

Abstract
The regulation of the inflammatory response is often viewed as very complex with many cellular players. The type of immune response generated is dependent upon the nature of the immune stimulation. In autoimmunity, one of the most important players is the CD4 T cell. The CD4 T cell lineage consists of a number of phenotypically and functionally distinct subsets. The unique functions of CD4 T cells are often mediated by soluble factors, which shape the nature of the immune response. In a T cell-mediated autoimmune response, such as in multiple sclerosis (MS), the CD4 T cell is thought to orchestrate and drive the immune response resulting in inflammation within the central nervous system (CNS). The extent of the inflammation must be tightly controlled or permanent tissue damage will occur. In MS, progressive debilitating disease is thought to be due to such damage. In addition to promoting inflammation, the CD4 T cell lineage also has the capacity to prevent and downmodulate inflammation. This is accomplished by specific CD4 T regulatory (Treg) cells and other regulatory feedback mechanisms. Thus although the complexity of the immune system is often viewed as too complicated for a nonimmunologist to fully understand, there are patterns that emerge that make the system clearer. One such pattern is the balance that the immune system must always maintain. A weak or slow immune response to a pathogen can lead to sickness and even death, while a too robust or uncontrolled immune response can lead to tissue damage, and for autoimmune diseases, ultimately death. How CD4 T cells maintain this balance will be discussed in the context of the CNS autoimmune disease MS.
Keywords: CD4 T cell, central nervous system, experimental autoimmune encephalomyelitis, inflammation, multiple sclerosis
1. Introduction
In order to break down the complexity of CD4 T cell-mediated autoimmunity one must first understand the cell lineage (Fig. 1). This section is meant as a general overview of the history of CD4 T cells with some license taken in regards to chronology. The information provided can be found and clarified in a number of immunological textbooks.
Figure 1. Naïve CD4+ cells emerge from the thymus and further differentiate into subtypes based on the cytokine microenvironment.

Each subtype of CD4 T cells exhibit unique functions largely based on the cytokines that they produce. Treg cells are both thymic derived and induced in the periphery (iTreg).
Some of us can remember a time when the immune system seemed simple. On a differential stain of peripheral blood there were red blood cells, platelets, macrophages, granulocytes and lymphocytes. Lymphocytes as they exist in the peripheral blood are rather nondescript cells with a large nucleus surrounded by a small amount of cytoplasm. Immunology became slightly more complicated when two types of lymphocytes were discriminated. One population emerged from the thymus called T lymphocytes or T cells, and a second population was found to emerge from the bursa of Fabricius in birds, and were called B lymphocytes or B cells. Since mammals lack a bursa of Fabricus, B cell production had to occur in a different organ, which was found to be the bone marrow (BM). The invention of flow cytometry and monoclonal antibodies (mAb) revolutionized immunology. With these technologies many laboratories generated mAb specific to leukocyte cell surface receptors. Often the new proteins discovered were named for the mAb and not for a particular function or phenotype. To compare and characterize the plethora of mAb produced, the First International Workshop and Conference on Human Leukocyte Differentiation Antigens was held in 1982 to generate a nomenclature system to generate cluster designations or cluster of differentiation (CD) groupings. Thus it became clear that there where two types of T cells: CD4 and CD8. CD4 and CD8 T cells were functionally distinguished based on recognition of the Major Histocompatibility Complex (MHC). CD8 T cells recognize peptide antigens in the context of MHC class I and CD4 T cells utilize MHC class II. Both CD4 and CD8 T cells recognize antigen/MHC complexes with a cell surface receptor called the T cell receptor (TCR), which is a heterodimer composed of an α and β chain, referred to as the αβ TCR.
A second lineage of T cells was discovered that expressed a similar TCR composed of γ and δ chains. Recognition of antigen by the γδ TCR is not as straightforward as for the αβ TCR, and thus much still needs to be learned regarding γδ T cell biology and function. As with αβ T cells, there are subpopulations of γδ T cells, which can often be distinguished by germ-line encoded TCR gene sequences and their site of residence in particular tissues. Very little is known about the function of the various subpopulations of γδ T cells.
