

Abstract
The basolateral nucleus of the amygdala (BLA) receives cholinergic innervation from the basal forebrain and nicotine, via activation of neuronal nicotinic acetylcholine receptors (nAChRs), can improve performance in amygdala-based learning tasks. We tested the hypothesis that acute and prenatal nicotine exposure modulates cortico-amygdala synaptic transmission. We found, that low dose, single trial exposures to nicotine can elicit lasting facilitation, the extent of which is dependent on the level of stimulation of the cortical inputs to the BLA. In addition, sustained facilitation is ablated by prenatal exposure to nicotine.
This study examined synaptic transmission in 238 patch clamp recordings from BLA neurons in acute slice from mouse brain. Pharmacological studies in wild type and nAChR subunit knock-out mice reveal that activation of presynaptic α7 containing (α7*) and non α7* nAChRs facilitates glutamatergic transmission in an activity-dependent manner. Without prior stimulation, application of nicotine elicits modest and transient facilitation of glutamatergic postsynaptic currents (PSCs) in ∼40% BLA neurons. With low frequency stimulation of cortical inputs nicotine elicits robust facilitation of transmission at ∼60% of cortico-BLA synapses and synaptic strength remains elevated at ∼40% of these connections for more than 15 minutes after nicotine washout. Following paired pulse stimulation nicotine elicits long-lasting facilitation of glutamatergic transmission at ∼70% of cortico-BLA connections. Nicotine reduces the threshold for activation of LTP of cortico-BLA synapses evoked by patterned stimulation. Prenatal exposure to nicotine reduced subsequent modulatory responses to acute nicotine application.
INTRODUCTION
Central cholinergic systems have been implicated in the modulation of a wide variety of behavioral functions including attention, anxiety, memory and learning (Levin 2002). In the CNS, cholinergic projections are diffuse, and nicotinic acetylcholine receptor (nAChR)-expressing target neurons are peppered through cortical, limbic, and central autonomic structures (Woolf 1991). Despite the widespread distribution of cholinoceptive sites, examples of direct nAChR-mediated fast synaptic transmission in the mammalian brain are relatively rare, possibly reflecting participation in functions other than the mediation of direct synaptic transmission per se(but see (Alkondon and Albuquerque 2001; 2002; Klein and Yakel 2006). It has been proposed that ACh, released via “volume transmission”, may act as a modulator of circuit excitability by interacting with pre-, post- and peri-synaptic nAChRs (Berg et al. 2006; Giocomo and Hasselmo 2005; Rousseau et al. 2005; Wonnacott et al. 2006; Zhang and Berg 2007). Indeed, numerous reports have documented the effects of presynaptic nAChR activation on the release of a variety of transmitters, including glutamate (Jo et al. 2005; Turner 2004; Wonnacott 1997; Zhang and Berg 2007).
The excitability of cortico-amygdala circuits, renowned for their role in consolidation of memories related to events of emotional salience, is modulated by cholinergic tone (Addy et al. 2003; Barros et al. 2005). In particular, the basolateral amygdala receives dense cholinergic inputs from the horizontal limb of the diagonal band nucleus and the ventral pallidum (Wainer et al. 1993; Woolf 1991). Vesicular acetylcholine transporter (VAChT) stained terminals are distributed throughout the amygdala with particularly high densities in the basal and lateral amygdala nuclei (Schafer et al. 1998).
Both α7* and β2* nAChRs are expressed in the basal and lateral amygdala nuclei (Hill et al. 1993; Seguela et al. 1993). In rat basolateral amygdala, infusions of the α7* nAChR antagonists and the α4β2* nAChR antagonists impair working memory performance (Addy et al. 2003; Levin 2002). Likewise, increased cholinergic input to the BLA is associated with increased arousal, improved performance in short term memory tasks and enhanced acuity in discrimination of somatosensory inputs (Barros et al. 2005; May-Simera and Levin 2003).
The effects of nAChR activation on synaptic transmission per se have been examined in dispersed neurons from mouse amygdala (Barazangi and Role 2001) and in slice preparations from rat (Zhu et al. 2005). The current work tests the effects of nicotine at concentrations similar to those in moderate-heavy smokers on cortico-BLA transmission. We find that brief exposure to nicotine result in sustained, activity dependent modulation of cortico-amygdala circuits and that prenatal exposure to nicotine results in long lasting changes in the extent of cholinergic modulation. To the best of our knowledge, this is the first report of activity dependent, nAChR- modulation of cortico-amygdala circuits in wild type vs. genetically modified mice.
METHODS
Slice preparation
Coronal brain slices were prepared from C57BL6 backcrossed mice at postnatal age 14-28 days (Jackson labs α7 genotype specifications). Mice used were +/+, +/− or −/− for the α7 nAChR subunit gene. Animals were anesthetized with a mixture of ketamine and xylazine (100 mg ketamine and 6 mg xylazine/kg body weight injected ip). After decapitation, the brain was transferred quickly into a sucrose-based ice-cold solution bubbled with 95%O2 and 5% CO2. This cutting solution contains (in mM): sucrose 230; KCl 2.5; MgSO4 10; CaCl2 0.5; NaH2PO4 1.25; NaHCO3 26 and glucose 10. Coronal brain slices (300μm) were prepared using a Leica VT1000S Vibratome. Slices were equilibrated with an oxygenated artificial cerebrospinal fluid (aCSF) at room temperature (24-26°C) for at least 1 hour prior to transfer to the recording chamber. The slices were continuously superfused with aCSF at a rate of 2ml/min containing (in mM); NaCl 126, KCl 2.5, NaH2PO4 1.25, NaHCO3 26, CaCl2 2, MgCl2 2 and glucose 10 bubbled with 95% O2 and 5% CO2 at room temperature (24-26°C).
In experiments on brain slices from older animals (e.g. older than 20 days), pyruvate (0.15mM to 0.75mM) was added to reduce oxidative damage and enhance survival (Gramsbergen et al. 2000; Matthews et al. 2003). With this protocol, slices are first incubated in a mixture of 50% cutting solution with pyruvate and 50% aCSF (in mM): sucrose 115, NaCl 63, KCl 2.5, NaH2PO4 1.25, MgSO4 5, CaCl2 1.25, MgCl2 1, NaHCO3 26, glucose 10 and sodium pyruvate 0.75 at 35°C for 30 minutes and then transferred to a mixture of 10% cutting solution and 90% aCSF (in mM): sucrose 23, NaCl 113.4, KCl 2.5, NaH2PO4 1.25, MgSO4 1, CaCl2 1.85, MgCl2 1.8, NaHCO3 26, glucose 10 and sodium pyruvate 0.15 at 35°C for 1∼4 hour prior to recording.
Electrophysiological recordings
BLA Pyramidal neurons were visualized on an Olympus BX51WI upright microscope (Olympus Optical) equipped with differential inference contrast (DIC) optics. Patch electrodes with a resistance of 4–6 MΩ were pulled with a P-97 Brown Flaming electrode puller (Sutter Instrument Company). Signals were recorded with a Multi Clamp 700A amplifier (Axon Instruments). The pipette solution contains (in mM) 130 K-gluconate, 2 KCl, 2 MgCl2, 10 HEPES, 0.5 EGTA, 1 ATP and 0.2 GTP (pH=7.3). The following criteria were applied for inclusion of recorded cells in the study; failure to meet all of these criteria resulted in exclusion from the sample population. (a) Seal resistances maintained throughout the recording period at >5 GΩ; (b) holding current in whole cell clamp configuration remained within 10% of the initial value and was ≤P100 pA (c) Series resistance; Rs measured every 10 minutes throughout the course of the experiment remained stable (i.e. <10% change from initial value).
