

Abstract
Insulin resistance, hyperglycemia, hyperinsulinemia, hyperlipidemia and oxidative stress are risk factors related to cardiovascular diseases including congestive heart failure, myocardial infarction, ventricular hypertrophy, endothelial nitric oxide impairment in systemic blood vessels and the heart, atherosclerosis, and hypercoagulability of blood. The traditional focus on insulin sensitivity and blood levels of markers of risk determined in the fasted state is inconsistent with the large volume of recent data that indicates that the metabolic defect in the pre-diabetic and diabetic condition relates more strongly to postprandial deficiency than to the fasting state. Risk factors for adverse cardiovascular events can be detected in the pre-diabetic insulin-resistant subject based upon the metabolic response to a test meal even in the absence of altered fasting parameters. The normal response to a mixed meal includes a doubling of insulin action secondary to insulin-induced release of a putative hepatic insulin sensitizing substance (HISS) that acts selectively on skeletal muscle. HISS is released only in the fed state and accounts for meal-induced insulin sensitization. Blockade of HISS release leads to a condition referred to as HISS-dependent insulin resistance, which is suggested as the primary postprandial metabolic defect, accounting for postprandial hyperglycemia, hyperinsulinemia, hyperlipidemia, and increased oxidative stress in the pre-diabetic and diabetic condition. HISS-dependent insulin resistance represents a novel hypothesis and suggests a new diagnostic and therapeutic target.
Keywords: HISS, hyperglycemia, hyperlipidemia, hyperinsulinemia, oxidative stress
Early diagnosis of diabetes was done by the diagnostician tasting the copious volumes of urine produced by their patients. This diagnostic approach led to a severe underestimation of the incidence of diabetes. Modern diagnosticians depend almost entirely upon determinations of fasting plasma levels of glucose and, less frequently, insulin. This approach also leads to a severe underestimation of the metabolic dysfunction that is typical of the type 2 diabetic. A state of insulin resistance is recognized to occur well prior to the development of diabetes. The pre-diabetic state of insulin resistance can be diagnosed by determining the response to a test meal. The theme of this brief review is that the metabolic dysfunction in the pre-diabetic state is quantifiable and represents a major risk factor for the development of cardiovascular disease. Postprandial hyperglycemia, hyperinsulinemia, hyperlipidemia, and oxidative stress are individually and collectively recognizable risk factors. An abnormal meal-induced insulin sensitization (AMIS) is proposed as a novel hypothesis to account for the pre-diabetic state of insulin resistance which, if uncorrected, progresses to diabetes when the pancreatic insulin release is no longer able to compensate for AMIS. In this review, I suggest that the “HISS story” may explain the mechanism of AMIS and affords new approaches to diagnosis and treatment of pre-diabetic and diabetic insulin resistance.
Postprandial hyperglycemia
In 1997 the American Diabetes Association (ADA) introduced the term “impaired fasting glucose” to describe individuals whose fasting plasma glucose concentration was between 110 to 125 mg/dl (ADA 1997). Since this diagnostic category was proposed, questions have been raised as to its clinical utility and it has been suggested that impaired glucose tolerance is a better predictor of both type 2 diabetes and cardiovascular disease (DECODE Study Group 2001, 2003). In an effort to respond to these concerns, the ADA has recently proposed that the glucose level used to identify individuals with impaired fasting glucose be lowered from 110 to 100 mg/dl (ADA 2003). Recent studies to evaluate the modified standard have indicated a 4-fold increase in individuals considered to have impaired fasting glucose following the new criteria. Serious questions related to the clinical and public health implications of this new impaired fasting glucose definition have been raised (Ford et al 2005).
The current approach of screening for type 2 diabetes using the fasted metabolic status, while convenient, is not effective. In a recent review evaluating the status of screening for type 2 diabetes, Engelgau et al (2000) stated that one of the criteria for appropriate screening is that the tests should detect the preclinical stage of disease and that the tests be shown to be acceptable and reliable. The conclusion that current screening recommendations are not consistent with available evidence was briefly reviewed. Evidence is accumulating that most people with a 54–67% range of impaired glucose tolerance have fasting glucose in the normal range. Meta-analysis of 20 different European studies showed as many as 31% of those who were diabetic according to post-challenge plasma glucose had normal fasting values and therefore would not have been detected by a screening procedure based upon fasting glucose measurements alone.
The majority of studies and diagnosis continue to focus on the fasted state. However, the importance of postprandial rather than fasting metabolic defects, as related to cardiovascular disease, is becoming increasingly recognized. The relationship between HbA1c and plasma glucose in patients with type 2 diabetes was determined at four time points during the day and HbA1c levels were found to be significantly predicted by plasma glucose levels measured only at post-lunch and extended post-lunch (5 hours) time points (Avignon et al 1997). The strongest age- and sex-adjusted relative risk for all-cause and cardiovascular mortality were associated with 2 hour post-load plasma glucose levels (de Vegt et al 1999). Increased mortality risk has been associated with 2 hour post-load plasma glucose levels to a much greater extent than with fasting plasma glucose (Hanefeld et al 1996; DECODE Study Group 1999). Isolated post-load hyperglycemia is a strong predictor of mortality (Simon et al 1987; DECODE Study Group 1999; Shaw et al 1999; Vaccaro et al 1999; Engelgau et al 2000; Simon and Brandenberger 2002).
It has been suggested that hyperglycemia-induced overproduction of superoxide by the mitochondrial electron transport chain accounts for the four main molecular mechanisms implicated in glucose-mediated vascular damage associated with blindness, renal failure, nerve damage, atherosclerosis, stroke, and hindlimb amputation (Brownlee 2001). Postprandial plasma glucose is an important determinant of both onset and development of nephropathy in type 2 diabetic patients (Schchiri et al 2000). Low density lipoprotein oxidation increases after meals (Diwadkar et al 1999) and directly relates to the degree of hyperglycemia (Ceriello et al 1999). Acute hyperglycemia is associated with an acute increase in clotting factor VII (Ceriello et al 1988) and enhanced thrombin activity that was proportional to the level of hyperglycemia (Ceriello et al 1996). The synthesis of fibrinogen, which is a strong risk factor for cardiovascular disease in both diabetic and non-diabetic subjects, increases during food intake in diabetic patients (Ceriello 1997; Bruttomesso et al 2001).
Acute hyperglycemia also stimulates increased expression of proadhesive proteins including ICAM-1 (Ceriello 2003). Dunn and Grant (2005) reviewed the relationship between the hypercoagulable prothrombic state that occurs in type 2 diabetes and cardiovascular disease risk. Hypertriglyceridemia and hyperglycemia have been shown to have an independent and cumulative effect on postprandial production of nitrotyrosine and adhesion molecules (Ceriello et al 2004).
In a review of the importance of postprandial hyperglycemia on the development of cardiovascular disease, Haffner (1998) suggested that atherosclerotic changes start to develop in the pre-diabetic state when postprandial blood glucose levels are only moderately elevated above normal levels. He suggested, however, that increased insulin resistance and hyperinsulinemia may be responsible for the atherosclerotic changes and that hyperglycemia may be a marker but not a cause of these changes.