Once the immunology community understood MHC restriction and it was established as fact, life as an immunologist was uncomplicated for some time with many laboratories undergoing the laborious process of understanding the functions of CD4 and CD8 T cells in the immune response. Often these immune responses were to innocuous proteins such as ovalbumin, hen egg lysosyme, or pigeon cytochrome C. Along the way, there were whisperings of regulatory cells that due to technical reasons could not be validated and were forgotten for a time or at least not spoken about in immunological gatherings. CD4 T cell functions were quickly separated into two functions called “helper” responses. Immunology textbooks refer to the two functions as cellular and humoral immunity. In cellular immunity, the classic function is the activation of macrophages, a process required for the clearance of intracellular pathogens such as Mycobacterium. These T cells are referred to as T helper 1 (Th1). In humoral immunity, T cells provide “help” to B cells in the generation of antigen-specific antibodies. These T cells are called Th2. CD8 T cells have the capacity to directly kill cells, such as in a viral infection, and were thus termed cytotoxic lymphocytes.
The investigation as to how T cells provided “help” paralleled that of the discovery of cytokines. Cytokines are generally small molecular weight proteins that are secreted and function by binding to their cognate receptors inducing a signaling cascade. Cytokines function in both an autocrine and paracrine manner and induce a number of cellular responses. One of the first cytokines discovered that impacted the immune system was called interleukin (IL)-1, with subsequent cytokines named numerically in order of discovery. There are currently 33 IL that have been cloned and characterized. Many of the cytokines were found to be members of a family based on sequence similarity, sharing of subunits, sharing of receptors or having cognate receptors that share subunits. For example, IL-2 is an important T cell cytokine produced at high levels by naïve CD4 T cells following antigen recognition that serves as a growth and survival factor for T cells. IL-2 binds to its receptor, called the IL-2 receptor (IL-2R), which can consist of up to three chains; α, β and γ. The combination of receptor components determines the affinity of IL-2 to its receptor. On naïve T cells, the α chain, also known as CD25, is rapidly upregulated following antigen recognition, and when in combination with the β and γ chains forms a high affinity receptor. The γ chain, also called the IL-2R common γ chain, is also a component of the IL-4, IL-7 and IL-15 receptors. There are a number of cytokine families that influence T cell biology.
For CD4 T cells, the Th1 and Th2 T cells are categorized by the cytokines that they produce. The signature cytokine for Th1 cells is interferon (IFN)-γ and for Th2 cells it is IL-4. Upon encounter with antigen/MHC complexes, naïve T cells become activated and have the capacity to polarize into either a Th1 or Th2 cell. The process is influenced by a variety of factors, the most important of which seems to be the cytokine environment. For Th1 cell commitment, IL-12 production by antigen presenting cells (APC) and the transcription factors STAT1, STAT4 and TBet are required. For Th2 cells, IL-4 and the transcription factors STAT6 and GATA-3 are required. Once polarized, on the single cell level, the CD4 T cell is committed and cannot revert back to a naïve phenotype or convert to the other lineage. Th1 and Th2 functions are largely driven by the cytokines they produce. Using the early definition of T cell functions, IFN-γ facilitates macrophage activation and IL-4 facilitates the production of certain immunoglobulin subtypes. However, as with most biological systems, the lines between Th1 and Th2 functions have become blurred. It is now known that IFN-γ also is required for the production of certain immunoglobulin subtypes, and that IL-4 can also be involved in macrophage activation. The immunoglobulins induced by IL-4 serve specific functions, thereby separating the activity of the two T cells. IL-4 is required for the production of IgG1 and IgE. IgE sensitizes mast cells, a consequence of which can be allergic reactions, and IgG1 is involved in opsonization of pathogens. For macrophages, the IFN-γ-induced or classically activated macrophages produce nitric oxide (NO), are proinflammatory and drive chronic inflammation and tissue injury. The IL-4-induced or alternatively activated macrophages, are thought to play a role in resolution of inflammation and promotion of wound healing (Gordon, 2003). Other cytokines produced by Th2 cells that influence the immune response include IL-5, IL-6, and IL-13. Th1 T cells also produce IL-2, IL-15, granulocyte macrophage-colony stimulating factor (GM-CSF), tumor necrosis factor (TNF)-α and others. In addition, like CD8 T cells, Th1 cells also have the capacity to induce cytotoxicity of target cells by several different mechanisms. Thus by controlling the phenotype of the responding CD4 T cell, the nature of the immune response can be shaped. The nature of the immune response can have detrimental consequences to human health, such as Th2-driven allergic responses and in the clearance of bacteria. For Mycobacterium tuberculosis infection, individuals with a “Th1-like” response have a better prognosis than those with a “Th2-like” response (Infante-Duarte and Kamradt, 1999).