To examine transmission at cortical-BLA inputs, EPSCs were evoked by field stimulation with a concentric bipolar stimulation electrode (FHC, Inc) placed in the external capsule (Figure 1A, left). A 0.1 Hz single stimulation was delivered via the stimulation electrode. Stimulation strength was adjusted to trigger ∼50% successful EPSCs. Paired pulse modulation was examined over a range of stimulus intervals, varying from 50ms to 200ms. Pairs of stimuli were delivered at 0.1Hz (interval 50ms; from −50 to −150 μA). The paired pulse modulation ratio (PPR) was defined as [(I2-I1)×100%]/I1. Where specified, a theta burst stimulation pattern (TBS), in which 4 pulses of stimulation are given at 50 Hz and repeated 10 times at a 5 Hz inter-bust interval. Spontaneous EPSCs of BLA pyramidal neurons were monitored in voltage clamp configuration, with a holding potential of −60mV. Bicuculline (10 μM) was used to block GABAA receptors, 50μM APV was used to block NMDA receptors, and 20 μM CNQX was used to block AMPA/kainate receptors. Miniature currents were recorded in the presence of 1μM TTX. Data were filtered at 2k Hz by Multi Clamp 700A and analyzed using pClamp9 (Axon instruments, Inc) and Mini Analysis 6.0 (Synaptosoft, Inc). All experiments were done in the presence of 200nM atropine, a concentration that blocks activation of muscarinic receptors without blocking nAChRs (Zwart and Vijverberg 1997).
Figure 1. Basic physiological properties and appearance of BLA pyramidal neurons in acute brain slice from P 14-25 mouse.

Schematic view and sample photomicrographs of the BLA region studied as viewed in coronal brain slices. Studies of evoked transmission involved placement of a stimulation electrode at the EC fork to excite cortical inputs. Post synaptic BLA neurons with pyramidal-shaped somas were chosen for patch clamp recording. Electrophysiological studies were followed by biocytin staining to assess morphological properties of physiologically characterized mouse BLA neurons
A: Left: Schematic of a coronal slice of adult mouse brain, including the basolateral amygdala region studied to illustrate the approximate placement of patch recording and extracellular stimulation electrodes. EC, external capsule; IC, internal capsule; Stim, stimulation electrode; Rec, recording electrode; BLA, basolateral nucleus of amygdala. Middle: enlarged DIC view of BLA. Right: Enlarged DIC view of BLA pyramidal neurons (scale bar, 10 μm).
B: More than 90% of BLA pyramidal neurons fired action potentials with modest adaptation of firing rate during a prolonged depolarizing current pulse. These neurons had more extensive dendritic arbors than the population of neurons described in part C.
C: About 10% of BLA pyramidal neurons showed strong adaptation of action potential firing during prolonged depolarization.
Nicotine delivery, doses and prenatal exposure
Prenatal nicotine exposure began at E14 of gestation. Pregnant female mice were given drinking water containing either nicotine hydrogen tartrate salt (200μg/ml) with sucrose (1%) or sucrose alone (control group). This dosing regimen continued from E14 through parturition and weaning. The effects of acute nicotine exposure were assayed in slice recordings with continuous superperfusion of aCSF plus local pressure application of nicotine at 100 nM to 1 μM. 500 nM nicotine was chosen as the optimal dose and was used in experiments shown, unless otherwise noted. Focal pressure application of drugs by a Picospritzer II (Parker Instrumentation) utilized 10 psi applied pressure for 2 to 3 minutes to a 2-3 μm diameter patch pipette that was positioned ∼15 μm from the soma of the recorded neurons.
Biocytin-staining
In a subset of studies we recorded from BLA pyramidal neurons with patch pipettes containing 0.2-0.5% biocytin solution (Sigma). After completion of the recordings, slices were perfused with 4% paraformaldehyde +0.2% picric acid +0.1% glutaraldehyde (pH = 7.4) for 10 min. Slices were then post-fixed overnight at 4°C. After repeated washes with 0.1 M phosphate-buffered saline (PBS), slices were treated with 0.3% H2O2 for 10 min at room temperature and then incubated in 0.2% Triton X-100 and 0.2% albumin for 1 h at 4°C. Biocytin staining was visualized by peroxidase -coupled anti-avidin/ biotin conjugation using an ABC kit (1:500, Pierce 32054) for 1 h at room temperature followed by incubation in 0.025% DAB plus 0.003% H2O2 Tris buffer solution for 15 minutes. Samples were mounted in Permount and relocalized and photographed with a CCD digital camera (DAGE-MTI) and DIC microscope.
RESULTS
This study reports findings from recordings in 238 neurons in the basolateral amygdala of the mouse.
Morphological and electrical properties of BLA pyramidal neurons
The basolateral nucleus of the amygdala (BLA) is easily identified within coronal brain slices by the landmark ‘forking’ appearance of the external capsule (EC) under DIC optics (Figure 1A, middle). BLA neurons with pyramidal shaped somas and evident apical dendrites were chosen for electrophysiological study (Figure 1A, right). In initial experiments, biocytin (0.5%) was included in the patch pipette solution, so that recordings could be followed by immunostaining to determine basic features of neuronal morphology and the precise location of the recorded neuron within the BLA. Depolarizing current steps of 1.5 s duration were delivered to the pyramidal neurons to elicit action potentials. BLA pyramidal neurons included two major groups based on the sub and super threshold response profiles. The majority of pyramidal neurons (∼90%) fire with moderate adaptation in action potential rate during a maintained depolarizing step (Figure 1B, left). About 10% of neurons subsequently identified as pyramidal neurons had strongly adapting action potential firing profile. (Figure 1C, left). Morphological analysis of biocytin labeled neurons established that neurons with a moderate spike adaptation had extensive perisomatic dendritic arbors (Figure 1B, right), while strongly adapting neurons had less branched perisomatic dendrites (Figure 1C, right).
Baseline synaptic transmission in the BLA
Recording in identified BLA neurons with K+-gluconate patch solution and holding at −60 mV revealed ongoing spontaneous synaptic events of both polarities and varying amplitudes and kinetic profile (Figure 2A, left). Application of the GABAA receptor antagonist bicuculline eliminated the slower, outward synaptic currents; the remaining inward synaptic currents were rapid in both rise and decay time course (Figure 2A, middle; Figure 2B and 2C, bicuculline). Recordings in the continued presence of bicuculline revealed a net disinhibition of faster spontaneous synaptic events (Figure 2B and 2C, bicuculline) and the addition of NMDA and AMPA receptor antagonists (see methods); abolished the bicuculline resistant currents (Figure 2A, right; Figures 2B and 2C). These findings are consistent with the idea that the majority of fast synaptic transmission detected under these recording conditions in the BLA involves activation of ionotropic GABA and glutamate receptors.
Figure 2. Fast synaptic transmission to BLA neurons is blocked by antagonists of GABAA, NMDA and AMPA receptors.

A: Sample traces of spontaneous PSCs (postsynaptic currents) recorded in BLA neurons in acute slice at a holding potential (Vh) of −60 mV from control conditions (no blockers) or, +bicuculline (10 μM) with and without APV ( 50 μM) and CNQX (20 μM) , as noted. The majority of smaller, slower PSCs are eliminated treatment with bicuculline; subsequent addition of CNQX + APV blocks all PSC activity at Vh=−60 mV.
B: PSC amplitude plotted vs. time of recording in sample BLA neuron in acute slice at a holding potential (Vh) of −60 mV from control conditions (no blockers) or, +bicuculline (10 μM ) with and without APV ( 50 μM) and CNQX (20 μM) , as noted. With bicuculline application, there is a decrease in the number of small amplitude PSCs, followed by an increase in the number of larger amplitude; faster (bicuculline-resistant) PSCs consistent with disinhibition .The subsequent addition of APV and CNQX blocked all remaining PSCs.
C: PSC frequency plotted vs. time of recording in same BLA neuron under control conditions (no blockers) or, +bicuculline (10 μM) with and without APV (50 μM) and CNQX (20 μM) as noted.