Postprandial lipidemia
The hypothesis that atherogenesis is a postprandial phenomenon (Zilversmit 1979) was largely ignored until fairly recently when re-emphasis on the acute response to test meals demonstrated a clear superiority in predicting cardiovascular risk factors. An oral glucose tolerance test resulted in hyperglycemia and similar cardiovascular responses in both healthy and diabetic subjects. However, the use of the more physiological mixed meal test revealed myocardial endothelial dysfunction in the diabetic subjects. Myocardial blood flow, which was similar in fasted diabetic and healthy subjects, increased significantly in response to a standardized mixed meal in the control subjects but decreased significantly in the diabetic patients (Scognamiglio et al 2005). The superiority of a mixed meal versus the oral glucose tolerance test, related to cardiac dysfunction, was proposed to relate to the postprandial hypertriglyceridemia which only occurred using the test meal (Scognamiglio et al 2005). A negative influence of postprandial hypertriglyceridemia on endothelial function has been reported in diabetic subjects (Bae et al 2001; Anderson et al 2001). Postprandial hypertriglyceridemia is a recognized independent predictor of cardiovascular pathology (Anderson et al 2001).
Postprandial dyslipidemia is prevalent in diabetic patients even with normal fasting triglyceride concentrations (Mero et al 1998). Insulin resistance causes increased flux of free fatty acids and thus enhanced VLDL synthesis in the liver. Postprandial hypertriglyceridemia contributes to the metabolic disturbances transforming lipoprotein subclasses into more atherogenic forms.
Healthy, male, first-degree relatives of patients with type 2 diabetes have insulin resistance and postprandial hypertriglyceridemia despite having normal fasting triglyceride levels (Axelsen et al 1999). Newly diagnosed hypertensive patients with hypertriglyceridemia were more insulin resistant and showed greater postprandial hypertriglyceridemia than hypertensive patients not showing elevated fasting levels of triglycerides (Hwu et al 2002). Atherosclerosis is an inflammatory disorder involving leukocytes. The postprandial triglyceride increment after a fat meal was paralleled by a leukocyte increment due to an increase in neutrophils in the first two postprandial hours (van Oostrom et al 2003). It has even been suggested that the cardiovascular risk recognized to occur with chronic smokers is related to insulin resistance and postprandial dyslipidemia that is not mirrored by fasting hypertriglyceridemia (Eliasson et al 1997). The recognized impact of various nutritional supplements or beverages, including tea, in lowering the incidence of cardiovascular disease may be related to the demonstration of attenuation of the postprandial increase in plasma triglyceride levels following a fat load (Unno et al 2005).
Postprandial hyperinsulinemia
Impaired glucose metabolism was demonstrated to be a predictor of development of congestive heart failure in a large community-based sample of elderly men (Ingelsson et al 2005). Surprisingly, when insulin sensitivity and the impaired glucose tolerance were analyzed as co-variates, obesity was no longer a significant predictor of the subsequent development of congestive heart failure. This suggests that obesity may be a coincidental predictor of congestive heart failure and may simply be a marker of insulin resistance, which is compensated for by hyperinsulinemia.
Ingelsson et al (2005) suggest that the hyperinsulinemia associated with impaired glucose metabolism represents a risk factor through several possible mechanisms. Insulin acts as a growth factor in the myocardium as shown by increased myocardial mass and decreased cardiac output in rats exposed to sustained hyperinsulinemia (Holmang et al 1996). Hyperinsulinemia leads to sodium retention (DeFronzo et al 1975) which may exacerbate subclinical myocardial dysfunction due to blood volume expansion. Hyperinsulinemia leads to sympathetic nervous system activation (Anderson et al 1991) which is a presumed causal factor for congestive heart failure (Kannel and Belanger 1991; Bell 2003). Insulin resistance is related to increased effectiveness of angiotensin II on blood pressure (Gaboury et al 1994) and cellular hypertrophy and collagen production (Sartori et al 2004) in individuals with hypertension, leading to myocardial hypertrophy and fibrosis (Bell 2003) and likely subsequent congestive heart failure.
The relationship between elevated insulin levels and heart disease remains controversial mainly because it is often not clear exactly what is being measured by insulin assays. Using an assay specific for insulin and proinsulin, Zethelius et al (2005) suggested that proinsulin levels rather than insulin levels were a better predictor of development of coronary heart disease in a study of older Swedish males. The literature supporting proinsulin levels as a better marker than insulin suggests a unique biological role for proinsulin and could also be interpreted to indicate that the fasting levels of proinsulin are not representative of postprandial responses, rather that the slow metabolic degradation of proinsulin may lead to greater fasting levels of proinsulin and thus reflect postprandial hyperinsulinemia.
Somogyi et al (2005) studied hyperglycemia-induced oxidative stress using the rat streptozotocin model. The diabetic rats not treated with insulin showed enhanced activity of antioxidant enzymes. Diabetic rats treated with insulin showed decreased total oxidant scavenger capacity and development of left ventricular hypertrophy. They suggested that increased activity of the antioxidant enzymes inhibits subcellular remodeling processes thereby inhibiting cardiac hypertrophy, whereas insulin decreases the activity of the antioxidant system and can enhance the function of other localized tissue-specific growth factors. Chronic hyperinsulinemia may contribute to pathologies related to unregulated growth stimulation.
Postprandial oxidative stress
Ceriello (2000) has suggested that increased postprandial oxidative stress may be the common pathway through which the majority of diabetic complications occur. As previously discussed, both hyperglycemia and hyperlipidemia lead to increased oxidative stress. Meal consumption in healthy subjects has been shown to acutely reduce antioxidant defenses (Ceriello et al 1998) and produces an oxidative stress (Ceriello et al 1998; Ursini et al 1998). Oxidative stress is linked to the pathogenesis of cardiovascular disease (Griendling and Alexander 1997) and hypertension (Nakazono et al 1991). Antioxidants produce vasodilation in patients with hypertension and coronary artery disease (Ceriello et al 1991; Levine et al 1996; Solzbach et al 1997). This is consistent with evidence that free radical production accompanies endothelial dysfunction (Griendling and Alexander 1997) and that antioxidants improve endothelial dysfunction (Anderson et al 1995). Ceriello, recognizing the strong relationship between processing a meal and the production of oxidative stress, concluded that “…paradoxically, the vast majority of the studies on cardiovascular disease risk factors have been conducted by measuring them in strictly fasting conditions. This simply means that most of the data available to date may not reflect the real situation” (Ceriello 2000, p128).
It has been suggested that nitric oxide is inactivated by excess superoxide production in the postprandial state in both healthy and diabetic subjects and that the subsequent impaired vascular reactivity can be predicted based upon the circulating level of nitrotyrosine (Ceriello 2002). Vitamin E and other antioxidants administered to diabetics increase tissue antioxidant levels, improve the action of insulin, promote endothelial function, prevent oxidation of serum lipids, suppress protein glycation and reduce the oxidative load (Rosen et al 1995). Drugs that have significant antioxidant activity may improve the risk factors for cardiovascular disease through the sequestration of superoxide, thereby reducing the nitric oxide-dependent cardiovascular and metabolic dysfunctions.
Whiteside (2005) reviewed the literature on vascular complications of diabetes and reactive oxygen species, concluding that attention must be directed to more effective antioxidants as few clinical trials indicate a major therapeutic benefit from current monotherapy approaches. A recent study of ours strongly supports this conclusion. Insulin resistance produced in response to the chemical hepatotoxin, thioacetamide, can be prevented from developing by the use of an antioxidant cocktail consisting of vitamin E, vitamin C, and S-adenosylmethionine, but not the individual components of the cocktail (Ming et al 2006). The insulin resistance produced by thioacetamide was shown to be represented by HISS-dependent insulin resistance with direct (HISS-independent) insulin action remaining unaltered (HISS and HISS-dependent insulin resistance are discussed later). The use of an antioxidant cocktail designed to protect multiple sites may offer a more rational approach to antioxidant therapy.