Enough time has now passed in the history of CD4 T cells for the story to become even more complicated, or enlightening depending upon ones perspective. It has now become clear that CD4 T cells can also differentiate into a regulatory population of T cells (Treg) capable of suppressing the function of other T cells. These T cells often express high levels of CD25 (IL-2Rα) and their development and function is dependent upon the transcription factor Foxp3 (Fontenot et al., 2003; Hori et al., 2003). Treg cells have been shown to play a role in regulating a variety of immune responses from autoimmunity to viral infection (Levings et al., 2006). The exact mechanism of Treg suppression is not clearly known (Miyara and Sakaguchi, 2007). Foxp3+ Treg cells can emerge from the thymus like other naïve CD4 T cells, but they can also be generated by the induction of Foxp3 by transforming growth factor (TGF)-β (Chen et al., 2003). Interestingly, the induction of Treg cells crosses paths with a newly described population of CD4 T cells that produce IL-17, which also requires TGF-β, as well as IL-6, for its production (Bettelli et al., 2006; Mangan et al., 2006; Veldhoen et al., 2006). IL-17 is a proinflammatory cytokine inducing the production of GM-CSF, G-CSF, a number of chemokines, metalloproteinases and IL-6 (Weaver et al., 2007). IL-17 cells are thought to propagate inflammation by the production of chemokines. Chemokines are used by the immune system as chemoattractants to promote the migration of immune cells into sites of inflammation. Thus the presence of IL-6, an inflammation induced cytokine, promotes IL-17 production that propagates inflammation. As with all aspects of the immune response, a balance exists between Treg cells and IL-17 production. This was demonstrated when IL-6 was shown to inhibit TGF-β induction of Foxp3 (Bettelli et al., 2006). Thus IL-6 production seems to control the extent of inflammation (Kishimoto, 2005).
Although the immune system seems complex, the receptors and soluble factors it uses in which to communicate and function fall into groups. For instance, there are pro- and anti-inflammatory cytokines. Pro-inflammatory cytokines include IFN-γ, IL-1, TNF-α and IL-17. Anti-inflammatory cytokines include TGF-β, IL-4 and IL-10. These two types of immune responses counterbalance the other so that the extent of the immune response is regulated. There are many such mechanisms by which the immune system exhibits self-control. A breakdown in the control mechanisms can result in uncontrolled or chronic immune responses that often have deleterious health effects. Autoimmunity is one such condition that occurs when the immune system inappropriately recognizes a self-protein as foreign leading tissue damage. In the CNS, the most prevalent autoimmune disease is multiple sclerosis (MS), which is widely believed to be driven by CD4 T cells. This review will discuss mechanisms of how CD4 T cells regulate the balance between promoting and regulating autoimmune inflammation in the CNS (Fig. 2).
Figure 2. The CD4 T cell lineage controls the balance between autoimmune inflammation and no disease in the CNS.
