then, we asked whether NFAT activation increases CCR2 promoter activity in F11 cells. F11 cells transfected with a CCR2 promoter-luciferase construct were treated with the Ca2+ ionophore ionomycin (IM). IM treatment increased CCR2 promoter activity which was blocked by a CN inhibitor cyclosporin A (CsA), indicating that the IM-mediated increase in CCR2 promoter activity was due to NFAT activation (Fig 4B). Next, we examined if IM treatment could also increase CCR2 mRNA. IM treatment increased CCR2 mRNA (Fig 5A), and this was due to de novo synthesis because the increase was completely blocked by the transcriptional inhibitor actinomycin D (ActD). Moreover, it was mediated by activation of the NFAT pathway since it was inhibited by CsA (Fig 5A). We reasoned that if the NFAT pathway is involved in the inducible regulation of CCR2 gene expression, it may also participate in the normal constitutive cell type-specific expression of CCR2. Accordingly, CCR2 mRNA levels were significantly decreased in WEHI265.1 by FK506 or CsA.
Figure 4. The components of the NFAT pathway in DRG neurons and F11 cells.
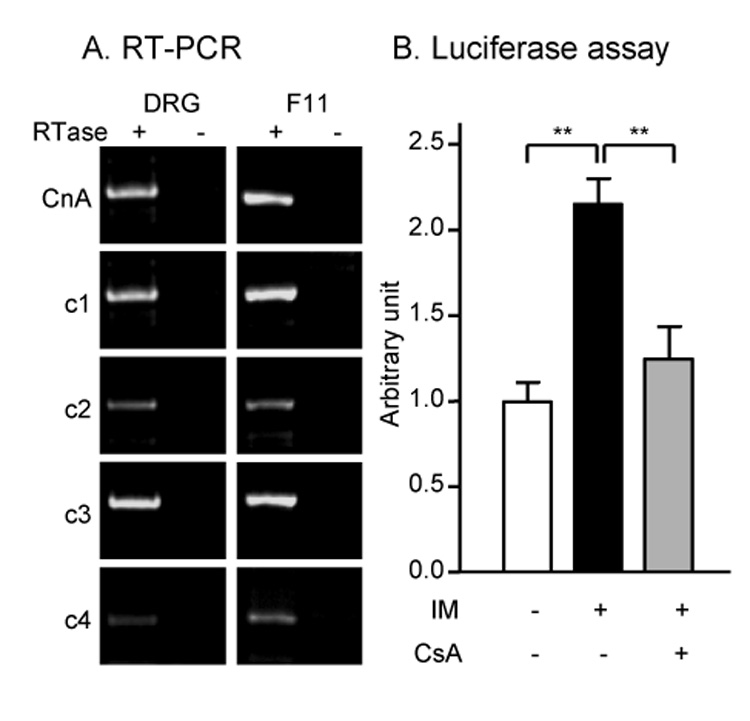
A) Calcineurin A (CnA) and NFATc1-4 were expressed by cultured DRG neurons and F11 DRG neurons (RTase: reverse transcriptase). RTase-negative control reaction was performed for each group. B) CCR2 promoter is activated by calcium ionophore ionomycin (IM: 1 µM) in F11 DRG neurons in a calcineurin inhibitor cyclosporin A (CsA: 1 µM)-sensitive way (**p<0.01, a post hoc Bonferroni’s multiple comparison test following one-way ANOVA).
Figure 5. NFAT-dependent transcription of CCR2 in F11 and WEHI265.1 cells.

A) NFAT-dependent de novo transcription of CCR2 in F11 DRG neurons. Ionomycin (IM: 1 µM) treatment induced CCR2 mRNA which was blocked by a transcriptional inhibitor actinomycin D (ActD: 1 µg/ml). The induction was significantly inhibited by a calcineurin inhibitor cyclosporin A (CsA: 1 µM). 28S rRNA was used as an internal control. B) NFAT-dependent transcription contributes to cell type-specific CCR2 expression. Calcineurin inhibitors CsA or FK506 (0.2 µM each) decreased baseline levels of CCR2 in WEHI265.1 monocytic cell line. GAPDH was used as an internal control.