Cardiovascular disease
Insulin resistance in type 2 diabetics is associated with increased prevalence of coronary heart disease, blood coagulability, and dislipidemia leading to the suggestion that reducing insulin resistance in diabetics may reduce their tendency to develop thrombosis and hence coronary heart disease risk (Wannamethee et al 2004). In post-myocardial infarction patients, the metabolic syndrome and diabetes were prevalent and associated with increased risk of cardiovascular events and death (Levantesi et al 2005). Impaired glucose regulation has been related to left ventricular systolic (Arnlov et al 2001) and diastolic (Arnlov et al 2005) dysfunction and left ventricular remodeling (Devereux et al 2000; Sundstrom et al 2000; Rutter et al 2003). The exaggerated cardiac damage in response to acute ischemia/reperfusion injury induced by repetitive hyperglycemia was prevented by co-administration of acarbose (Frantz et al 2005).
Chronic inflammatory diseases such as rheumatoid arthritis (Dessein et al 2005), polycystic ovarian syndrome (Dokras et al 2005; Kravariti et al 2005), and obesity (Karelis et al 2005) represent a high risk factor for development of insulin resistance. Primary renal disease is associated with postprandial hypertriglyceridemia (Charlesworth et al 2005). C reactive protein has recently been demonstrated to be associated with high incidence of insulin resistance (Festa et al 2000; Lee et al 2004; Rutter et al 2004) and cardiovascular disease (Koenig et al 2004; Pai et al 2004) and is also elevated in the above chronic inflammatory disease conditions. Low grade systemic inflammation, identified by elevated C reactive protein, is present in normal weight subjects who show insulin resistance but no other metabolic abnormalities (Bo et al 2005). Testing for high sensitivity C reactive protein has recently (Pearson et al 2003) been recommended by the American Heart Association for the assessment of the risk for developing cardiovascular disease (Rifai 2005). Although one study suggests that the association between C reactive protein and the metabolic syndrome disappears after adjustment for fitness level (Lamonte et al 2005).
Postprandial nutrient processing and the HISS hypothesis
We have recently demonstrated (Lautt et al 2001) that the glucose disposal effect of a bolus of insulin is approximately doubled in the early postprandial state and this meal-induced insulin sensitization (MIS) progressively decreases to a baseline level of insulin sensitivity after 24 hours of fasting. As a brief overview, the MIS process is proposed to occur as a result of insulin acting on the liver to release a hepatic insulin sensitizing substance (HISS) that acts selectively on skeletal muscle to stimulate glucose uptake. HISS action thus accounts for the large postprandial glucose sequestration in skeletal muscle (Xie and Lautt 1996a, 1996b; Moore et al 2002). The HISS hypothesis further proposes that blockade of HISS release results in postprandial hyperglycemia, which leads to postprandial compensatory hyperinsulinemia, with the increased insulin acting primarily on the liver and adipose tissue and resulting in increased formation of triglycerides and VLDL as well as increased free radical production (i.e. the metabolic syndrome). The sequence of discoveries leading to the current status of the HISS hypothesis is reviewed (Lautt 2004) as are methods to evaluate HISS-dependent and HISS-independent insulin action (Lautt 2003).
Methods used to quantitate the MIS process rely on an index of insulin sensitivity determined before and after administration of a mixed test meal. Although the original discovery of the dependence of whole body insulin action on hepatic nerves was made in cats using a standard insulin tolerance test (Xie et al 1993), the problems associated with the hypoglycemia that ensues following a bolus of insulin led us to develop the rapid insulin sensitivity test (RIST). The RIST is simply a rapidly sampled transient euglycemic clamp (Lautt et al 1998) and has been used in cats, mice, rats, pigs, and humans and with a wide range of physiological and pathophysiological models (for review see Lautt 2003). The RIST and the insulin tolerance test are equally able to detect HISS action and different insulin effectiveness in the fed versus fasted state (Reid et al 2002). The RIST is highly reproducible and can be repeated several times consecutively in the same animal.
Abnormal insulin delivery, such as produced by a single large bolus of 300 mU/kg, results in an inability of subsequent normal doses of insulin to cause HISS release. This would appear to be protective in nature. Further, when insulin is administered as pulses, subsequent insulin action is demonstrated to be unaffected whereas administration of the same dose of insulin by constant infusion results in an impairment of HISS release in subsequent tests with the degree of impairment related to the duration of the constant infusion (Reid and Lautt 2004). Similar endocrine dependence on pulsatile stimulation is seen with the gonadotropins, which, when delivered in pulses results in testosterone secretion but when delivered in the same dose but at a constant rate results in chemical castration (Belchetz et al 1978). In our hands, the 3-hour hyperinsulinemic euglycemic clamp utilizing continuous insulin infusion was not capable of determining differences in insulin sensitivity before and after a meal except for an increased response in the fed state within the first 30 minutes of infusion (Reid et al 2002). Others (Zsuga et al 2004) however have studied aspects related to HISS and have found similar results using a prolonged clamp or the RIST. Studies in the conscious dog (Moore et al 2002) were able to detect significant HISS-like responses using a prolonged clamp. HISS release is dependent on hepatic parasympathetic tone. Cardiovascular parasympathetic dysfunction is associated with diabetes and is detectable at the pre-diabetic stage of insulin resistance (Lautt 1999). A 90 minute hyperinsulinemic euglycemic clamp resulted in a decrease in cardiac parasympathetic tone and an increase in sympathetic nerve activity in skeletal muscle (Van de Borne et al 1999) suggesting that the prolonged, constant-infusion euglycemic clamp may not be the most appropriate index of insulin sensitivity under conditions, such as diabetes and obesity, where normal parasympathetic tone is important. Use of the euglycemic clamp also resulted in elevated levels of cytokines, beta amyloid and norepinephrine in cerebrospinal fluid (Fishel et al 2005). It is crucial that any methods used to study MIS, and the postprandial defect we refer to as HISS-dependent insulin resistance (HDIR), must be able to quantify insulin effect in the fed and fasted state.
HISS release can be acutely blocked by surgical parasympathetic denervation of the liver which results in decreased postprandial insulin sensitivity that is indistinguishable from that measured in the fasted state. Further, after hepatic denervation, mimicking the hepatic parasympathetic permissive signal through intraportal venous infusion of acetylcholine does not result in a direct impact on glycemia but the restored parasympathetic signal allows insulin to cause HISS release. Both the defect in insulin response subsequent to hepatic denervation and its restoration subsequent to intraportal but not intravenous acetylcholine has been demonstrated to occur in skeletal muscle (Xie and Lautt 1996a, 1996b; Moore et al 2002).
The ability of surgical denervation of the liver to result in a selective inhibition of skeletal muscle response to glucose could suggest either a neural or hormonal link between the liver and skeletal muscle. However, the observation that restoration of the parasympathetic permissive signal can be achieved by intraportal but not intravenous continuous administration of acetylcholine proves the hormonal nature of the regulatory process. It is important to indicate that the parasympathetic signal in the liver is “permissive” in nature as demonstrated by the observation that the intraportal acetylcholine infusion does not result in an alteration of baseline glycemia but does allow insulin to have a restored effect in skeletal muscle. Similar studies have been done related to the hepatic parasympathetic requirement for sequential activation of hepatic muscarinic receptors and nitric oxide production. Sadri and Lautt (1999) reported that nitric oxide synthase antagonists were much more effective at blocking HISS release than were intravenous doses. Similarly, in the presence of blockade of hepatic nitric oxide synthase, the permissive parasympathetic signal could be restored by intraportal but not intravenous administration of nitric oxide donors. Guarino et al (2004) demonstrated that the sequence of parasympathetic nerve activation involves sequential muscarinic and secondary nitric oxide synthase activation as demonstrated by the ability of a nitric oxide donor to reverse atropine-induced HISS-dependent insulin resistance and the inability of intraportal acetylcholine to reverse HISS-dependent insulin resistance produced by prior blockade of hepatic nitric oxide synthase.