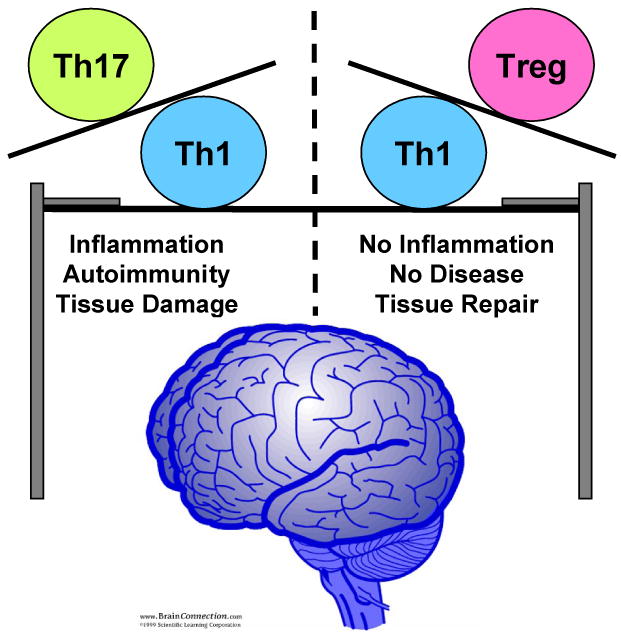
Th1 and Th17 are CD4 T cells that produce pro-inflammatory cytokines, which when produced in response to a CNS self-antigen can result in tissue damage resulting in MS disease symptoms. CD4 Treg cells can regulate autoimmunity by suppressing Th1 cell effector functions.
2. MS an Autoimmune Disease of the CNS
MS is a devastating human autoimmune disease that is manifested in the CNS, and is thought to be the result of severe inflammation driven by T cells specific for self-antigens expressed in the myelin sheath (Sospedra and Martin, 2005). Inflammatory lesions in the CNS are largely composed of macrophages and lymphocytes. These lymphocytes are thought to damage both oligodendrocytes and axons resulting in demyelination and neuronal dysfunction. This cell damage causes a large variety of neurological symptoms including problems with muscle weakness/paralysis, vision, sphincter control and even psychological manifestations. The majority of MS patients exhibit a relapsing remitting form of disease that in general, becomes increasingly progressive overtime with patients becoming more and more debilitated.
Although the consensus is that MS is a T cell-mediated autoimmune disease, what is not clear is how the autoreactive T cells become activated and why the inflammation can reoccur overtime. Since myelin-specific T cells can be detected in normal controls, one hypothesis is that they cross-react with epitopes from infectious agents and become inappropriately activated (Wucherpfennig and Strominger, 1995). Once activated, they have the capacity to enter the CNS and encounter self-antigen putting in motion a cascade of immunological events that results in the accumulation of inflammatory cells with the capacity to induce CNS tissue damage targeting both oligodendrocytes and neurons. This review will focus on the activity of CD4 T cells within the CNS in the induction and downmodulation of inflammation.
3. Experimental Autoimmune Encephalomyelitis as a Model for MS
The understanding of the T cell-mediated pathological process leading to MS has been advanced by the development of an animal model known as experimental autoimmune encephalomyelitis (EAE). EAE in mice mimics the inflammatory infiltrate, the neurological paralytic symptoms and demyelination observed in MS. EAE is mediated by CD4 T cells and can be induced actively by immunization with myelin antigens or their immunodominant peptides emulsified in complete Freund's adjuvant in combination with pertussis toxin injections. The myelin components myelin basic protein (MBP), proteolipid protein and myelin oligodendrocyte glycoprotein are the most studied encephalitogenic self-antigens. EAE can also be induced by the adoptive transfer of activated self-reactive CD4 T cells into irradiated host mice. The encephalitogenic T cells are generated by immunizing susceptible mice with the self-antigen of choice, harvesting the primed cells from the lymph nodes and reactivating them with the same antigen in vitro prior to adoptive transfer. Encephalitogenic T cells can also be generated from TCR transgenic mice with specificity for a myelin antigen epitope. In particular, the MBP-TCR transgenic mouse with specificity to MBP has been used to generate encephalitogenic T cells following in vitro activation of splenic T cells with the immunodominant MBP peptide (Dittel et al., 1999). For both methods of EAE induction, the quintessential clinical symptom of EAE is ascending paralysis, starting with tail and hind limb weakness that commonly spreads to paralysis of the hind and then the forelimbs. The paralytic episodes vary from a monophasic disease course that can be acute or chronic, or a chronic relapsing remitting course, depending upon the antigen and strain of mouse used.