Next, we asked if activation of NFAT could lead to the CCR2 upregulation in DRG neurons. We developed a semi-acute DRG culture system to measure mRNA selectively from neurons while minimizing contamination from non-neuronal cells (supplementary figure 2A). Dissociated DRG cells were allowed to attach on an uncoated tissue culture dish for several hours and then neurons were depolarized using a medium containing high concentration of potassium (high K). Twenty four hours later, neurons which are loosely attached to dishes were dislodged by gentle pipetting, total RNA was isolated, and RT-PCR was performed to measure CCR2 mRNA. Neurons incubated in high K media had an elevated level of basal [Ca2+]i due to activation of voltage-dependent Ca2+ channels (supplementary figure 2B). In the unstimulated control group, where neurons were incubated in a medium containing a low concentration of potassium while overall osmorality was maintained, CCR2 mRNA was undetectable (Fig 6A). However, high K stimulation increased CCR2 mRNA which was completely blocked by CsA/FK506, indicating that a depolarization-induced increase in CCR2 mRNA was mediated by activation of the NFAT pathway (Fig 6A and B).
Figure 6. Depolarization-induced transcription of CCR2 in DRG neurons.

A) NFAT-dependent transcription of CCR2 is induced in DRG neurons by depolarization. Acutely isolated DRG neurons were depolarized by high external potassium (43K: 43 mM) for 24 hr with or without calcineurin inhibitors (CsA/FK506: 0.2 µM each), which were added 30 min prior to depolarization. Control culture medium contained 3 mM of potassium (3K). Osmorality of different media was identical. Total RNA was isolated from neurons (see supplementary figure 1) and subjected to RT-PCR. B) Real-time quantitative RT-PCR was used to quantify mRNA levels of different chemokine receptors in conditions described in A (n=3–5 independent DRG cultures) (**p<0.01, *p<0.05 vs. 3K and ††p<0.01, †p<0.05 vs. 43K, a post hoc Bonferroni’s multiple comparison test following one-way ANOVA).
Expression of several other chemokine receptors has also been closely associated with the genesis of neuropathic pain (Bhangoo et al., 2006; Bhangoo et al., 2007). We therefore examined whether additional chemokine receptors are targets of NFAT activation in DRG neurons (Fig 6B). Among several chemokine receptors we examined (CCR1, CCR2, CCR5, CXCR4 and CX3CR1), expression of CCR2 and CCR5 was induced by high K depolarization in a CN inhibitor-sensitive manner (Fig 6B), suggesting that a group of chemokine receptor genes could be induced synchronously using the same mechanism. CXCR4 expression, however, was increased by CN inhibitors indicating that the same pathway might have opposite effects on different genes. Expression of CCR1 and CX3CR1 remained unchanged. Overall, these data suggest that the transcription of different groups of chemokine receptors is differentially regulated at the transcriptional level in DRG neurons.
Discussion
Sustained pain hypersensitivity and neuronal hyperexcitability associated with neuropathic pain is believed to result from long-term changes in gene expression in DRG and central dorsal horn neurons. Many genes have been identified whose expression changes in association with the development of neuropathic pain using several animal models, although the molecular mechanisms responsible for such changes are largely unknown (Mogil et al., 2000). In particular, the CCR2 chemokine receptor appears to play an important role in the genesis of neuronal hyperexcitability in states of chronic pain (Abbadie et al., 2003; White et al., 2005a; White et al., 2005b). This receptor is not expressed by DRG neurons under normal circumstances but its expression is induced under pathological conditions. Accordingly, CCR2 expression was undetectable in cultured DRG neurons or F11 DRG neuronal cells. By examining the promoter sequence of CCR2, we found that its expression in sensory neurons can be induced by activation of the NFAT pathway.