Cats (Xie and Lautt 1995), rats (Lautt et al 2001), and dogs (Moore et al 2002) that have been fasted for 16 hours continue to show a small HISS-dependent component of insulin action of approximately 25%–35%. The HISS-independent component (direct insulin action) is not altered by feeding or fasting nor is it altered by administration of atropine or hepatic denervation or blockade of hepatic nitric oxide (Lautt et al 2001). The HISS-dependent component of insulin action represents approximately 55% of the glucose disposal action of boluses of insulin over a wide range of insulin doses, from a barely detectable stimulation of glucose uptake induced by 5 mU/kg up to 100 mU/kg.
HISS-dependent insulin resistance (HDIR) accounts for the insulin resistance seen in a wide range of animal models including the spontaneously hypertensive rat, sucrose-fed rats, animals with liver disease induced by chronic bile duct ligation or chemical toxins, adult offspring of fetal alcohol exposure, acute stress and ageing, physical interruption of hepatic parasympathetic nerves, and pharmacological blockade of hepatic muscarinic cholinergic receptors, nitric oxide production or cyclooxygenase (reviewed Lautt 2003).
The permissive feeding signal (Figure 1), delivered to the liver through the parasympathetic nerves acts via muscarinic receptor activation and generation of hepatic nitric oxide which then results in the insulin dose-related release of HISS (Sadri and Lautt 1999; Guarino et al 2003). The nitric oxide acts through hepatic guanylyl cyclase (Correia et al 2002; Guarino et al 2004). Hepatic glutathione level, which decreases significantly with fasting and rapidly increases following re-feeding is also an essential component of the feeding response in rats (Guarino et al 2003; Lautt et al 2005; Sadri et al 2006). The mechanism of regulation of hepatic glutathione in response to feeding is unknown.
Figure 1.
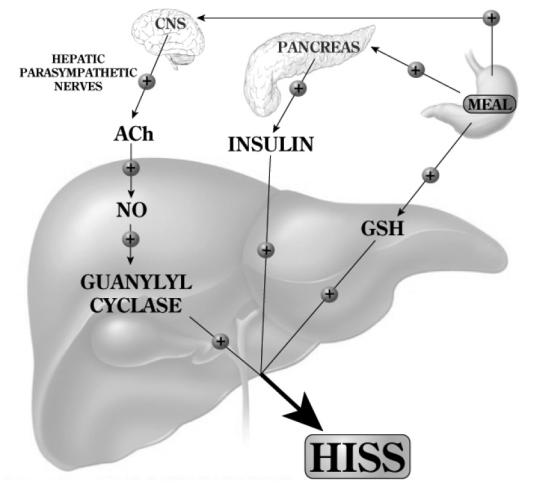
Feeding results in an increase of hepatic glutathione (GSH) and a parasympathetic signal to the liver that acts, via acetylcholine, on muscarinic receptors to activate NO release which, in turn, activates adenylyl cyclase. Both of these signals are permissive and both are needed in order that insulin can cause the release of hepatic insulin sensitizing substance (HISS). HISS acts selectively on skeletal muscle. Blockade of any portion of these pathways leads to blockade of HISS release and a state of HISS-dependent insulin resistance which is physiologically regulated to occur in the fasted state but, when not activated by feeding, is suggested to account for postprandial hyperglycemia, hyperinsulinemia, hyperlipidemia, and increased oxidative stress.
In a recent study we utilized a liquid test meal administered through a gastric catheter to quantitate MIS in both conscious and anesthetized rats by determining insulin sensitivity before and after the meal (Sadri et al 2006). A mixed liquid test meal, but not sucrose or glucose, resulted in MIS that could be completely reversed by atropine administration (Figure 2). The insulin sensitivity index increased in response to feeding and was reversed completely back to fasting levels of sensitivity by atropine. Similarly, prior denervation of the liver completely prevented the MIS process from occurring, thereby confirming the central role of hepatic parasympathetic nerves in the feeding signal. MIS has also recently been shown to occur in healthy male volunteers and blockade of MIS has been confirmed to occur in humans subsequent to atropine administration (Patarrao et al 2005).
Figure 2.
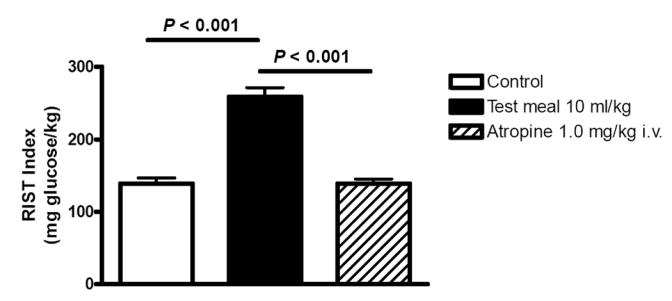
The RIST index (mg glucose/kg body weight required to maintain euglycemia after a bolus of 50 mU/kg of insulin) increased 90 minutes after administration of a mixed liquid test meal via an indwelling gastric catheter in conscious unrestrained rats. Atropine (1 mg/kg), which blocks release of hepatic insulin sensitizing substance (HISS), completely eliminated the meal-induced insulin sensitization (MIS). The same report showed that sucrose or glucose were ineffective in activating MIS. Prior surgical denervation of the liver blocked MIS from developing (Sadri et al 2006).
Therapeutic potential
The HISS hypothesis is suggested to account for meal-induced insulin sensitization. Absence of HISS action, or HISS-dependent insulin resistance (HDIR), is suggested to account for the major postprandial metabolic dysfunctions seen in the pre-diabetic insulin resistant state. The conclusions reached in many of the studies referred to previously suggest that therapeutic intervention should be focused on correction of the postprandial insulin resistance. In a recent review evaluating new drug targets for type 2 diabetes and the metabolic syndrome, Moller (2001 p821) emphasized that current therapies for type 2 diabetes were developed in the absence of defined molecular targets or an understanding of disease pathogenesis: “These therapies have limited efficacy, limited tolerability, and significant mechanism-based side effects. Of particular concern is the tendency for most treatments to enhance weigh gain. Several current approaches are also associated with episodes of hypoglycemia, and few of the available therapies adequately address underlying defects such as obesity and/or insulin resistance.” He further concluded that “Thus, newer approaches are desperately needed. Particular emphasis should be placed on finding and using mechanisms that are dependent on physiological responses and that result in weight loss.” With the recent understanding of the physiology and pathology related to HISS, targeting of this mechanism offers a novel approach to restoring normal postprandial physiological insulin sensitivity in skeletal muscle, the major site where the insulin resistance is generally acknowledged to occur in many insulin resistant states.
Conclusion
Postprandial hyperglycemia, hyperinsulinemia, hyperlipidemia and increased oxidative stress are strongly related to cardiovascular disease. Meal-induced insulin sensitization (MIS) normally occurs in response to a mixed meal, and absence of MIS is based on inability of the liver to secrete HISS in response to insulin. The argument has been made that HDIR could account for the primary endocrine defect in the pre-diabetic and diabetic state. Absence of MIS results in altered postprandial nutrient processing away from skeletal muscle and toward lipid production by liver and fat cells. These are all hypotheses that should be tested in the clinical setting.
The absence of MIS in response to glucose or sucrose “meals” in rats has major implications for interpretation of a large body of research, based on the oral glucose tolerance test, if confirmed in humans.