The ability to activate myelin-specific encephalitogenic T cells in vitro allows the generation of T cell lines from genetically modified mice to study the function of individual T cell effector mechanisms in the pathogenesis of EAE. Because the adoptive transfer model does not require immunization of the recipient mouse, immune effector mechanisms that are involved in T cell priming can be specifically studied in the CNS. For instance, if a mouse lacks MHC class II, T cell priming will not occur in the lymph node and the animal will not succumb to EAE. In this situation, nothing would be learned regarding the role of MHC class II in the CNS. Another advantage of using the adoptive transfer system to study T cell effector mechanisms is that the phenotype of the T cell can be manipulated and controlled. This review focuses on T cell effector functions once they have entered the CNS.
4. Antigen Presentation
In the adoptive transfer model, the T cell is stimulated in vitro with its cognate antigen resulting in an activated phenotype. Thus, upon entry into the CNS, the T cell is armed with the capacity to proliferate, produce cytokines, interact with APC and induce cytotoxicity. The T cell uses all these mechanisms to promote inflammation in the CNS. The exact series of events that occur in the CNS upon entry of the activated T cell is unclear; however, it is clear that T cells produce cytokines and begin to expand prior to overt disease symptoms (Ponomarev et al., 2004; Ponomarev et al., 2007b). Presumably, the cell expansion is due to reactivation of the T cells by APC within the CNS. Since the encephalitogenic T cell is CD4+ the MHC restriction is to class II. This same restriction is also thought to be important in MS, as it is the MHC class II locus that has the highest gene linkage to MS susceptibility (Haines et al., 1998). In addition to recognition of the peptide/MHC class II complex by the TCR, effective antigen presentation also requires engagement of costimulatory molecules. The T cell costimulatory molecule is CD28, which binds to the B7 molecules B7.1 (CD80) and B7.2 (CD86) expressed by APC (Greenwald et al., 2005). T cell expression of CD28 is constitutive and does not require activation. In addition, T cells provide costimulation to APC by engaging CD40 with CD40-ligand (L) (Grewal and Flavell, 1998). CD40L is expressed on activated T cells. Thus upon entry into the CNS, activated T cells have the capacity to engage APC providing them costimulation, which in turn results in the upregulation of B7, enhancing T cell costimulation. A role for CD40, and perhaps B7, within the CNS has been shown to be important for EAE pathogenesis (Becher et al., 2001; Chang et al., 1999; Gerritse et al., 1996; Ponomarev et al., 2006). Costimulation of T cells via CD28 binding to the B7 molecules generates a positive signal important for the activation of T cells. However, the same B7 molecules are used to downmodulate T cell functions by binding to the inhibitory molecule CTLA-4, expressed by activated T cells. It is clear that downmodulating inflammation with treatments such as steroids can result in the diminishing of disease symptoms in MS during an inflammatory episode. In other words, if the T cell pathogenic effector population could be controlled or eliminated, individuals with MS would not undergo relapses. CTLA-4 serves to control the extent of the T cell effector response, and in its absence, mice develop a fatal lymphoproliferative disorder (Tivol et al., 1995). Several studies have linked polymorphisms in the CTLA-4 gene to MS susceptibility (Ligers et al., 1999; Suppiah et al., 2005).