Transcriptional regulation of chemokine receptors in neurons has not been extensively studied, since most research on this topic has been focused on immune cells. Previous reports using immune cells have suggested that tissue specific expression of CCR2 was attributable to differential Oct1 and C/EBP activities (Yamamoto et al., 1999). In the mouse promoter, Oct1 and C/EBP binding sites are also conserved. In accordance with this, we have observed that co-transfection of C/EBPβ could also activate the CCR2 promoter, suggesting that multiple transcriptional pathways may regulate CCR2 gene expression under different circumstances (Jung and Miller, unpublished data). In addition to these transcription factors, constitutive activity of NFAT signaling may also contribute to tissue specific expression, since inhibition of the NFAT pathway by FK506 and CsA decreased constitutive levels of CCR2 mRNA in WEHI265.1 monocyte cells (Fig 5B).
NFAT is a particularly pertinent candidate pathway for regulating activation of genes responsible for neuropathic pain, because it can be activated by neuronal depolarization in a Ca2+-dependent manner (Graef et al., 1999). It was recently shown that repetitive action potentials in sympathetic neurons resulted in nuclear translocation of NFAT (Hernandez-Ochoa et al., 2007). Since the ectopic discharge of action potentials in DRG neurons is a typical feature of neuropathic pain (Basbaum and Woolf, 1999), tonic excitation of DRG neurons under pathological conditions might induce the activity and Ca2+-dependent transcription of NFAT target genes, including CCR2 receptors. Because CCR2 signaling in DRG neurons is itself excitatory (Sun et al., 2006; White et al., 2005b), the net effect would be amplification of hyperexcitability and pain. It is interesting to note that other forms of chemokine signaling are also upregulated in the DRG in association with neuropathic pain including CCR5 and CXCR4 receptors and their ligands (Bhangoo et al., 2006; Bhangoo et al., 2007). As we have observed (Fig 6B), transcription of several chemokine receptor genes in DRG neurons appears to involve diverse signaling pathways. For example, whereas upregulation of CCR2 and CCR5 in response to depolarization appears to proceed in an NFAT-dependent manner, CXCR4 was upregulated by inhibition of the NFAT pathway. As the promoters for CCR2 and these other chemokine receptors contain binding sites for multiple transcription factors it is likely that they can be regulated by different signaling pathways under different circumstances- in addition to depolarization. For example, chemokine receptor upregulation in response to inflammatory cytokines such as TNF-α may also play a role in reshaping the gene expression profile of DRG neurons in states of pain hypersensitivity and may function through pathways in addition to NFAT (White et al., 2005a). Interestingly, we have demonstrated that in ganglia from animals exhibiting nociceptive pain behavior, both CCR2 and its ligand MCP-1/CCL2 are upregulated by the same neurons (Jung et al., in revision). As we have shown here CCR2 can be upregulated by depolarization in a Ca2+/NFAT-dependent manner, but this is not the case for MCP-1 upregulation which does not respond to these stimuli. On the other hand MCP-1 can be upregulated in a TNF-α/NF-kB-dependent manner (Supplementary figure 3). Thus, the entire panoply of important gene transcriptional changes occurring in the DRG neurons in association with states of chronic pain presumably result from a spectrum of transcriptional events.
It is known that NFAT-mediated transcription plays a role in neuronal development and synaptic plasticity. With respect to DRG neurons in particular, Thayer and colleagues reported that bradykinin, a pronociceptive peptide secreted from the site of injury, activates NFAT signaling in DRG neurons resulting in NFAT-dependent transcription of COX2 (Jackson et al., 2007). Seybold and colleagues reported that NGF and BDNF, pronociceptive neurotrophins, also activate NFAT-dependent transcription (Groth et al., 2007). It is likely that the NFAT pathway might not only be activated in the development phase of neuropathic pain as a result of the action of multiple proinflammatory molecules, but also in the maintenance phase where activity and Ca2+-dependent transcription takes place. Activation of diverse transcriptional pathways, including NFAT might turn on the transcription of a cohort of genes responsible for sustained hyperexcitability of DRG neurons in neuropathic pain. Therefore, the NFAT pathway may represent a novel point for therapeutic intervention in the treatment of neuropathic pain. Because inhibition of transcriptional pathways such as NFAT may regulate expression of several target genes, its inhibition may be more efficacious than individual receptor antagonists in long-term reversal of chronic pain hypersensitivity.