Nicotine enhances glutamatergic transmission in the BLA
Activation of nicotinic receptors facilitates glutamatergic transmission in the BLA. The effects of a range of low concentrations of nicotine (from 100nM to 1μM) were initially assessed using either focal application or bath application as described in Methods. Both spontaneous synaptic activity (data not shown) and TTX (1 μM) resistant synaptic transmission were enhanced by local application of nicotine in 40% (16/40) of BLA neurons tested (Figure 3A and 3B). Enhancement of glutamatergic transmission was reflected in an increase in miniature postsynaptic current (mPSC) frequency (Figure 3C and 3F) without alteration of mPSC amplitude (Figure 3D). Thus inter-event intervals were significantly reduced (Figure 3C, Kolmogorov-Smirnov test, P<0.001, n=16) without a change in amplitude (Figure 3D, Kolmogorov-Smirnov test, P=0.7, n=16). Amplitude histograms of the mPSCs were well fit by log normal distributions with a unit mode of 8.8 ± 0.2 pA (r2 =0.96; X2/df <0.0001 (Figure 3H). The observed increases in mPSC frequency without altered PSC amplitude is consistent with a presynaptic mechanism contributing to nicotinic facilitation.
Figure 3. Nicotine elicits a transient facilitation of TTX-resistant glutamatergic transmission in BLA neurons.

In the absence of direct stimulation and in the presence of TTX to block spontaneous suprathreshold activity, a single application of nicotine elicits brief facilitation of glutamatergic PSCs in 40% of BLA neurons tested (16/40). Nicotine significantly increased the frequency of TTX- resistant, miniature PSCs,(mPSCs) without effecting mPSC amplitude. Following nicotine application, the PSC frequency returned to pre-nicotine levels. Sustained facilitation of mPSCs was never observed, in contrast to findings at other glutamatergic synapses in mouse (LWR unpublished).
A: Representative recordings of mPSC recorded from a BLA pyramidal neuron under control conditions plus TTX (see Methods).
B: Representative recordings of miniature PSCs during the application of nicotine (0.5μM). A single (2 minute) application and washout of nicotine caused a transient increase in the frequency of mPSCs without alteration of mPSC amplitude.
C: Cumulative distribution of the inter-event interval times from 16 separate experiments in which nicotine increased mPSC frequency. The cumulative plot is significantly left-shifted during nicotine exposure, indicating reduced inter-event intervals, i.e. an increase in mPSC frequency (solid circle●, control; open circle, ○: + nicotine; ). Control vs. + nicotine are significantly different at P<0.001 Kolmogorov-Smirnov test.
D: Cumulative distribution of the mPSC amplitudes from the 16 separate experiments in which nicotine elicited significant effects on mPSC frequency. The cumulative distribution of mPSC amplitudes was slightly right-shifted with nicotine; Control vs. + nicotine mPSCs are not significantly different in this or other +/− nicotine comparison groups (solid circle ●, control; open circle ○, nicotine; Kolmogorov-Smirnov test, P>0.6). Inset: Box plot of mPSC amplitudes from the population data. The horizontal lines in each box denote (starting from the lower bar) the range of the data that fall within the 25th, 50th and 75th percent values, respectively. The whiskers indicate the 5th and 95th percentile values. The square symbol within the box is the mean of the entire population.
E: Bar graph of the percent of BLA neurons tested in which nicotine elicited a significant facilitation of spontaneous synaptic transmission. (Left to right) Under control conditions, 40% of the 40 BLA neurons tested were nicotine responsive, When nicotine was applied in the presence of the α7* selective antagonist α-BgTx significant facilitation was only seen in 25%; nicotine plus the, α4β2 antagonist DhβE decreased the percent responsive to 19% and in the presence of the non-selective nAChR antagonist mecamylamine synaptic facilitation was never observed (0/12).
F: Box plot of the mPSC frequency of the population data during the conditions indicated. The horizontal lines in each box denote (starting from the lower bar) the range of the data that fall within the 25th, 50th and 75th percent values, respectively. The whiskers indicate the 5th and 95th percentile values. The square symbol within the box is the mean of the entire population. Nicotine alone significantly increases mPSC frequency (P=0.001, KS test); Nicotine treatment in the presence of either α-BgTx, DhβE or mecamylamine was without effect.
G: The net shift in the mPSC cumulative ISI plot was assessed from the integrated area between the control and nicotine (± antagonist) curves in panel C. Nicotine alone shifted the cumulative intervals curve by 340 units; mecamylamine rendered the curve indistinguishable from control (i.e. the net shift ∼0) In the presence of α-BgTx, nicotine shifted the cumulative interval curve by less than a third of the nicotine alone value DhβE, was somewhat less effective, decreasing the shift in the cumulative intervals curve to half that of nicotine alone.
H: A plot of the amplitudes of miniature PSCs under control conditions was best fit by a lognormal distribution with a peak at 8.8± 0.2 pA (r2=0.96, χ2/df<0.0001)
Both α7* and non α7* type nAChRs contribute to short term synaptic facilitation
Examination of the relative contribution of α7* vs. non-α7* nAChR was tested using both pharmacological and genetic deletion techniques. First, we monitored nicotine-induced facilitation of TTX-resistant glutamatergic synaptic transmission with and without nicotine or the selective α7* receptor antagonist α bungarotoxin (α-BgTx). In the presence of 100nM α-BgTx, increased mPSC frequency by nicotine was observed in only 1/4 recordings (Figure 3E, F and G, Table 1). The α4β2* selective nAChR antagonist DHβE (5μM) was similarly effective; detectable facilitation occurred in <1/4 recordings (Figure 3E, F, G and Table 1). The nonselective nAChR antagonist, mecamylamine (100 μM), abolished all facilitatory effects of nicotine (Figure 3E, F, G Table 1).
Table 1.
Activation of nAChRs by nanomolar nicotine facilitates glutamatergic transmission and /or elicits direct post synaptic current (Inic)
| WT |
α7 KO |
||||
|---|---|---|---|---|---|
| +α-BgTx | +DHβE | +MEC | |||
| n | 40 | 12 | 16 | 12 | 11 |
| Facilitation | 40% | 25% | 19% | 0% | 0% |
| Inic | 40% | 25% | 19 % | 0% | 0% |
| Both | 25% | 16% | 6% | 0% | 0% |
Recent work by Yakel and colleagues revealed a role of somatic nAChRs in direct responses of BLA neurons to ACh (Klein and Yakel 2006). Although the current study examined 100-1000 fold lower concentrations of agonist with the goal of testing “smokers' concentrations” of nicotine, we did observe small (5-10pA) shifts in the steady state current during the application of nicotine in ∼40% of recordings. A comparison of the α-BgTx, DHβE and mecamylamine sensitivity of the direct inward currents with that of nicotine elicited facilitation revealed a similarly mixed profile of both α7* and non-α7* nAChRs. Likewise, assay of nAChR-stimulated facilitation in slices from α7 mutant mice, by either nicotine application or by treatment with the acetylcholine esterase inhibitor), ambenonium 500 nM, confirmed involvement of both α7* and non α7* nAChRs in the modulatory effects (Tables 1 & 2).
Table 2.