Several groups have used a number of strategies to demonstrate that antigen presentation by MHC class II within the CNS is required for the propagation of EAE. However, the phenotype of the APC was more difficult to identify. There a number of cells within the CNS capable of presenting antigen via MHC class II that the activated T cells have the opportunity to encounter. These include astrocytes, microglial cells, perivascular macrophages and dendritic cells. The APC that was shown to be sufficient for EAE induction was a vessel-associated CD11c+ dendritic cell (Greter et al., 2005). Thus the T cells seem to encounter antigen essentially upon entry into the CNS, a process that will result in the reactivation of the T cells inducing cytokine production and proliferation; thereby promoting T cell-mediated inflammation in the CNS.
5. Microglial Cell Activation
Although the initial antigen presentation event appears to be mediated by a dendritic cell, encephalitogenic T cells also interact with and activate a population of CNS resident myeloid-lineage cells, called microglial cells. Microglial cells are located throughout the CNS and send out processes constantly surveying their microenvironment (Nimmerjahn et al., 2005). In the steady state, microglial cells are fairly quiescent expressing very low levels of both CD45 and antigen presentation molecules, and exhibit a ramified morphology (Ponomarev et al., 2005; Sedgwick et al., 1991). A variety of stimuli can rapidly induce microglial cell activation resulting the upregulation of CD45 and the antigen presentation molecules MHC class II, CD40 and B7.2; and morphological changes (Sedgwick et al., 1991). The activated microglial cell both phenotypically and morphologically resembles a macrophage (Ponomarev et al., 2005). Thus the study of microglial cells is difficult because they cannot be differentiated from macrophages that migrate into the CNS from the periphery during the EAE/MS disease course. Using CD45 expression as a marker of resting and activated microglial cells, they were first differentiated from peripheral macrophages by the use of BM chimeras (Sedgwick et al., 1991). Since microglial cells are long lived and radiation resistant, the peripheral immune system can be replaced by a BM transplant leaving the microglial cell population intact within the CNS. Using this strategy, we showed that microglial cells rapidly become activated in the CNS prior to both the onset of EAE disease symptoms and the infiltration of peripheral macrophages into the CNS (Ponomarev et al., 2005). Although the exact role of microglial cells in EAE or MS pathogenesis is not completely known, these data suggest that they play a role in the onset of EAE. Further evidence of their importance in disease onset was indicated when their elimination resulted in less severe EAE disease (Heppner et al., 2005).
The activated encephalitogenic T cell upon entry into the CNS is poised to interact with and activate microglial cells by the production of both soluble factors and direct cell-cell contact. Soluble factors include GM-CSF and IFN-γ. GM-CSF is highly expressed by activated CD4 T cells that are of the Th1 or Th17 phenotype. We showed that GM-CSF production in the CNS was very high just prior to EAE disease symptoms (Ponomarev et al., 2007b). Mice deficient in GM-CSF were shown to be resistant to EAE induction (McQualter et al., 2001). However, this study did not specifically examine the role of GM-CSF in the CNS. Using the MBP-TCR transgenic system, we showed that when EAE was induced in GM-CSF-deficient mice with wild-type encephalitogenic T cells, the EAE disease course was indistinguishable from EAE in wild-type mice (Ponomarev et al., 2007b). When EAE was induced with GM-CSF-deficient T cells, the mice were not susceptible to EAE induction (Ponomarev et al., 2007b). These data indicate that GM-CSF production by the pathogenic T cell population is required for EAE onset. In the absence of T cell-derived GM-CSF, the microglial cells failed to upregulate MHC class II, CD40 and B7.2 (Ponomarev et al., 2007b). In contrast, the expression of these molecules was not altered on the peripheral macrophage population in the absence of T cell-derived GM-CSF (Ponomarev et al., 2007b). In addition to promoting microglial cell activation, T cell-derived GM-CSF also likely plays a role in the function of dendriitc cells in the CNS (Fleetwood et al., 2005). These data demonstrate that GM-CSF functions early in microglial cell activation (Ponomarev et al., 2007b), a two-step process that requires their expression of CD40 for completion (Ponomarev et al., 2006). These data further indicate that interaction of the pathogenic CD4 T cell with microglial cells is essential for EAE pathogenesis.