Although we advocate the use of the rapid insulin sensitivity test, any method used to evaluate the mechanisms and consequence of abnormal meal-induced insulin sensitization must clearly demonstrate a difference in insulin effect in the fed and fasted state.
HDIR is suggested to occur in association with a wide range of chronic inflammatory states and to contribute negatively as a strong risk factor for development of diabetes, obesity, cardiac and vascular disease, and asthma. If the “HISS story” is fundamentally correct, it represents a new paradigm impacting diagnosis and therapy of a broad spectrum of metabolic disorders.
Acknowledgments
The HISS-related work of the author cited here has been supported by operating grants from the Canadian Institutes of Health Research and the Canadian Diabetes Association. The HISS-related studies were carried out mainly by graduate students, technologists and trainees in the laboratories of Lautt (Winnipeg, Canada) and Macedo (Lisbon, Portugal). Manuscript preparation was by Karen Sanders. The author discloses that intellectual property related to diagnosis, prevention, and therapy of HISS-dependent insulin resistance has been licensed by the University of Manitoba to DiaMedica Inc.
References
- [ADA] ADA Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Report of the Expert Committee on the diagnosis and classification of diabetes mellitus. Diabetes Care. 1997;20:1183–97. doi: 10.2337/diacare.20.7.1183. [DOI] [PubMed] [Google Scholar]
- [ADA] ADA Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Report of the Expert Committee on the diagnosis and classification of diabetes mellitus. Diabetes Care. 2003;26:S5–20. doi: 10.2337/diacare.26.2007.s5. [DOI] [PubMed] [Google Scholar]
- Anderson RA, Evans ML, Ellis GR, et al. The relationship between post-prandial lipeemia, endothelial function and oxidative stress in healthy individuals and patients with type 2 diabetes. Atherosclerosis. 2001;154:475–83. doi: 10.1016/s0021-9150(00)00499-8. [DOI] [PubMed] [Google Scholar]
- Anderson EA, Hoffman RP, Balon TW, et al. Hyperinsulinemia produces both sympathetic neural activation and vasodilation in normal humans. J Clin Invest. 1991;87:2246–52. doi: 10.1172/JCI115260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson TJ, Meredith IT, Yeung AC, et al. The effect of cholesterol-lowering and antioxidant therapy on endothelial-dependent coronary vasomotion. N Engl J Med. 1995;332:488–93. doi: 10.1056/NEJM199502233320802. [DOI] [PubMed] [Google Scholar]
- Arnlov J, Lind L, Zethelius B, et al. Several factors associated with the insulin resistance syndrome are predictors of left ventricular systolic dysfunction in a male population after 20 years of follow-up. Am Heart J. 2001;142:720–4. doi: 10.1067/mhj.2001.116957. [DOI] [PubMed] [Google Scholar]
- Arnlov J, Lind L, Sundstrom J, et al. Insulin resistance, dietary fat intake and blood pressure predict left ventricular diastolic dysfunction 20 years later. Nutr Metab Cardiovasc Dis. 2005;15:242–9. doi: 10.1016/j.numecd.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Avignon A, Radauccanu A, Monnier L. Nonfasting plasma glucose is a better marker of diabetic control than fasting plasma glucose in type 2 diabetes. Diabetes Care. 1997;20:1822–6. doi: 10.2337/diacare.20.12.1822. [DOI] [PubMed] [Google Scholar]
- Axelsen M, Smith U, Eriksson JW, et al. Postprandial hypertriglyceridemia and insulin resistance in normoglycemic first-degree relatives of patients with type 2 diabetes. Ann Intern Med. 1999;131:27–31. doi: 10.7326/0003-4819-131-1-199907060-00006. [DOI] [PubMed] [Google Scholar]
- Bae JH, Bassenge E, Kim KB, et al. Postprandial hypertriglyceridemia impairs endothelial function by enhanced oxidant stress. Atherosclerosis. 2001;155:517–23. doi: 10.1016/s0021-9150(00)00601-8. [DOI] [PubMed] [Google Scholar]
- Belchetz PE, Plant TM, Nakai Y, et al. Hypophysial responses to continuous and intermittent delivery of hypothalamic gonadotropin-releasing hormone. Science. 1978;202:631–3. doi: 10.1126/science.100883. [DOI] [PubMed] [Google Scholar]
- Bell DS. Heart failure: the frequent, forgotten, and often fatal complication of diabetes. Diabetes Care. 2003;26:2433–41. doi: 10.2337/diacare.26.8.2433. [DOI] [PubMed] [Google Scholar]
- Bo S, Gambino R, Uberti B, et al. Does C-reactive protein identify a subclinical metabolic disease in healthy subjects? Eur J Clin Invest. 2005;35:265–70. doi: 10.1111/j.1365-2362.2005.01490.x. [DOI] [PubMed] [Google Scholar]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–19. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- Bruttomesso D, Iori E, Kiwanuka E, et al. Insulin infusion normalizes fasting and post-prandial albumin and fibrinogen synthesis in type 1 diabetes mellitus. Diabet Med. 2001;18:915–20. doi: 10.1046/j.1464-5491.2001.00606.x. [DOI] [PubMed] [Google Scholar]
- Ceriello A. Fibrinogen and diabetes mellitus. Is it time for intervention trials? Diabetologia. 1997;40:731–4. doi: 10.1007/s001250050741. [DOI] [PubMed] [Google Scholar]
- Ceriello A. The post-prandial state and cardiovascular disease: relevance to diabetes mellitus. Diabetes Metab Res Rev. 2000;16:125–32. doi: 10.1002/(sici)1520-7560(200003/04)16:2<125::aid-dmrr90>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Ceriello A. Nitrotyrosine: new findings as a marker of postprandial oxidative stress. Int J Clin Pract Suppl. 2002;129:51–8. [PubMed] [Google Scholar]
- Ceriello A. The possible role of postprandial hyperglycaemia in the pathogenesis of diabetic complications. Diabetologia. 2003;46:M9–16. doi: 10.1007/s00125-002-0931-5. [DOI] [PubMed] [Google Scholar]
- Ceriello A, Bortolotti N, Motz E, et al. Meal-induced oxidative stress and low-density lipoprotein (LDL) oxidation in diabetes: the possible role of hyperglycemia. Metabolism. 1999;48:1503–8. doi: 10.1016/s0026-0495(99)90237-8. [DOI] [PubMed] [Google Scholar]
- Ceriello A, Bortolotti N, Motz E, et al. Meal-generated oxidative stress in type 2 diabetic patients. Diabetes Care. 1998;21:1529–33. doi: 10.2337/diacare.21.9.1529. [DOI] [PubMed] [Google Scholar]
- Ceriello A, Giugliano D, Quatraro A, et al. Anti-oxidants show an anti-hypertensive effect in diabetic and hypertensive subjects. Clin Sci (Colch) 1991;81:739–42. doi: 10.1042/cs0810739. [DOI] [PubMed] [Google Scholar]
- Ceriello A, Giugliano D, Quatraro A, et al. Blood glucose may condition factor VII levels in diabetic and normal subjects. Diabetologia. 1988;31:889–91. doi: 10.1007/BF00265372. [DOI] [PubMed] [Google Scholar]