Stimulus dependence of Nicotine-induced facilitation
| Stimulation | Nicotine - induced facilitation |
||
|---|---|---|---|
| n | n (percent ) | ||
| total | positive | sustained* | |
| None (WT) | 40 | 16 (40%) | 0 |
| 0.1 Hz single (WT) | 51 | 35 (68%) | 15 (44%) |
| 0.1 Hz PPS (WT) | 23 | 13 (57 %) | 9 (69%) |
| None (α7−/−) | 11 | 0 | 0 |
| 0.1 Hz single (α7+/−) | 35 | 17(49%) | - |
Sustained facilitation is defined as an increase of more than 50% control for >10-30 min
Nicotine enhances evoked cortical input to the BLA
The TTX resistant glutamatergic mPSCs recorded reflect inputs to the BLA neurons from several different sources that may (or may not) have different profiles of nAChR mediated modulation. To examine the effects of nAChR activation on specific inputs to BLA pyramidal neurons, we stimulated cortical fibers within the external capsule, at the point that the tract bifurcates along the lateral and basolateral nucleus. Stimulus-evoked synaptic current responses were recorded in whole cell voltage clamp of 129 BLA pyramidal neurons. In the presence of bicuculline, stimulation of cortical inputs evoked PSCs with rapid rise (τ=∼1 ms) and decay (τ=∼3-5 ms) kinetics. Modulation of stimulus- evoked PSCs by nicotine was examined by decreasing the magnitude of the stimulus delivered to the external capsule so that approximately 50% of stimuli elicited post synaptic currents (Figure 4A, control). Under these conditions, a single application of nicotine (500nM; 2 minutes) increased both the success probability and the average amplitude of the stimulus-evoked PSC's (Figures 4B1 and 4B2). Nicotinic facilitation of evoked transmission was significantly increased in 68% (35/51) of the BLA neurons tested with single stimuli. In contrast to the transient effects of nicotine on spontaneous transmission, the facilitation of evoked corticoamygdala transmission was often sustained well after the wash out of nicotine from the perfusion , persisting for >10 to 30 minutes (see Table 2).
Figure 4. Nicotine increases the number of successes vs. failures of evoked PSCs at cortical –BLA inputs without altering EPSC amplitude.
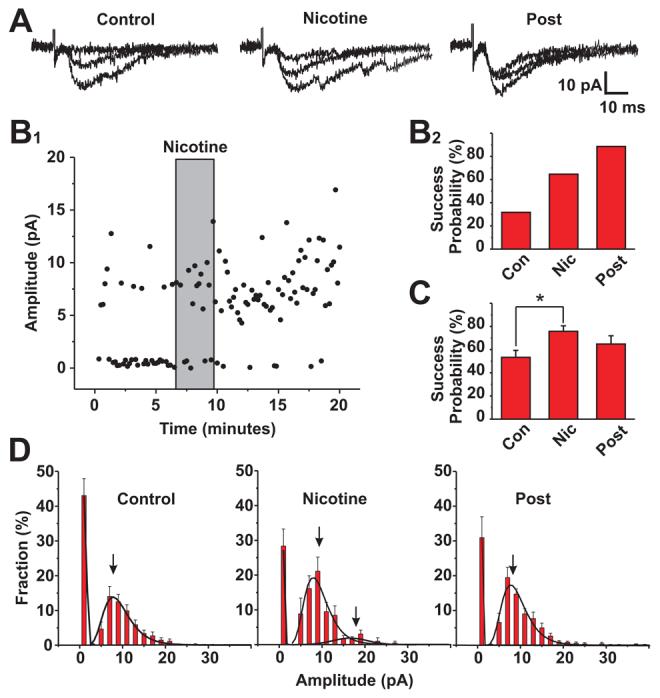
The effect of nicotine on evoked transmission at cortical –BLA inputs was tested using a minimal stimulation and low frequency (0.1Hz) paradigm. Stimulus intensity was adjusted such that under control conditions, ∼50% of the stimuli elicited stimulus-locked, post synaptic currents. Under these conditions nicotine elicited robust facilitation of EPSCs at 68% of the cortico-amygdala synapses tested (35/51). At 44% (15/35) of these nicotine's effects long outlasted the application and removal of the drug resulting in a statistically significant increases for > 10-30 minutes.
A: Three overlapping traces of evoked PSC during control conditions, during the application of nicotine (500nM) and after the washout of nicotine from the perfusion media. Note that only the first, stimulus-locked PSCs were included in determinations of EPSC success vs. failure and in EPSC amplitude analyses.
B1: Plot of EPSC amplitude before, during and after a single 2 minute nicotine (500 nM) application. This example (representative of 15 of the 35 nicotine facilitated synapses) both the number of stimuli that evoked EPSCs was increased and the facilitation lasted for 20 minutes. B2: Plot of success probability under control, + nicotine and post nicotine conditions for the neuron illustrated in B1. In this neuron the ratio of success to failures continued to increase for at least 20 minutes after the washout of nicotine from perfusion.
C: Summary plot for success probabilities for all neurons tested with the minimal stimulus paradigm with EPSCs under control, + nicotine and >10 minutes post nicotine conditions. The overall effect of a single application of nicotine (500nM) was statistically significant (paired t-test, P,0.05, n=35).
D: Histograms of the fraction of responses vs. the amplitude of the stimulus locked postsynaptic currents for control, nicotine and post nicotine conditions. All distributions were best fit by lognormal curves (n=12, r2 from 0.92 to 0.98, χ2/df<0.001). The fraction of events that were failures (plotted as 0 amplitude) was significantly reduced by nicotine (P<0.04, n=12). Likewise the number of events in the first peak of the amplitude histogram increased with nicotine and post nicotine (P<0.04, n=12; A1: 1.08 ± 0.03 control; 1.37 ± 0.07 +nicotine), but there are no statistically significant changes in the average amplitude of events before, during or after nicotine (x1 control=9.3 ± 0.1 pA vs. +nicotine 8.9 ± 0.2 pA and, post8.8 ± 0.2 pA, n=12). In some recordings a small 2nd cluster of events could be discerned in the amplitude histogram with nicotine application (x2= 18.47 ± 0.86 pA, n=5).
The observed facilitation of cortical input-evoked glutamatergic transmission by nicotine could arise from enhanced release and/or increased postsynaptic responses to glutamate per se. As expected, the minimum stimulation paradigm used typically resulted in PSCs of 1-2 times the unit amplitude of ∼9 pA (see Figure 4D). The relative contributions of increased release probability vs. enhanced postsynaptic glutamate receptor sensitivity can be assessed by comparing the distribution of evoked PSC amplitudes before and after nicotine application. Under control conditions (i.e. prior to nicotine application), evoked PSC amplitudes were distributed between the peak at zero (i.e., failures) and a peak at ∼10 pA (Figure 4D). The failure rate under control conditions was ∼ 42%. During and immediately after the application of nicotine, the number of failures decreased to less than 30%, the number of events in the first non-zero peak increased (Acontrol =1.08 ±0.03 vs. Anicotine = 1.37 ± 0.07; P<0.05, n=12, paired t-test), without alteration in the average amplitude of the unit events (9.3 ± 0.1, pA vs. 8.9± 0.2, NS; n=12, paired t-test). A small second amplitude cluster is also apparent following nicotine application (I2= 18.4 ± 0.08 pA ; n=5). Overall these data are consistent with a nicotine induced facilitation of glutamatergic transmission due to an increase in the probability of release without a change in postsynaptic glutamate responsiveness.