The function of microglial cells in initiating and/or promoting inflammation in the CNS is not clear. Possible mechanisms of disease propagation include antigen presentation, cytokine and chemokine production, nitric oxide (NO) production and damage to neurons via glutamate (Carson, 2002). There is experimental evidence that microglial cells can also play a role in the downmodulation of inflammation in the CNS. Some potential microglial cell pathogenic mechanisms could also be anti-inflammatory. For instance, microglial cells could interact with and activate Treg cells and the cytokines they produce could be anti-inflammatory. Recently, we showed that microglial cells produce the anti-inflammatory cytokine IL-4 in the CNS (Ponomarev et al., 2007a), which promotes an alternatively activated macrophage phenotype (Ponomarev et al., 2007a; Stein et al., 1992). Alternatively activated macrophages are anti-inflammatory have been associated with tissue repair, thus they are poised to be neuroprotective in the CNS (Gordon, 2003). Indeed, in the normal CNS and during EAE, microglial cells exhibited markers of alternatively activated macrophages and did not produce NO (Ponomarev et al., 2007a). When IL-4 or its receptor were absent from the CNS, EAE disease was more severe and the microglial cells lost the alternatively activated phenotype (Ponomarev et al., 2007a). Thus CD4 T cell interactions with microglial cells, resulting in their activation, is likely to promote both pro- and anti-inflammatory processes. As with T cell costimulation, it is the balance between the two that likely determines whether the inflammatory cascade can be controlled before permanent tissue damage occurs.
6. Cytokine Production
There are a number of T cell-derived cytokines that have been implicated in the pathogenesis of EAE besides GM-CSF. One of the best studied is the hallmark Th1 cytokine IFN-γ. Since IFN-γ upregulates MHC class II molecules as well as adhesion molecules on endothelial cells important for the migration of T cells into the CNS it was thought the IFN-γ would be required for the initiation of EAE and MS. However, IFN-γ was found to be dispensable for EAE onset and its absence resulted in more severe EAE (Ferber et al., 1996; Krakowski and Owens, 1996; Willenborg et al., 1996). Thus IFN-γ seems to be important not in the induction of disease, but in the downregulation of inflammation. Although the mechanisms involved are not completely clear, it has been suggested that IFN-γ exerts its effects by a reduction in macrophage activation resulting in less NO production, a molecule that could function to downregulate T cells responses in the CNS (Willenborg et al., 1999). In addition, a study using IFNR-γ-deficient mice provided evidence that the CD4 cellular infiltrate was more expansive, while MHC class II expression was induced and several chemokines were upregulated (Tran et al., 2000). Thus IFN-γ seems to be important for the anti-inflammatory side of T cell effector functions in the CNS.
Since IFN-γ was not found to be essential for CD4 T cells to induce inflammation in the CNS, the search for other important cytokines lead to IL-17 (Weaver et al., 2007). Mice deficient in IL-17 exhibit less severe EAE, indicating its importance in EAE pathogenesis (Komiyama et al., 2006). Although little is known regarding the function of IL-17 in MS, one study did detect its expression in lesions from MS patients (Lock et al., 2002). A number of studies have now culminated in the concept that CD4 T cells can also become IL-17 producing cells independent of IFN-γ or IL-4 production (Weaver et al., 2007). The production of IL-17 is dependent upon TGF-β and IL-6 and is enhanced by IL-23 and IL-21 (Aggarwal et al., 2003; Bettelli et al., 2006; Korn et al., 2007; Mangan et al., 2006; Nurieva et al., 2007; Veldhoen et al., 2006; Wei et al., 2007; Zhou et al., 2007). IL-17 also induces chemokine production, so it likely plays a role in propagating inflammation by promoting infiltration of immune cells into the CNS (Weaver et al., 2007). Little is known regarding how Th17 cells interact with other cells in the CNS and whether they also have anti-inflammatory mechanisms.