Experimental Methods
Plasmid constructions and materials
The fragments of 5’-flanking region of CCR2 was amplified from mouse genomic DNA (from a CD-1 mouse) by polymerase chain reaction (PCR) and cloned into pGL3-Basic (Promega). A point mutation in the NFAT binding site was introduced using QuickChange site-directed mutagenesis kit (Stratagene). The constitutively active calcineurin A construct (caCnA) was a kind gift from Dr. Neil A. Clipstone (Loyola University Chicago, Maywood, IL) (Clipstone et al., 1994). All PCR products were cloned into pCR2.1 vector (Invitrogen) and sequence identities were confirmed by dideoxy-sequencing methods. Primers used in cloning are listed in Table 1.
Table 1.
| Primers for promoter cloning | |
| 1.0kb forward | 5′-TTGGTACCTGCTTAGCTAGCATGTGT-3′ |
| 0.5kb forward | 5′-TTGGTACCAACTCACACAAAGGGATC-3′ |
| reverse | 5′-AACTCGAGAATTTTGAAATCTTGCTTATGC-3′ |
| mutNFAT forward | 5′-GAAGAGAACAGAGAGTGGTTTAGCTATGAAAGACTTA-3′ |
| mutNFAT reverse | 5′-TAAGTCTTTCATAGCTAAACCACTCTCTGTTCTCTTC-3′ |
| Primers for RT-PCR | |
| CCR2 exon1 forward | 5′-AATCCTGTAAAGACCTCAGCC-3′ |
| CCR2 exon2 forward | 5′-TGCAAAGACCAGAAGAGG-3′ |
| CCR2 exon3 reverse | 5′-CAGGATCCAAGCTCCAAT-3′ |
| NFATc1 forward | 5′-CAACGCCCTGACCACCGATAG-3′ |
| NFATc1 reverse | 5′-GGCTGCCTTCCGTCTCATAGT-3′ |
| NFATc2 forward | 5′-TGGCCCGCCACATCTACCCT-3′ |
| NFATc2 reverse | 5′-TGGTAGAAGGCGTGCGGCTT-3′ |
| NFATc3 forward | 5′-TGGATCTCAGTATCCTTTAA-3′ |
| NFATc3 reverse | 5′-CACACGAAATACAAGTCGGA-3′ |
| NFATc4 forward | 5′-CATTGGCACTGCAGATGAG-3′ |
| NFATc4 reverse | 5′-CGTAGCTCAATGTCTGAAT-3′ |
| CnA forward | 5′-CAGGGTGGTGAAAGCCGTTC-3′ |
| CnA reverse | 5′-GGATGTCCCCACAAACTGTG-3′ |
| MCP-1 forward | 5′-ATGCAGGTCCCTGTCATGCTT-3′ |
| MCP-1 reverse | 5′-GTTCACTGTCACACTGGTCA-3′ |
| PPT-A forward | 5′-TGCCAACGATGATCTAAATT-3′ |
| PPT-A reverse | 5′-CTTCTTTCGTAGTTCTGCAT-3′ |
| CGRP forward | 5′-AGATCCTGCAACACTGCCAC-3′ |
| CGRP reverse | 5′-CCACATTGGTGGGCACAAAGTTG-3′ |
| GAPDH forward | 5′-TTGACCTCAACTACATGG-3′ |
| GAPDH reverse | 5′-ATTGAGAGCAATGCCAGC-3′ |
| Taqman gene expression assays for qRT-PCR | |
| CCR1 | Mm00438260_s1 |
| CCR2 | Mm00438270_m1 |
| CCR5 | m01216171_m1 |
| CXCR4 | m99999055_m1 |
| CX3CR1 | m00438354_m1 |
| GAPDH | m99999915_g1 |
| Primers for the probes used in Northern blot | |
| CCR2 3′UTR forward | 5′-GCAAGAGGTCTCGGTTGGGTTG-3′ |
| CCR2 3′UTR reverse | 5′-TCCTCCTTCTCACTCAGTCCTG-3′ |
| Primer for the primer extension | |
| CCR2 exon1 reverse | 5′-TCTACAGTTCTTCTTTTCCAGCCAGG-3′ |
| Oligomers for EMSA | |
| NFAT forward | 5′-CTAGAGAGAGTGGAAAAGCTAT-3′ |
| NFAT reverse | 5′-CTAGATAGCTTTTCCACTCTCT-3′ |
| Mutant NFAT forward | 5′-CTAGAGAGAGTGGTTTAGCTAT-3′ |
| Mutant NFAT reverse | 5′-CTAGATAGCTAAACCACTCTCT-3′ |
Cell culture
F11 rat DRG neuron X mouse neuroblastoma hybrid cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 0.5% penicillin-streptomycin (P/S) at 37°C under 5% CO2. WEHI265.1 mouse monocytic cells were grown in the same condition except that the medium was further supplemented with 0.05 mM 2-mercaptoethanol. DRG neurons were isolated as described (Oh et al., 2001).