- Ceriello A, Taboga C, Tonutti L, et al. Post-meal coagulation activation in diabetes mellitus: the effect of Acarbose. Diabetologia. 1996;39:469–73. doi: 10.1007/BF00400679. [DOI] [PubMed] [Google Scholar]
- Ceriello A, Quagliaro L, Piconi L, et al. Effect of postprandial hypertriglyceridemia and hyperglycemia on circulating adhesion molecules and oxidative stress generation and the possible role of simvastatin treatment. Diabetes. 2004;53:701–10. doi: 10.2337/diabetes.53.3.701. [DOI] [PubMed] [Google Scholar]
- Charlesworth JA, Kriketos AD, Jones JE, et al. Insulin resistance and postprandial triglyceride levels in primary renal disease. Metab Clin Exp. 2005;54:821–8. doi: 10.1016/j.metabol.2005.01.028. [DOI] [PubMed] [Google Scholar]
- Correia NC, Guarino MP, Paposo J, et al. Hepatic guanylyl cyclase inhibition induces HISS-dependent insulin resistance. Proc West Pharmacol Soc. 2002;45:57–8. [PubMed] [Google Scholar]
- DECODE Study Group on behalf of the European Diabetes Epidemiology Group. Glucose tolerance and mortality comparison of WHO and American Diabetes Association diagnostic criteria. Lancet. 1999;354:617–21. [PubMed] [Google Scholar]
- DECODE Study Group the European Diabetes Epidemiology Group. Glucose tolerance and cardiovascular mortality: comparison of fasting and 2-hour diagnostic criteria. Arch Intern Med. 2001;161:397–404. doi: 10.1001/archinte.161.3.397. [DOI] [PubMed] [Google Scholar]
- DECODE Study Group the European Diabetes Epidemiology Group. Is the current definition for diabetes relevant to mortality risk form all causes and cardiovascular and noncardiovascular disease? Diabetes Care. 2003;26:688–96. doi: 10.2337/diacare.26.3.688. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Cooke CR, Andres R, et al. The effect of insulin on renal handling of sodium, potassium, calcium, and phosphate in man. J Clin Invest. 1975;55:845–55. doi: 10.1172/JCI107996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessein PH, Joffe BI, Stanwix AE. Editorial: Should we evaluate insulin sensitivity in rheumatoid arthritis? Sem Arth Rheum. 2005;35:5–7. doi: 10.1016/j.semarthrit.2005.03.008. [DOI] [PubMed] [Google Scholar]
- De Vegt F, Dekker JM, Ruhe HG, et al. Hyperglycaemia is associated with all-cause and cardiovascular mortality in the Hoorn population: the Hoorn study. Diabetologia. 1999;42:926–31. doi: 10.1007/s001250051249. [DOI] [PubMed] [Google Scholar]
- Devereux RB, Roman MJ, Paranicas M, et al. Impact of diabetes on cardiac structure and function: the strong heart study. Circulation. 2000;101:2271–6. doi: 10.1161/01.cir.101.19.2271. [DOI] [PubMed] [Google Scholar]
- Diwadkar VA, Anderson JW, Bridges SR, et al. Postprandial low density lipoproteins in type 2 diabetes are oxidized more extensively than fasting diabetes and control samples. Proc Soc Exp Biol Med. 1999;222:178–84. doi: 10.1046/j.1525-1373.1999.d01-129.x. [DOI] [PubMed] [Google Scholar]
- Dokras A, Bochner M, Hollinrake E. Screening women with polycystic ovary syndrome for metabolic syndrome. Obstet Gynecol. 2005;106:131–7. doi: 10.1097/01.AOG.0000167408.30893.6b. [DOI] [PubMed] [Google Scholar]
- Dunn EJ, Grant PJ. Type 2 diabetes: an atherothrombotic syndrome. Curr Molec Med. 2005;5:323–32. doi: 10.2174/1566524053766059. [DOI] [PubMed] [Google Scholar]
- Eliasson B, Mero N, Taskinen M-R, et al. The insulin resistance syndrome and postprandial lipid intolerance in smokers. Atherosclerosis. 1997;129:79–88. doi: 10.1016/s0021-9150(96)06028-5. [DOI] [PubMed] [Google Scholar]
- Engelgau MM, Narayan KMV, Herman WH. Screening for type 2 diabetes. Diabetes Care. 2000;23:1563–80. doi: 10.2337/diacare.23.10.1563. [DOI] [PubMed] [Google Scholar]
- Festa A, D’Agostino R, Jr, Howard G, et al. Chronic subclinical inflammation as part of the insulin resistance syndrome: the Insulin Resistance Atherosclerosis Study (IRAS) Circulation. 2000;102:42–7. doi: 10.1161/01.cir.102.1.42. [DOI] [PubMed] [Google Scholar]
- Fishel MA, Watson S, Montine TJ, et al. Hyperinsulinemia provokes synchronous increases in central inflammation and β-amyloid in normal adults. Arch Neurol. 2005;62:1539–44. doi: 10.1001/archneur.62.10.noc50112. [DOI] [PubMed] [Google Scholar]
- Ford ES, Abbasi F, Reaven GM. Prevalence of insulin resistance and the metabolic syndrome with alternative definitions of impaired fasting glucose. Atherosclerosis. 2005;181:143–8. doi: 10.1016/j.atherosclerosis.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Frantz S, Calvillo L, Tillmanns J, et al. Repetitive postprandial hyperglycemia increases cardiac ischemia/reperfusion injury: prevention by the α-glucosidase inhibitor acarbose. FASEB J. 2005;19:591–3. doi: 10.1096/fj.04-2459fje. [DOI] [PubMed] [Google Scholar]
- Gaboury CL, Simonson DC, Seely EW, et al. Relation of pressor responsiveness to angiotensin II and insulin resistance in hypertension. J Clin Invest. 1994;94:2295–300. doi: 10.1172/JCI117593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griendling KK, Alexander RW. Oxidative stress and cardiovascular disease. Circulation. 1997;96:3264–5. [PubMed] [Google Scholar]
- Guarino MP, Afonso RA, Raimundo N, et al. Hepatic glutathione and nitric oxide are critical for hepatic insulin-sensitizing substance action. Am J Physiol Gastrointest Liver Physiol. 2003;284:G588–94. doi: 10.1152/ajpgi.00423.2002. [DOI] [PubMed] [Google Scholar]
- Guarino MP, Correia NC, Lautt WW, et al. Insulin sensitivity is mediated by the activation of the Ach/NO/cGMP pathway in rat liver. Am J Physiol Gastrointest Liver Physiol. 2004;287:G527–32. doi: 10.1152/ajpgi.00085.2004. [DOI] [PubMed] [Google Scholar]
- Haffner SM. The importance of hyperglycemia in the nonfasting state to the development of cardiovascular disease. Endocrine Rev. 1998;19:583–92. doi: 10.1210/edrv.19.5.0343. [DOI] [PubMed] [Google Scholar]
- Hanefeld M, Fischer S, Julius U, et al. Risk factors for myocardial infarction and death in newly detected NIDDM: the Diabetes Intervention Study, 11-year follow-up. Diabetologia. 1996;39:1577–83. doi: 10.1007/s001250050617. [DOI] [PubMed] [Google Scholar]
- Holmany A, Yoshida N, Jennische E, et al. The effects of hyperinsulinaemia on myocardial mass, blood pressure regulation and central haemodynamics in rats. Eur J Clin Invest. 1996;26:973–8. doi: 10.1046/j.1365-2362.1996.2880577.x. [DOI] [PubMed] [Google Scholar]