Modulation of paired pulse evoked glutamatergic transmission at cortico-BLA synapses
To further examine the pre- vs. postsynaptic loci of nicotine induced facilitation, we analyzed glutamatergic transmission at cortico-BLA synapses evoked by paired-pulse stimulation (PPS). Responses to paired stimuli were tested over a range of inter-stimulus intervals from 50 to 200ms (Figure 5A), and stimulation strength was adjusted such that the first pulse of each pair evoked stimulus locked responses. Although all inter-stimulus intervals tested elicited paired-pulse facilitation, the 50ms interval appeared the most robust (Figure 5B, C and D). As such, subsequent experiments used a 50 ms interval. The effects of nicotine on paired-pulse evoked responses were assessed at 23 cortico-amygdala inputs in 10 different slice preparations. Nicotine increased the total integrated current at 57% and elicited a sustained facilitation at 69% of the synapses. Paired pulse stimulation of cortical inputs coupled with nicotine-treatment facilitated synaptic responses for more than 30 minutes, longer than ever observed with single stimuli (Table 2). Profiles of the two major types of paired pulse evoked responses observed before and after nicotine application are presented in Figure 6A, B and Figure 6 C, D, respectively. The majority of PPS responses were facilitated in a sustained manner and synaptic enhancement was manifest as a greater increase in the amplitude of the second (as opposed to the first) postsynaptic current response (Figure 6 A, B). Short-term facilitation was seen in ∼30% of the nicotine responsive synapses, (e.g. in Figure 6 C, D) with a larger effect of nicotine on the first PSC of the PPS responses. Analysis of all PPS recordings with nicotinic facilitation revealed that nicotine modulated the paired pulse ratio by more than 10% in the majority of pairs tested (11/13, Figure 6E and 6F) with a net average increase in synaptic strength (based on amplitude and total integrated current, Figure 6G and 6H), of ∼ 35%. These data are also consistent with the involvement of a presynaptic mechanism in nicotine-induced synaptic facilitation.
Figure 5. Paired pulse stimulation of mouse cortico-amygdala synapses over a range of inter-stimulus intervals.

The intensity of stimuli to cortical inputs was adjusted such that stimulus-locked EPSCs were recorded in individual basolateral amygdala neurons in response to both the first and the second pulse. Varying the inter stimulus interval between 50 and 200 ms. revealed significant paired pulse facilitation at 50 and 100 ms intervals; longer delays were associated with less prominent facilitation. Paired pulse depression was never detected using this stimulus paradigm.
A: Overlapped traces of EPSCs at a typical cortico- BLA synapse in mouse Pairs of stimuli were separated by 50, 100, 150 and 200 ms.
B: Comparison of paired pulse response profile from multiple experiments to assess the optimal inter stimulus interval paradigm revealed that the most robust facilitation was obtained with stimulus intervals of 50 ms.
C, D: Sample profile of 100 paired-pulse evoked responses using a 50 ms interval: Overlap of 10 individual traces (C: top); 10 averaged traces (C: bottom). D: Amplitude distribution of all responses to the first (P1, closed ▎) and second (P2; open ▯) pulse of each pair from the recorded neuron shown in panel C.
Figure 6. Nicotine modulates the ratio paired-pulse -EPSCs at cortico-amygdala inputs.

Nicotine elicited a statistically significant increase in the net integrated post synaptic current of ∼ 60% (13/23) of BLA neurons tested with a paired pulse stimulation, with an inter-stimulus interval of 50 ms; sustained facilitation was observed at ∼70% (9/13) of these pairs. The fraction of neurons showing sustained facilitation was higher and the duration of this facilitation was longer than single evoked stimulation. Nicotine elicited a statistically significant modulation of the ratio of paired-pulse responses at 11/13 pairs tested, consistent with a contribution of changes in presynaptic function to the net facilitatory effect of nicotine.
A: Sample traces of paired-pulse stimulation EPSC responses (5 traces overlaid; top) and the averaged waveforms (below) from one of the 9/13 cortico-BLA pairs where an acute application of nicotine elicited a sustained (>10-45 min) facilitation of synaptic transmission.
B: Sustained facilitation was typically manifest, as in A, with a stronger increase in the amplitude of P2 (open ○) vs. P1 (closed ●). The time of nicotine application is indicated by the shaded area (500 nM; 3 min; 20 pA × 25 ms.
C: Sample traces of paired-pulse stimulation EPSC responses (5 traces overlaid; top) and the averaged waveforms (below) from one of the 4/13 cortico-BLA pairs where an acute application of nicotine elicited only a transient facilitation of synaptic transmission.
D: Transient facilitation was manifest, as in C, with a greater increase in the amplitude of P1(closed ●) vs. P2 (open ○) during and immediately after the time of nicotine application (as indicated by the shaded area; 500 nM; 3 min; 20 pA × 25 ms).
E: The distribution of the paired pulse ratios before, during and after nicotine application in those 13 nicotine responsive neurons. In 5 of the 11, nicotine increased the ratio of the second relative to the first paired-pulse response (closed ●) and in 6 of the 11 cases the increase in P1 exceeded the increase in P2 (open ○).
F: Summary plot of the extent of change of 13 cortico-amygdala pairs assayed for modulation of paired pulse EPSC responses by nicotine; The paired pulse ratio was significantly affected by nicotine in 11/13 pairs tested (>10% change, t≥ 3 minutes). Closed ●, nicotine increased the PPR; open ○, nicotine decreased the PPR.
G: Amplitude distribution histogram from the representative neuron in panel A and B in which nicotine elicited a sustained increase in the amplitude of second EPSC. P2 amplitude histogram under control conditions (closed ▎) vs. post nicotine application (open ▯). Nicotine caused a significant and sustained right-shift of the entire amplitude histogram of P2 responses.
H: The synaptic strength as measured by the differences in the integrated EPSC s was increased by nicotine (P< 0.008, paired t-test, n=11).
Nicotine lowers the threshold for patterned (theta burst) stimulation triggering long lasting potentiation of cortical-BLA synaptic transmission
Stimulation of cortical inputs to the BLA with a theta burst stimulation pattern (TBS; see Methods) elicited brief (<10 minutes) vs. sustained potentiation (>10 minutes to∼90 minutes) in about half of the pairs tested (Figure 7A and 7B). From a total of 16 cortico-BLA pairs, TBS elicited brief potentiation in 7 pairs and sustained potentiation in 8 pairs (TBS failed to potentiate 1 of the tested pairs). When 4 of the 7 pairs that were only transiently potentiated with TBS alone were exposed to a single application of nicotine, subsequent TBS elicited in sustained potentiation. That is, nicotine paired with TBS facilitated glutamatergic transmission for as long as the recordings could be maintained (up to 90 minutes, Figure 7C). For the 1 pair that was unaffected by TBS alone, the combination of TBS + nicotine administration elicited a transient potentiation. In the 8 pairs in which long lasting potentiation was elicited by TBS itself, addition of nicotine did not elicit any further potentiation.
Figure 7. Nicotine lowers the threshold for long lasting facilitation of cortico-BLA synapses triggered by theta burst stimulation.
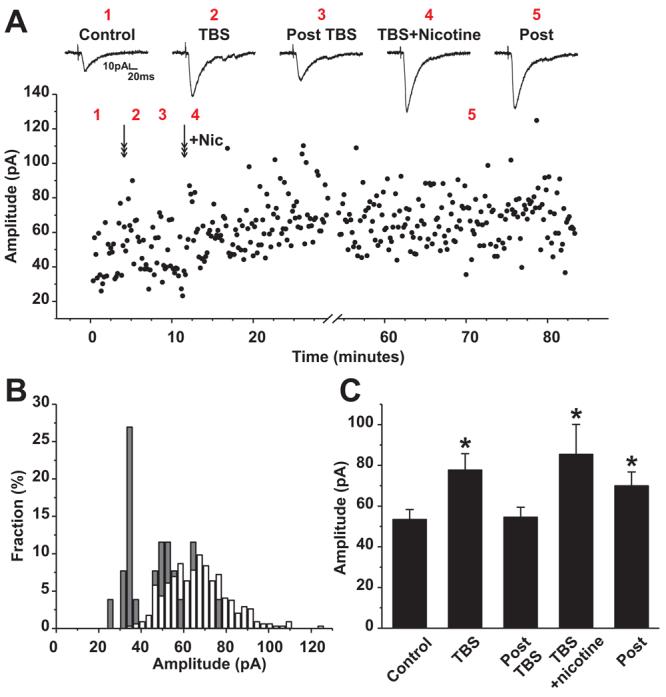
Theta burst stimulation (TBS) of cortical inputs elicits a brief post TBS potentiation of synaptic transmission at cortico-BLA inputs. A single application of nicotine (500nM; 3 minutes) with TBS resulted in significant long -lasting facilitation of responses to subsequent single stimuli in 6 of 10 pairs tested. The increase in synaptic strength in response to TBS + nicotine persisted for up to 90 minutes.