Northern blot analysis
Total RNA was extracted with Trizol reagent (Life Technologies). RNA was dissolved in deionized water and denatured in 50% formamide, 6.2% formaldehyde, 20mM MOPS (3-[N-morpholino]propanesulfonic acid), 5 mM sodium acetate, and 1 mM EDTA at 60 °C for 5 min. Electrophoresis was performed at 100V for 1.5 hr in a submarine 1.2% formalehyde agarose gel. RNA was transferred to Nytran filter (Amersham) for 18 hr by capillary transfer. CCR2 and GAPDH probes labeled with 32P-dCTP (Amersham) were prepared by random labeling of mouse CCR2 cDNA 397 bp fragment and mouse GAPDH cDNA 800 bp fragment cloned into pGEM-Teasy (Promega). Hybridization procedures and rehybridization with GAPDH cDNA were performed using ExpressHyb hybridization solution (BD biosciences) according to the manufacturer’s instructions. Primers used are listed in Table 1.
Reverse transcription-PCR (RT-PCR)
RNA samples (1 µg) were applied to the reverse transcription (RT) reaction containing 100 U of Superscript II, 0.5 µg of oligo(dT) primers, 20 U of RNase inhibitor, and 1 mM dNTPs in a buffer supplied by the manufacturer (Invitrogen). The RT reaction was carried out at 42°C for 50 min followed by inactivation at 70°C for 15 min, after which RNase H was added and the reaction mixture was incubated at 37°C for 30 min. Subsequently, 1/50 of reaction product was added to 20 µl PCR reaction mixture containing 2.5 U of Taq DNA polymerase (Qiagen) in a buffer supplied by the manufacturer. Thirty-two to forty-two cycles of PCR amplification were carried out with the condition of denaturation at 95°C from 1 min, primer annealing at 55–60°C for 1 min, and primer extension at 72°C for 40 sec. Five µl aliquots of PCR products were electrophoresed on 1.2 – 2.0 % agarose gel in Tris-acetate-EDTA buffer, stained with ethidium bromide, and photographed under UV illumination. For quantitative PCR, real-time PCR was performed using Taqman probe-based chemistry on a sequence detector (7900HT, Applied Biosystems). Taqman gene expression assays containing two primers and one Taqman probe for each gene were purchased from Applied Biosystems (listed in Table 1). The most assays (CCR2, CCR5, CXCR4, and CX3CR1) were designed to span an exon-exon boundary to exclude false positive amplification of contaminated genomic DNA. Also, experiments with the RTase-negative controls confirmed negligible levels of genomic DNA contamination in cDNA samples (ΔCt ≈ 10). Relative quantification analyses were performed by a comparative Ct method using GAPDH as an internal control.
Primer extension
Twenty pmole of primer (Table 1) was end-labeled with 32P-γ-dATP using T4 polynucleotide kinase (PNK). One hundred fmole of primer was hybridized to 10 µg of total RNA isolated from WEHI265.1 cells by incubating the sample at 58 °C for 20 min and slowly cooling to 10 °C, after which standard RT reaction mixture was added. Reaction products were resolved in a 5% polyacrylamide gel in denaturing condition (7M urea and 1X Tris-borate-EDTA buffer).