- Hwu CN, Kwok CF, Kuo CS, et al. Exacerbation of insulin resistance and postprandial triglyceride response in newly diagnosed hypertensive patients with hypertriglyceridaemia. J Human Hypertens. 2002;16:487–93. doi: 10.1038/sj.jhh.1001426. [DOI] [PubMed] [Google Scholar]
- Ingelsson E, Sundstrom J, Arnlow J, et al. Insulin resistance and risk of congestive heart failure. JAMA. 2005;294:334–41. doi: 10.1001/jama.294.3.334. [DOI] [PubMed] [Google Scholar]
- Kannel WB, Belanger AJ. Epidemiology of heart failure. Am Heart J. 1991;121:951–7. doi: 10.1016/0002-8703(91)90225-7. [DOI] [PubMed] [Google Scholar]
- Karelis AD, Faraj M, Bastard JP, et al. The metabolically healthy but obese individual presents a favorable inflammation profile. J Clin Endocrinol Metab. 2005;90:4145–50. doi: 10.1210/jc.2005-0482. [DOI] [PubMed] [Google Scholar]
- Koenig W, Lowel H, Baumert J, et al. C-reactive protein modulates risk prediction based on the Framingham Score: implications for future risk assessment: results from a large cohort study in southern Germany. Circulation. 2004;109:1349–53. doi: 10.1161/01.CIR.0000120707.98922.E3. [DOI] [PubMed] [Google Scholar]
- Kravariti M, Naka KK, Kalantaridou SN, et al. Predictors of endothelial dysfunction in young women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2005;90:5088–95. doi: 10.1210/jc.2005-0151. [DOI] [PubMed] [Google Scholar]
- Lamonte MJ, Ainsworth BE, Durstine JL. Influence of cardiorespiratory fitness on the association between C-reactive protein and metabolic syndrome prevalence in racially diverse women. J Women’s Hlth. 2005;14:233–9. doi: 10.1089/jwh.2005.14.233. [DOI] [PubMed] [Google Scholar]
- Lautt WW. The HISS story overview: A novel hepatic neurohumoral regulation of peripheral insulin sensitivity in health and diabetes. Can J Physiol Pharmacol. 1999;77:553–62. [PubMed] [Google Scholar]
- Lautt WW. Practice and principles of pharmacodynamic determination of HISS-dependent and HISS-independent insulin action: methods to quantitate mechanisms of insulin resistance. Med Res Rev. 2003;23:1–14. doi: 10.1002/med.10022. [DOI] [PubMed] [Google Scholar]
- Lautt WW. A new paradigm for diabetes and obesity: the hepatic insulin sensitizing substance (HISS) hypothesis. J Pharmacol Sci. 2004;95:9–17. doi: 10.1254/jphs.95.9. [DOI] [PubMed] [Google Scholar]
- Lautt WW, Legare DJ, Reid MAG, et al. Alcohol suppresses meal-induced insulin sensitization (MIS) Metab Synd Rel Disord. 2005;3:51–9. doi: 10.1089/met.2005.3.51. [DOI] [PubMed] [Google Scholar]
- Lautt WW, Macedo MP, Sadri P, et al. Hepatic parasympathetic nerve-dependent control of peripheral insulin sensitivity is determined by feeding and fasting: dynamic control of HISS-dependent insulin action. Am J Physiol Gastrointest Liver Physiol. 2001;281:G29–36. doi: 10.1152/ajpgi.2001.281.1.G29. [DOI] [PubMed] [Google Scholar]
- Lautt WW, Wang X, Sadri P, et al. Rapid insulin sensitivity test (RIST) Can J Physiol Pharmacol. 1998;76:1080–6. doi: 10.1139/cjpp-76-12-1080. [DOI] [PubMed] [Google Scholar]
- Lee WY, Park JS, Noh WY, et al. C-reactive protein concentrations are related to insulin resistance and metabolic syndrome as defined by the ATP III report. Int J Cardiol. 2004;97:101–6. doi: 10.1016/j.ijcard.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Levantesi G, Macchia A, Marfisi RM, et al. Metabolic syndrome and risk of cardiovascular events after myocardial infarction. J Am Coll Cardiol. 2005;46:277–83. doi: 10.1016/j.jacc.2005.03.062. [DOI] [PubMed] [Google Scholar]
- Levine GN, Frei B, Koulouris SN, et al. Ascorbic acid reverses endothelial vasomotor dysfunction in patients with coronary artery disease. Circulation. 1996;93:1107–13. doi: 10.1161/01.cir.93.6.1107. [DOI] [PubMed] [Google Scholar]
- Mero N, Syvanne M, Taskinen M-R. Postprandial lipid metabolism in diabetes. Atherosclerosis. 1998;141:S53–5. doi: 10.1016/s0021-9150(98)00218-4. [DOI] [PubMed] [Google Scholar]
- Ming Z, Fan Y-J, Yang X, et al. Synergistic protection by S-adenosylmethionine with vitamins C and E on liver injury induced by thioacetamide in rats. Free Rad Biol Med. 2006;40:617–24. doi: 10.1016/j.freeradbiomed.2005.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller DE. New drug targets for type 2 diabetes and the metabolic syndrome. Nature. 2001;414:821–7. doi: 10.1038/414821a. [DOI] [PubMed] [Google Scholar]
- Moore MC, Satake S, Baranowski B, et al. Effect of hepatic denervation on peripheral insulin sensitivity in conscious dogs. Am J Physiol Endocrinol Metab. 2002;282:E286–96. doi: 10.1152/ajpendo.00201.2001. [DOI] [PubMed] [Google Scholar]
- Nakazono K, Watanabe N, Matsuno K, et al. Does superoxide underlie the pathogenesis of hypertension? Proc Natl Acad Sci USA. 1991;88:10045–8. doi: 10.1073/pnas.88.22.10045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai JK, Pischon T, Ma J, et al. Inflammatory markers and the risk of coronary heart disease in men and women. N Engl J Med. 2004;351:2599–610. doi: 10.1056/NEJMoa040967. [DOI] [PubMed] [Google Scholar]
- Patarrao RS, Lautt WW, Guarino MP, et al. Atropine inhibits postprandial insulin sensitivity in a dose dependent manner. Diabetologia. 2005;48:A211. [Google Scholar]
- Pearson TA, Mensah GA, Alexander RW, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107:499–511. doi: 10.1161/01.cir.0000052939.59093.45. [DOI] [PubMed] [Google Scholar]
- Reid MAG, Latour MG, Legare DJ, et al. Comparison of the rapid insulin sensitivity test (RIST), the insulin tolerance test (ITT), and the hyperinsulinemic euglycemic clamp (HIEC) to measure insulin action in rats. Can J Physiol Pharmacol. 2002;80:811–18. doi: 10.1139/y02-102. [DOI] [PubMed] [Google Scholar]
- Reid MAG, Lautt WW. Pattern of insulin delivery affects HISS action and insulin resistance. Can J Physiol Pharmacol. 2004;82:1068–1074. doi: 10.1139/y04-111. [DOI] [PubMed] [Google Scholar]
- Rifai N. High-sensitivity C-reactive protein: a useful marker for cardiovascular disease risk prediction and the metabolic syndrome. Clin Chem. 2005;51:504–5. doi: 10.1373/clinchem.2004.044990. [DOI] [PubMed] [Google Scholar]
- Rosen PT, Ballhausen T, Bloch W, et al. Endothelial relaxation is disturbed by oxidative stress in the diabetic rat heart: influence of tocopherol as antioxidant. Diabetologia. 1995;38:1157–68. doi: 10.1007/BF00422364. [DOI] [PubMed] [Google Scholar]
- Rutter MK, Meigs JB, Sullivan LM. C-reactive protein, the metabolic syndrome, and prediction of cardiovascular events in the Framingham Offspring Study. Circulation. 2004;110:380–5. doi: 10.1161/01.CIR.0000136581.59584.0E. [DOI] [PubMed] [Google Scholar]