A: (upper panel) Representative EPSC responses to 1 Hz stimulation during control conditions (1: Control), immediately after theta burst stimulation (2: TBS), 5 minutes after TBS (3: Post TBS), immediately after concurrent administration of nicotine and TBS (4: TBS + Nicotine) and >1 hour after TBS + Nicotine (5: Post). (from left to right) Sample records shown were taken at the time points as indicated in the lower panel. Lower panel: Plot of EPSC amplitudes over ∼1.5 hour recording from a BLA neuron with 1 Hz stimulation of cortical inputs. Arrows indicate the time of delivery of theta burst stimulation alone (first arrow) and the time of delivery of TBS + nicotine (second arrow; + Nic). The numbers 1-5 in the plot correspond to the conditions shown in the upper panel.
B: Plot of the fraction of events at the indicated EPSC amplitude during control period (solid bars; t=0-5 min) and after TBS plus nicotine (open bars, >20 min). TBS + nicotine is sufficient to elicit a sustained increase in the EPSC amplitude reflected in a right shift of the amplitude histogram. The predominant peak under control condition which includes ∼30% of all EPSCS is centered at ∼36 pA. Second and third peaks of the histogram (each ∼20% of the EPSCS) are evident at ∼45pA and ∼63 pA. Following theta burst stimulation + nicotine, less than 2% of the EPSCs are <45pA.
C: The averaged amplitude for neurons that nicotine can switch brief potentiation to sustained potentiation induced by TBS (Paired t-test, P<0.05, n=4).
Prenatal nicotine exposure reduced subsequent modulation of cortical-BLA transmission
The above experiments are consistent with the idea that the efficacy of nicotine facilitation is related to the level of synaptic activity at glutamatergic cortico-BLA inputs. In view of these findings and the results of prior studies on prenatal nicotine effects on amygdala-related behaviors, we examined whether exposure to nicotine in the pre- and perinatal period to long term changes in cortico-amygdala circuits.
Six days of prenatal nicotine exposure and same dosing regimen continued through weaning, followed by recording at postnatal day 15-22 revealed a broadening of the (non normal) distribution of mPSC frequency compared with control littermates. However, statistically significant changes in baseline glutamatergic transmission were not observed.
Subsequent assays of the nicotinic modulation of cortico-BLA transmission in prenatal nicotine vs. control mice revealed that significant effects. Miniature PSC's recorded under control conditions and following acute nicotine application in prenatal nicotine treated animals are shown in Figures 8A and 8B. The frequencies of mPSC's recorded under control conditions were similar regardless of prenatal nicotine exposure. However, subsequent challenge with an acute application of nicotine facilitated spontaneous transmission in half as many BLA neurons from prenatal nicotine treated vs. control animals. Furthermore, the nicotine elicited facilitation that was detected in prenatal nicotine exposed animals was less than that in untreated siblings (Figures 8C and 8D, solid line, control animal control; broken line, control animal with acute nicotine; solid circle, prenatal exposed animals control; open circle, prenatal exposed animals with acute nicotine). Figure 8C is the cumulative probabilities of inter-event intervals illustrating that spontaneous transmission in control vs. prenatal nicotine exposed animals were similar though non identical. In amygdala slices from animals exposed to prenatal nicotine, facilitation of transmission by an acute application of nicotine was considerably less than that seen in controls but still significant, (Kolmogorov-Smirnov test, P=0.004, n=6). No statistically significant changes in direct, nicotine elicited currents were seen in prenatal nicotine treated vs. untreated controls (Figure 8E and 8F).
Figure 8. Exposure to nicotine during the prenatal period ablates post natal synaptic facilitation in BLA.

A, B: Sample traces of mPSCs recorded in BLA from acute brain slice preparations from postnatal day 20 control mouse (A) vs. mPSCs recorded from age matched slice from mouse that was exposed to nicotine in utero since ED 14, (B). Although the mPSC frequency range is broader and there is a trend toward increased mPSC frequency in prenatal nicotine exposed animals, these effects are not statistically significant.
C: Cumulative histogram of mPSC frequency: acute nicotine increased the frequency of m EPSCs in control animals shifting cumulative interval curves to the left. Prenatal nicotine-exposed animals had somewhat higher baseline mPSC frequency than prenatal controls and the effects of acute nicotine treatment is significantly less, (KS test, P=0.004, n=6). Note that the dashed line (left most curve) in C is the pooled data from Figure 3C (acute nicotine). Compare with open circles (prenatal nicotine exposed animals + acute nicotine) vs. solid circle (prenatal nicotine exposed animals, no acute nicotine).
D: There was no statistically significant change in mPSC amplitude under any of the conditions tested
E: Box plot of all determinations of mPSC frequency in postnatal recordings from animals that were exposed to nicotine during the prenatal period (E14 on; Prenatal nic +; see Methods) vs. those that were under the prenatal control conditions (Prenatal nic −;). Despite a trend toward increased mPSC frequency in control vs. prenatally-exposed animals the difference was not statistically significant. Acute applications of nicotine (acute nic +) elicits significant increase in mPSC frequency in the prenatal-control group. In contrast, acute nicotine is without effect on the mPSC frequency in BLA neurons from prenatal nicotine treated mice. (KS test, p>0.3; n=6).
F: Box plot of all determinations of mPSC amplitude in postnatal recordings from animals that were exposed to nicotine during the prenatal period (E14 on; Prenatal nic +; see Methods) vs. those that were under the prenatal control conditions (Prenatal nic −;). Neither prenatal nor acute nicotine application (acute nic +) alters mPSC amplitude.
DISCUSSION
The major finding of this study pertains to the mixed nAChR receptor profile and activity dependent nature of glutamatergic facilitation of mouse cortico-amygdala synapses. Patch clamp recordings in nearly 300 WT and α7* mutant mice were assessed to delineate the effect of low level nicotine exposure on glutamatergic inputs to BLA neurons in mouse. Basic characteristics of BLA neurons in mouse appear similar to those described in the rat (Sah et al. 2003). Most BLA pyramidal neurons fire action potentials with relatively little spike adaptation; fewer than 10% displayed strongly adapting spike profiles in response to prolonged depolarizing current injection. The latter neurons, which also appeared to have a less elaborate profile of primary dendritic arborization, were not included in subsequent analysis.
Our studies focused on cortico-amygdala glutamatergic synaptic transmission by examining the bicuculline resistant post synaptic currents. The bicuculline resistant synaptic activity was deemed to be largely, if not exclusively, glutamatergic as all currents were blocked by a cocktail of AMPA and NMDA receptor antagonists.
Prior tract tracing and immunohistochemical studies have demonstrated that the major cholinergic input to the rodent BLA is from basal forebrain (Carlsen et al. 1985; Nagai et al. 1982; Woolf and Butcher 1982). Despite robust ACh-positive projections, primary cholinergic neurons are not present in the BLA (Schafer et al. 1998). Prior studies in rat have demonstrated a direct post-synaptic response of BLA neurons to relatively high concentrations of nAChR agonists. Most nicotinic effects are consistent with nAChR mediated modulation of transmission although direct cholinergic synaptic connections are also indicated by recent reports (Alkondon et al. 1998; Klein and Yakel 2006; Zhu et al. 2005).
The current studies focused on the effects of low (“smokers' level”) concentrations of nicotine on inputs to BLA neurons to examine the possible modulatory role of acute and prenatal nicotine receptor activation in the developing BLA. In the presence of bicuculline, TTX and atropine activation of nicotinic receptors increased the frequency, but not the amplitude of spontaneous glutamatergic PSCs, consistent with the involvement of presynaptic nAChRs.