Electrophoretic mobility shift assay (EMSA)
Four pmole of annealed oligonucleotides were labeled with 20 µCi of [α-32P] dCTP (Amersham) using Klenow fragment (Promega). Labeled probes were diluted to 40 fmole/µl. A total of 40 fmole of 32P-probe was incubated with 2–5 µg of nuclear extracts, which were prepared using a commercial kit (Nuclear/Cytosol Fractionation Kit, BioVision, Mountain View, CA), in gel shift assay buffer (20mM HEPES, pH 7.6, 0.1 mM KCl, 10% Glycerol, 0.1% Nonidet P-40, 2 mM DTT) containing 0.5 µg of polydeoxyinosinic-deoxycytidylic acid in a 20-µl reaction. Binding reactions were performed on ice for 20 min and at 30 °C for 30 min. The reaction mixture was run on a 4 % native polyacrylamide (37.5:1) gel in 0.5x TGE (25 mM Trizma base, 20 mM Glycine, 1 mM EDTA) to resolve protein-DNA complexes. For competition assays, a 100-fold molar excess of unlabeled double-stranded competitor DNA was added. For supershift assays, a pan-specific NFATc antibody which recognizes NFATc1 to c4 (sc-1149X) and an isotype-matched control antibody (Santa Cruz Biotechnology, Santa Cruz, CA) were added at the beginning of the incubation at 30 °C. Oligomers used are listed in Table 1.
Transient transfection and luciferase assay
Luciferase assay was performed to measure CCR2 promoter activities. DNA was transfected using TranIT-LT1 (Mirus, WI) according to the manufacturer’s instructions. One hundred ng of CCR2 promoter construct was co-transfected with 900 ng of transactivator construct or empty vector along with 10 ng of pRL-TK, which served as an internal control. Renilla and firefly luciferase assays were performed in 24–48 hr after the transfection using commercial enzyme assay kits (Dual-Luciferase Reporter Assay System, Promega).
Supplementary Material
Supplementary figure 1. F11 cells express the neuropeptides of DRG neurons. The expression of CGRP and PPT-A, known neuropeptides expressed by DRG neurons, was examined in F11 DRG neurons by RT-PCR (RTase: reverse transcriptase). RTase-negative control reactions were performed for each group. F11 neurons expressed both CGRP and PPT-A.
Supplementary figure 2. Semi-acute culture of DRG neurons. A) DRG isolated from 5–6 CD1 pups (postnatal day 3–5) were dissociated and plated on an uncoated and tissue culture treated dish for 2 hr in F12 medium supplemented with 0.5% FBS, 1% N2, 50 ng/ml NGF and 0.5% P/S. After cell attached, 0.25 volume of 200 mM KCl or 200 mM NaCl solution was added to culture media (final concentrations: for high K medium, 101 mM NaCl and 43 mM KCl; for control medium, 141 mM NaCl and 3 mM KCl). Calcineurin inhibitors (0.2 µM each) were added 30 min prior to the addition of the salt solutions. After 24 hr, neurons which are loosely attached to the dish (the upper panel: asterisk) were dislodged by gentle pipetting (the lower panel). Isolated neurons were subjected to RT-PCR. B) High K culture increased baseline levels of [Ca2+]i. After 24 hr culture in high potassium (43K) or control (3K) media, intracellular calcium level was examined by Fura-2 based Ca2+ imaging as previously described (Bhangoo et al., 2007) (***p<0.001 vs. 3K, unpaired t-test).
Supplementary figure 3. Differential regulation of MCP-1 and CCR2 in F11 DRG neurons. F11 DRG neurons were stimulated with various chemicals (TNF: 20 ng/ml TNF-α; I: 30 nM NF-kB activation inhibitor; DMOG: 1 nM dimethyloxalylglycine; CoCl2: 1 nM CoCl2; BSO: 200 µM buthionine-L-sulfoximine; IM: 1 µM ionomycin), and RT-PCR was performed to measure MCP-1 and CCR2 mRNA levels. Note that IM treatment only induced CCR2 expression and TNF-α only MCP-1. TNF-α-mediated MCP-1 induction was completely blocked by the NF-kB activation inhibitor.