- Rutter MK, Parise H, Benjamin EJ, et al. Impact of glucose intolerance and insulin resistance on cardiac structure and function: sex-related differences in the Framingham Heart Study. Circulation. 2003;107:448–54. doi: 10.1161/01.cir.0000045671.62860.98. [DOI] [PubMed] [Google Scholar]
- Sadri P, Lautt WW. Blockade of hepatic nitric oxide synthase causes insulin resistance. Am J Physiol. 1999;277:G101–8. doi: 10.1152/ajpgi.1999.277.1.G101. [DOI] [PubMed] [Google Scholar]
- Sadri P, Reid MAG, Afonso RA, et al. Meal-induced insulin sensitization in conscious and anesthetized rat models comparing liquid mixed meal with glucose and sucrose. Br J Nutr. 2006;95:288–95. doi: 10.1079/bjn20051644. [DOI] [PubMed] [Google Scholar]
- Sartori M, Ceolotto G, Papparella I, et al. Effects of angiotensin II and insulin on ERK1/2 activation in fibroblasts from hypertensive patients. Am J Hypertens. 2004;17:604–10. doi: 10.1016/j.amjhyper.2004.02.017. [DOI] [PubMed] [Google Scholar]
- Schchiri M, Kishikawa H, Ohkubo Y, et al. Long-term results of the Kumamoto Study on optimal diabetes control in type 2 diabetic patients. Diabetes Care. 2000;23:B21–9. [PubMed] [Google Scholar]
- Scognamiglio R, Negut C, Vigili De Kreutzenberg S, et al. Postprandial myocardial perfusion in healthy subjects and in type 2 diabetic patients. Circulation. 2005;112:179–84. doi: 10.1161/CIRCULATIONAHA.104.495127. [DOI] [PubMed] [Google Scholar]
- Shaw JE, Hodge AM, de Courten M, et al. Isolated post-challenge hyperglycemia confirmed as a risk factor for mortality. Diabetologia. 1999;42:1050–4. doi: 10.1007/s001250051269. [DOI] [PubMed] [Google Scholar]
- Simon C, Brandenberger G. Ultradian oscillations of insulin secretion in humans. Diabetes. 2002;51:S258–61. doi: 10.2337/diabetes.51.2007.s258. [DOI] [PubMed] [Google Scholar]
- Simon C, Follenius M, Brandenberger G. Postprandial oscillations of plasma glucose insulin and C-peptide in man. Diabetologia. 1987;30:769–73. doi: 10.1007/BF00275742. [DOI] [PubMed] [Google Scholar]
- Solzbach U, Hornng B, Jeserich M, et al. Vitamin C improves endothelial dysfunction of epicardial coronary arteries in hypertensive patients. Circulation. 1997;96:1513–19. doi: 10.1161/01.cir.96.5.1513. [DOI] [PubMed] [Google Scholar]
- Somogyi A, Ruzicska E, Blazovics A, et al. Insulin treatment decreases the antioxidant defense mechanism in experimental diabetes. Med Sci Monit. 2005;11:BR206–11. [PubMed] [Google Scholar]
- Sundstrom J, Lind L, Nystrom N, et al. Left ventricular concentric remodeling rather than left ventricular hypertrophy is related to the insulin resistance syndrome in elderly men. Circulation. 2000;101:2595–600. doi: 10.1161/01.cir.101.22.2595. [DOI] [PubMed] [Google Scholar]
- Takayama S, Legare DJ, Lautt WW. Dose-related atropine-induced insulin resistance: comparing intraportal versus intravenous administration. Proc West Pharmacol Soc. 2000;43:33–4. [PubMed] [Google Scholar]
- Tse TF, Clutter WE, Shah SD, et al. Neuroendocrine responses to glucose ingestion in man: specificity, temporal relationships, and quantitative aspects. J Clin Invest. 1983;72:270–7. doi: 10.1172/JCI110966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unno T, Tago M, Suzuki Y, et al. Effect of tea catechins on postprandial plasma lipid responses in human subjects. Br J Nutr. 2005;93:543–7. doi: 10.1079/bjn20041379. [DOI] [PubMed] [Google Scholar]
- Ursini F, Zamburlini A, Cazzolato G, et al. Postprandial plasma lipid hydroperoxides: a possible link between diet and atherosclerosis. Free Rad Biol Med. 1998;25:250–2. doi: 10.1016/s0891-5849(98)00044-6. [DOI] [PubMed] [Google Scholar]
- Vaccaro O, Ruffa G, Imperatore G, et al. Risk of diabetes in the new diagnostic category of impaired fasting glucose. Diabetes Care. 1999;22:1490–3. doi: 10.2337/diacare.22.9.1490. [DOI] [PubMed] [Google Scholar]
- Van De Borne P, Hausberg M, Hoffman RP, et al. Hyperinsulinemia produces cardiac vagal withdrawal and nonuniform sympathetic activation in normal subjects. Am J Physiol. 1999;276:R178–83. doi: 10.1152/ajpregu.1999.276.1.R178. [DOI] [PubMed] [Google Scholar]
- Van Oostrom AJHHM, Sijmonsma TP, Rabelink TJ, et al. Postprandial leukocyte increase in healthy subjects. Metabolism. 2003;52:199–202. doi: 10.1053/meta.2003.50037. [DOI] [PubMed] [Google Scholar]
- Wannamethee SG, Lowe GDO, Shaper AG, et al. Insulin resistance, haemostatic and inflammatory markers and coronary heart disease risk factors in Type 2 diabetic men with and without coronary heart disease. Diabetologia. 2004;47:1557–65. doi: 10.1007/s00125-004-1491-7. [DOI] [PubMed] [Google Scholar]
- Whiteside CI. Cellular mechanisms and treatment of diabetes vascular complications converge on reactive oxygen species. Curr Hypertens Rep. 2005;7:148–54. doi: 10.1007/s11906-005-0090-4. [DOI] [PubMed] [Google Scholar]
- Xie H, Lautt WW. Induction of insulin resistance by cholinergic blockade with atropine in the cat. J Auton Pharmacol. 1995;15:361–9. doi: 10.1111/j.1474-8673.1995.tb00402.x. [DOI] [PubMed] [Google Scholar]
- Xie H, Lautt WW. Insulin resistance of skeletal muscle produced by hepatic parasympathetic interruption. Am J Physiol. 1996a;270:E858–63. doi: 10.1152/ajpendo.1996.270.5.E858. [DOI] [PubMed] [Google Scholar]
- Xie H, Lautt WW. Insulin resistance caused by hepatic cholinergic interruption and reversed by acetylcholine administration. Am J Physiol. 1996b;271:E587–92. doi: 10.1152/ajpendo.1996.271.3.E587. [DOI] [PubMed] [Google Scholar]
- Xie H, Tsybenko VA, Johnson MV, et al. Insulin resistance of glucose response produced by hepatic denervations. Can J Physiol Pharmacol. 1993;71:175–8. doi: 10.1139/y93-024. [DOI] [PubMed] [Google Scholar]
- Zethelius B, Lithell H, Hales CN, et al. Insulin sensitivity, proinsulin and insulin as predictors of coronary heart disease. A population-based 10-year, follow-up study in 70-year-old men using the euglycaemic insulin clamp. Diabetologia. 2005;48:862–7. doi: 10.1007/s00125-005-1711-9. [DOI] [PubMed] [Google Scholar]
- Zilversmit DB. Atherogenesis: a post-prandial phenomenon. Circulation. 1979;60:473–85. doi: 10.1161/01.cir.60.3.473. [DOI] [PubMed] [Google Scholar]
- Zsuga J, Tory K, Jaszlits L, et al. Pre-clinical methods for the determination of insulin sensitivity. J Biochem Biophys Meth. 2004;61:253–8. doi: 10.1016/j.jbbm.2004.06.006. [DOI] [PubMed] [Google Scholar]