Pharmacological studies comparing the effects of nicotine in the presence of selective nAChR antagonists and assessing nicotine-induced facilitation of cortico-amygdala transmission in WT vs. α7* nAChR mutant mice indicate that both α7* and (α4β2)* nAChRs contribute to the modulatory effects of low level nicotine in the BLA. These findings, considered in the context of elegant behavioral studies of Levin and colleagues (Addy et al. 2003; Levin 2002; Levin et al. 2006) support the contention that nAChR activation may constitute an important mechanism for the modulation of contextual learning associated with fearful stimuli.
Changes in the strength of synaptic connections are fundamental to the process of information storage with activity-dependent modulation of pre and postsynaptic components as key mediators of such plasticity. The input from somato-sensory cortical areas provides the amygdala with highly detailed sensory information pertinent to discrimination via cortico-amygdalaloid projections to the lateral and basolateral nuclei. Rapid, less finely-tuned input from thalamic areas provides information related to gross stimulus discrimination (Armony et al. 1997).
Modulation of cortical inputs to the BLA by stimulation of the external capsule to evoke PSCs, could arise from either single or multiple fiber inputs to the BLA. If originating from multiple fibers, a facilitation of release would be predicted to manifest in both increased release probability and increased amplitude of evoked responses. To decrease the contribution of activating multiple cortical afferents, we used a minimum stimulation paradigm (Stevens and Wang 1995). Single fiber activation is indicated by the predominance of single quantal-coefficient evoked amplitudes in the response histogram profiles. With minimum stimulation, single evoked EPSCs showed an increase in success probability but unchanged mean amplitudes (not including failures). In view of the consistent value of the quantal size of ∼9 pA before, during and after nicotine application, it is unlikely that alterations in postsynaptic responsiveness to glutamate contribute to the observed nicotinic modulation (Korn and Faber 1998).
Paired pulse stimulation has been widely used as an assay of synaptic plasticity and the relative contributions of pre- and postsynaptic mechanisms (Jiang et al. 2000). Alterations in the paired pulse ratio are consistent with changes in synaptic strength due, at least in part, to changes in presynaptic function. Paired pulse stimulation studies of cortico-amygdala synapses revealed an overall increase in synaptic efficacy following nicotine treatment. An increased paired pulse ratio following nicotine, the predominant observed effect, reflected a greater increase of peak 2 relative to peak 1. Decreased paired pulse ratio was typically due to a greater increase in amplitude of the first peak relative to the second. In a minority of cases (4/13) the amplitude of both P1 and P2 increased proportionally, so that there was no change in PPR. The latter result underscores that a contribution of a postsynaptic component to the overall effect of nicotine on facilitating glutamatergic transmission cannot be ruled out.
We found that the extent and duration of nicotine-induced facilitation of cortico-BLA synapses was directly related to the pattern and intensity of stimulation of the cortical afferents. Without prior stimulation, nicotine elicited a moderate facilitation of mini PSCs. Forty percent (16/40) of neurons showed facilitation, but the facilitation was never sustained after nicotine removal and wash out. With minimal stimulation of cortical inputs at a low frequency (0.1 Hz), nicotine elicited considerably more robust facilitation at ∼ 60% of corticoamygdala synapses), with 50% showing sustained facilitation that out-lasted the nicotine administration by at least 10 minutes. With stronger, paired pulse stimulation of cortico-BLA synapses nicotine facilitates ∼ 60% of the inputs and elicits a sustained facilitation at 70% of cortico-BLA synapses tested. The sustained facilitation elicited by paired-pulse stimulation often lasted longer than that detected with nicotine plus single shock stimulation of cortical inputs (>30').
The activity dependence of the extent of nicotine-induced facilitation is even observed with a subset of synapses that are below threshold for LTP in response to theta burst stimulation (TBS) alone. The addition of nicotine to TBS was sufficient to convert transient, post TBS potentiation to long lasting facilitation of glutamatergic transmission. Nicotinic modulation of synaptic transmission is thought to be mediated, at least in part, by Ca++ influx through activated presynaptic nAChRs. Activity dependent modulation also causes presynaptic Ca++ accumulation. The Ca++ accumulated by repetitive activity in the presynaptic terminal could therefore be amplifying the nicotinic modulation.
Current evidence in the hippocampus indicates a role for postsynaptic changes during potentiation including increased number of synaptic AMPA receptors and/or an increase in single-channel conductance. However, there is also evidence that synaptic facilitation and depression in hippocampus can involve alterations in presynaptic function. Such changes are thought to arise from enhanced probability of release or in the quantity of glutamate released as a result of an alteration in fusion pore kinetics or quantal content.
Nicotine administration has long been associated with enhancement of attention, short term memory function and in some cases, more prolonged aspects of cognitive processing. We tested the hypothesis that activation of nicotinic receptors at cortico-amygdala synapses might contribute to such effects of nicotine. The observation that the degree of modulation and the duration of nicotine induced synaptic facilitation are related to the activity of corticoamygdala inputs is, to the best of our knowledge, without precedent.
Nicotine readily crosses the placental barrier (Luck et al. 1985), and fetal exposure to nicotine damages the developing brain (Mansvelder et al. 2006). Prenatal exposure to nicotine increases the risk for cognitive deficits, attention deficits, behavioral problems and learning disabilities. Several studies have shown that nicotinic receptor expression levels and inactivation profiles could be changed by prenatal nicotine exposures. Prior studies of the effects of prenatal nicotine exposure on post natal CNS transmission revealed dramatic increases in baseline GABAergic transmission in appetite related circuits (Jo et al. 2005). The effects of prenatal nicotine exposure on postnatal glutamatergic inputs to BLA appear relatively modest, but the subsequent modulation of cortico-amygdala synaptic transmission by nAChR activation is virtually abolished in prenatal nicotine exposed animals.
Acknowledgements
We thank Michael Lieberman, Sarah Marshall and David Talmage for critiques of the manuscript. These studies were supported by funds from the NIH and a Philip Morris Extramural Research Group award. In addition, LWR receives support from the Sidney Baer Foundation as a Distinguished Investigator of NARSAD.