Acknowledgements
This work was supported by National Institutes of Health Grants DA013141, NS043095, and MH040165 to RJM. The authors would like to thank Dr. Fletcher A. White (Loyola University Chicago, Maywood, IL) for his advice when carrying out these studies and with preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci U S A. 2003;100:7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Woolf CJ. Pain. Curr Biol. 1999;9:R429–R431. doi: 10.1016/s0960-9822(99)80273-5. [DOI] [PubMed] [Google Scholar]
- Bhangoo SK, Jung H, Chan DM, Ripsch M, Miller RJ, White FA. Neuroscience Meeting Planner. Atlanta, GA: Society for Neuroscience, 2006; 2006. Peripheral demyelination injury induces upregulation of chemokine/receptor expression and neuronal signaling in a model of neuropathic pain. Online. 250.3. [Google Scholar]
- Bhangoo SK, Ren D, Miller RJ, Chan DM, Ripsch MS, Weiss C, McGinni C, White FA. CXCR4 chemokine receptor signaling mediates pain hypersensitivity in association with antiretroviral toxic neuropathy. Brain Behav Immun. 2007;21:581–591. doi: 10.1016/j.bbi.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clipstone NA, Fiorentino DF, Crabtree GR. Molecular analysis of the interaction of calcineurin with drug-immunophilin complexes. J Biol Chem. 1994;269:26431–26437. [PubMed] [Google Scholar]
- Fan SF, Shen KF, Scheideler MA, Crain SM. F11 neuroblastoma x DRG neuron hybrid cells express inhibitory mu- and delta-opioid receptors which increase voltage-dependent K+ currents upon activation. Brain Res. 1992;590:329–333. doi: 10.1016/0006-8993(92)91116-v. [DOI] [PubMed] [Google Scholar]
- Francel PC, Harris K, Smith M, Fishman MC, Dawson G, Miller RJ. Neurochemical characteristics of a novel dorsal root ganglion X neuroblastoma hybrid cell line, F-11. J Neurochem. 1987;48:1624–1631. doi: 10.1111/j.1471-4159.1987.tb05711.x. [DOI] [PubMed] [Google Scholar]
- Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, Crabtree GR. L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature. 1999;401:703–708. doi: 10.1038/44378. [DOI] [PubMed] [Google Scholar]
- Groth RD, Coicou LG, Mermelstein PG, Seybold VS. Neurotrophin activation of NFAT-dependent transcription contributes to the regulation of pro-nociceptive genes. J Neurochem. 2007 doi: 10.1111/j.1471-4159.2007.04632.x. [DOI] [PubMed] [Google Scholar]
- Hernandez-Ochoa EO, Contreras M, Cseresnyes Z, Schneider MF. Ca(2+) signal summation and NFATc1 nuclear translocation in sympathetic ganglion neurons during repetitive action potentials. Cell Calcium. 2007;41:559–571. doi: 10.1016/j.ceca.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson JG, Usachev YM, Thayer SA. Bradykinin-Induced NFAT-Dependent Transcription in Rat Dorsal Root Ganglion Neurons. Mol Pharmacol. 2007 doi: 10.1124/mol.107.035048. [DOI] [PubMed] [Google Scholar]
- Jahnel R, Bender O, Munter LM, Dreger M, Gillen C, Hucho F. Dual expression of mouse and rat VRL-1 in the dorsal root ganglion derived cell line F-11 and biochemical analysis of VRL-1 after heterologous expression. Eur J Biochem. 2003;270:4264–4271. doi: 10.1046/j.1432-1033.2003.03811.x. [DOI] [PubMed] [Google Scholar]
- Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Sci STKE 2004. 2004:reE14. doi: 10.1126/stke.2522004re14. [DOI] [PubMed] [Google Scholar]
- Jung H, Toth PT, White FA, Miller RJ. Monocyte chemoattractant protein-1 functions as a neuromodulator in the dorsal root ganglia neurons. J Neurochem. doi: 10.1111/j.1471-4159.2007.04969.x. in revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara T, Bravo R. Cloning and functional expression of mCCR2, a murine receptor for the C-C chemokines JE and FIC. J Biol Chem. 1996;271:11603–11607. doi: 10.1074/jbc.271.20.11603. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Yu L, Basbaum AI. Pain genes?: natural variation and transgenic mutants. Annu Rev Neurosci. 2000;23:777–811. doi: 10.1146/annurev.neuro.23.1.777. [DOI] [PubMed] [Google Scholar]