Literature Cited
- Addy NA, Nakijama A, Levin ED. Nicotinic mechanisms of memory: effects of acute local DHbetaE and MLA infusions in the basolateral amygdala. Brain Res Cogn Brain Res. 2003;16:51–57. doi: 10.1016/s0926-6410(02)00209-4. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX. Nicotinic acetylcholine receptor alpha7 and alpha4beta2 subtypes differentially control GABAergic input to CA1 neurons in rat hippocampus. J Neurophysiol. 2001;86:3043–3055. doi: 10.1152/jn.2001.86.6.3043. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX. A non-alpha7 nicotinic acetylcholine receptor modulates excitatory input to hippocampal CA1 interneurons. J Neurophysiol. 2002;87:1651–1654. doi: 10.1152/jn.00708.2001. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Albuquerque EX. alpha-bungarotoxin- and methyllycaconitine-sensitive nicotinic receptors mediate fast synaptic transmission in interneurons of rat hippocampal slices. Brain research. 1998;810:257–263. doi: 10.1016/s0006-8993(98)00880-4. [DOI] [PubMed] [Google Scholar]
- Armony JL, Servan-Schreiber D, Romanski LM, Cohen JD, LeDoux JE. Stimulus generalization of fear responses: effects of auditory cortex lesions in a computational model and in rats. Cereb Cortex. 1997;7:157–165. doi: 10.1093/cercor/7.2.157. [DOI] [PubMed] [Google Scholar]
- Barazangi N, Role LW. Nicotine-induced enhancement of glutamatergic and GABAergic synaptic transmission in the mouse amygdala. J Neurophysiol. 2001;86:463–474. doi: 10.1152/jn.2001.86.1.463. [DOI] [PubMed] [Google Scholar]
- Barros DM, Ramirez MR, Izquierdo I. Modulation of working, short- and long-term memory by nicotinic receptors in the basolateral amygdala in rats. Neurobiology of learning and memory. 2005;83:113–118. doi: 10.1016/j.nlm.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Berg DK, Conroy WG, Liu Z, Zago WM. Nicotinic signal transduction machinery. J Mol Neurosci. 2006;30:149–152. doi: 10.1385/JMN:30:1:149. [DOI] [PubMed] [Google Scholar]
- Carlsen J, Zaborszky L, Heimer L. Cholinergic projections from the basal forebrain to the basolateral amygdaloid complex: a combined retrograde fluorescent and immunohistochemical study. The Journal of comparative neurology. 1985;234:155–167. doi: 10.1002/cne.902340203. [DOI] [PubMed] [Google Scholar]
- Giocomo LM, Hasselmo ME. Nicotinic modulation of glutamatergic synaptic transmission in region CA3 of the hippocampus. The European journal of neuroscience. 2005;22:1349–1356. doi: 10.1111/j.1460-9568.2005.04316.x. [DOI] [PubMed] [Google Scholar]
- Gramsbergen JB, Sandberg M, Kornblit B, Zimmer J. Pyruvate protects against 3-nitropropionic acid neurotoxicity in corticostriatal slice cultures. Neuroreport. 2000;11:2743–2747. doi: 10.1097/00001756-200008210-00027. [DOI] [PubMed] [Google Scholar]
- Hill JA, Jr., Zoli M, Bourgeois JP, Changeux JP. Immunocytochemical localization of a neuronal nicotinic receptor: the beta 2-subunit. J Neurosci. 1993;13:1551–1568. doi: 10.1523/JNEUROSCI.13-04-01551.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Sun S, Nedergaard M, Kang J. Paired-pulse modulation at individual GABAergic synapses in rat hippocampus. The Journal of physiology. 2000;523(Pt 2):425–439. doi: 10.1111/j.1469-7793.2000.t01-1-00425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo YH, Wiedl D, Role LW. Cholinergic modulation of appetite-related synapses in mouse lateral hypothalamic slice. J Neurosci. 2005;25:11133–11144. doi: 10.1523/JNEUROSCI.3638-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RC, Yakel JL. Functional somato-dendritic alpha7-containing nicotinic acetylcholine receptors in the rat basolateral amygdala complex. The Journal of physiology. 2006;576:865–872. doi: 10.1113/jphysiol.2006.118232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn H, Faber DS. Quantal analysis and long-term potentiation. Comptes rendus de l'Academie des sciences. 1998;321:125–130. doi: 10.1016/s0764-4469(97)89811-3. [DOI] [PubMed] [Google Scholar]
- Levin ED. Nicotinic receptor subtypes and cognitive function. J Neurobiol. 2002;53:633–640. doi: 10.1002/neu.10151. [DOI] [PubMed] [Google Scholar]
- Levin ED, McClernon FJ, Rezvani AH. Nicotinic effects on cognitive function: behavioral characterization, pharmacological specification, and anatomic localization. Psychopharmacology. 2006;184:523–539. doi: 10.1007/s00213-005-0164-7. [DOI] [PubMed] [Google Scholar]
- Luck W, Nau H, Hansen R, Steldinger R. Extent of nicotine and cotinine transfer to the human fetus, placenta and amniotic fluid of smoking mothers. Dev Pharmacol Ther. 1985;8:384–395. doi: 10.1159/000457063. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, van Aerde KI, Couey JJ, Brussaard AB. Nicotinic modulation of neuronal networks: from receptors to cognition. Psychopharmacology. 2006;184:292–305. doi: 10.1007/s00213-005-0070-z. [DOI] [PubMed] [Google Scholar]
- Matthews CC, Zielke HR, Parks DA, Fishman PS. Glutamate-pyruvate transaminase protects against glutamate toxicity in hippocampal slices. Brain research. 2003;978:59–64. doi: 10.1016/s0006-8993(03)02765-3. [DOI] [PubMed] [Google Scholar]
- May-Simera H, Levin ED. NMDA systems in the amygdala and piriform cortex and nicotinic effects on memory function. Brain Res Cogn Brain Res. 2003;17:475–483. doi: 10.1016/s0926-6410(03)00163-0. [DOI] [PubMed] [Google Scholar]
- Nagai T, Kimura H, Maeda T, McGeer PL, Peng F, McGeer EG. Cholinergic projections from the basal forebrain of rat to the amygdala. J Neurosci. 1982;2:513–520. doi: 10.1523/JNEUROSCI.02-04-00513.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau SJ, Jones IW, Pullar IA, Wonnacott S. Presynaptic alpha7 and non-alpha7 nicotinic acetylcholine receptors modulate [3H]d-aspartate release from rat frontal cortex in vitro. Neuropharmacology. 2005;49:59–72. doi: 10.1016/j.neuropharm.2005.01.030. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiological reviews. 2003;83:803–834. doi: 10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- Schafer MK, Eiden LE, Weihe E. Cholinergic neurons and terminal fields revealed by immunohistochemistry for the vesicular acetylcholine transporter. I. Central nervous system. Neuroscience. 1998;84:331–359. doi: 10.1016/s0306-4522(97)00516-2. [DOI] [PubMed] [Google Scholar]
- Seguela P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Wang Y. Facilitation and depression at single central synapses. Neuron. 1995;14:795–802. doi: 10.1016/0896-6273(95)90223-6. [DOI] [PubMed] [Google Scholar]
- Turner TJ. Nicotine enhancement of dopamine release by a calcium-dependent increase in the size of the readily releasable pool of synaptic vesicles. J Neurosci. 2004;24:11328–11336. doi: 10.1523/JNEUROSCI.1559-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainer BH, Steininger TL, Roback JD, Burke-Watson MA, Mufson EJ, Kordower J. Ascending cholinergic pathways: functional organization and implications for disease models. Prog Brain Res. 1993;98:9–30. doi: 10.1016/s0079-6123(08)62378-x. [DOI] [PubMed] [Google Scholar]
- Wonnacott S. Presynaptic nicotinic ACh receptors. Trends Neurosci. 1997;20:92–98. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]
- Wonnacott S, Barik J, Dickinson J, Jones IW. Nicotinic receptors modulate transmitter cross talk in the CNS: nicotinic modulation of transmitters. J Mol Neurosci. 2006;30:137–140. doi: 10.1385/JMN:30:1:137. [DOI] [PubMed] [Google Scholar]
- Woolf NJ. Cholinergic systems in mammalian brain and spinal cord. Prog Neurobiol. 1991;37:475–524. doi: 10.1016/0301-0082(91)90006-m. [DOI] [PubMed] [Google Scholar]
- Woolf NJ, Butcher LL. Cholinergic projections to the basolateral amygdala: a combined Evans Blue and acetylcholinesterase analysis. Brain research bulletin. 1982;8:751–763. doi: 10.1016/0361-9230(82)90102-2. [DOI] [PubMed] [Google Scholar]
- Zhang J, Berg DK. Reversible inhibition of GABAA receptors by alpha7-containing nicotinic receptors on the vertebrate postsynaptic neurons. The Journal of physiology. 2007;579:753–763. doi: 10.1113/jphysiol.2006.124578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PJ, Stewart RR, McIntosh JM, Weight FF. Activation of nicotinic acetylcholine receptors increases the frequency of spontaneous GABAergic IPSCs in rat basolateral amygdala neurons. J Neurophysiol. 2005;94:3081–3091. doi: 10.1152/jn.00974.2004. [DOI] [PubMed] [Google Scholar]
- Zwart R, Vijverberg HP. Potentiation and inhibition of neuronal nicotinic receptors by atropine: competitive and noncompetitive effects. Molecular pharmacology. 1997;52:886–895. doi: 10.1124/mol.52.5.886. [DOI] [PubMed] [Google Scholar]