- Oh SB, Tran PB, Gillard SE, Hurley RW, Hammond DL, Miller RJ. Chemokines and glycoprotein120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J Neurosci. 2001;21:5027–5035. doi: 10.1523/JNEUROSCI.21-14-05027.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JH, Yang B, Donnelly DF, Ma C, LaMotte RH. MCP-1 enhances excitability of nociceptive neurons in chronically compressed dorsal root ganglia. J Neurophysiol. 2006;96:2189–2199. doi: 10.1152/jn.00222.2006. [DOI] [PubMed] [Google Scholar]
- White FA, Bhangoo SK, Miller RJ. Chemokines: integrators of pain and inflammation. Nat Rev Drug Discov. 2005a;4:834–844. doi: 10.1038/nrd1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Sun J, Waters SM, Ma C, Ren D, Ripsch M, Steflik J, Cortright DN, Lamotte RH, Miller RJ. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc Natl Acad Sci U S A. 2005b;102:14092–14097. doi: 10.1073/pnas.0503496102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Takeshima H, Hamada K, Nakao M, Kino T, Nishi T, Kochi M, Kuratsu J, Yoshimura T, Ushio Y. Cloning and functional characterization of the 5′-flanking region of the human monocyte chemoattractant protein-1 receptor (CCR2) gene. Essential role of 5′-untranslated region in tissue-specific expression. J Biol Chem. 1999;274:4646–4654. doi: 10.1074/jbc.274.8.4646. [DOI] [PubMed] [Google Scholar]
- Zanzouri M, Lauritzen I, Duprat F, Mazzuca M, Lesage F, Lazdunski M, Patel A. Membrane potential-regulated transcription of the resting K+ conductance TASK-3 via the calcineurin pathway. J Biol Chem. 2006;281:28910–28918. doi: 10.1074/jbc.M606092200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1. F11 cells express the neuropeptides of DRG neurons. The expression of CGRP and PPT-A, known neuropeptides expressed by DRG neurons, was examined in F11 DRG neurons by RT-PCR (RTase: reverse transcriptase). RTase-negative control reactions were performed for each group. F11 neurons expressed both CGRP and PPT-A.
Supplementary figure 2. Semi-acute culture of DRG neurons. A) DRG isolated from 5–6 CD1 pups (postnatal day 3–5) were dissociated and plated on an uncoated and tissue culture treated dish for 2 hr in F12 medium supplemented with 0.5% FBS, 1% N2, 50 ng/ml NGF and 0.5% P/S. After cell attached, 0.25 volume of 200 mM KCl or 200 mM NaCl solution was added to culture media (final concentrations: for high K medium, 101 mM NaCl and 43 mM KCl; for control medium, 141 mM NaCl and 3 mM KCl). Calcineurin inhibitors (0.2 µM each) were added 30 min prior to the addition of the salt solutions. After 24 hr, neurons which are loosely attached to the dish (the upper panel: asterisk) were dislodged by gentle pipetting (the lower panel). Isolated neurons were subjected to RT-PCR. B) High K culture increased baseline levels of [Ca2+]i. After 24 hr culture in high potassium (43K) or control (3K) media, intracellular calcium level was examined by Fura-2 based Ca2+ imaging as previously described (Bhangoo et al., 2007) (***p<0.001 vs. 3K, unpaired t-test).
Supplementary figure 3. Differential regulation of MCP-1 and CCR2 in F11 DRG neurons. F11 DRG neurons were stimulated with various chemicals (TNF: 20 ng/ml TNF-α; I: 30 nM NF-kB activation inhibitor; DMOG: 1 nM dimethyloxalylglycine; CoCl2: 1 nM CoCl2; BSO: 200 µM buthionine-L-sulfoximine; IM: 1 µM ionomycin), and RT-PCR was performed to measure MCP-1 and CCR2 mRNA levels. Note that IM treatment only induced CCR2 expression and TNF-α only MCP-1. TNF-α-mediated MCP-1 induction was completely blocked by the NF-kB activation inhibitor.