It is clear that the inflammatory response must be controlled in order to maintain healthy tissues. When it is not, permanent tissue damage can occur resulting in very debilitating disease. This balance must be constant, even in the absence of infectious disease. It has become clear that Treg cells are essential in the everyday control of immune responses. The best-studied Treg cell is the Foxp3+ CD4+ T cell. Mice that have a mutated form of Foxp3 or are rendered genetically deficient exhibit a lethal lymphoproliferative disorder (Fontenot et al., 2003). Children that lack Foxp3 succumb to immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome; in which activated CD4 T cells persist resulting in type 1 diabetes, enteropathy, eczema, hypothyroidism, and other autoimmune disorders (Le Bras and Geha, 2006; Wildin and Freitas, 2005). Although the exact mechanisms Treg cells utilize to regulate and suppress immune responses are not always clear, one method is through the production of the anti-inflammatory cytokine IL-10 (Taylor et al., 2006). IL-10 controls inflammation by regulating the expression of cytokines and molecules involved in antigen presentation (Moore et al., 2001). In EAE, the transfer of IL-10-deficient Treg cells abolished their ability to suppress EAE, and correlatively, Treg cells are readily detectable in the CNS with suppressive capabilities (Mann et al., 2007; McGeachy et al., 2005).
The heterogeneity of the entire CD4 T cell population is essential for a properly functioning inflammatory response, and their differential production of cytokines is one method by which they exert their unique functions.
7. Mechanisms of Oligodendrocyte Damage by CD4 T Cells
In addition to the propagation of inflammation, CD4 T cells have the potential to directly mediate tissue damage within the CNS. Inflammatory lesions in MS and EAE contain areas of demyelination that indicates oligodendrocyte loss. It is still unclear exactly how oligodendrocytes are targeted by the immune system and whether their loss is due to cell death. Assuming that oligodendrocyte cell death occurs at some level, CD4 T cells possess mechanisms in which they could induce such a response. The best-studied possible mechanism is the induction of oligodendrocyte apoptosis by several TNF-family members.
The cell death-inducing ligand pair Fas and FasL are expressed in the CNS. Cytotoxicity is mediated by the enagagment of FasL with Fas, inducing the apoptotic cell death of the Fas expressing cell (Nagata and Suda, 1995). Upon activation, FasL is upregulated on CD4 T cells, thus upon entry into the CNS they have the capacity to engage Fas on oligodendrocytes inducing their death. A number of groups demonstrated that Fas was important for the induction of EAE (Dittel, 2000; Sabelko et al., 1997; Waldner et al., 1997). Using the adoptive transfer model, we showed that Fas expression in the recipient mouse, but not by T cells, was essential for EAE induction (Dittel et al., 1999). By inducing EAE with T cells carrying a dysfunctional FasL our data showed that the encephalitogenic T cell population delivered the apoptotic hit (Dittel et al., 1999; Sabelko-Downes et al., 1999). Activated T cells expressing both Fas and FasL also have the capacity to induce apoptosis within the T cell population, thereby controlling their numbers and the extent of inflammation. This lack of regulation within the T cell population is observed in mice lacking functional Fas or FasL genes, which exhibit a lymphoproliferative disorder and are susceptible to autoimmunity (Nagata and Suda, 1995). In EAE, experimental evidence highly suggests that Fas expression by the encephalitogenic T cell population is essential for their elimination, and by inference control of disease (Dittel, 2000; Suvannavejh et al., 2000). Thus the Fas/FasL system maintains balance by controlling the T cell population. In addition to self-population control via Fas/FasL, other cells in the CNS expressing FasL also have the capacity to control the pathogenic T cell population (Dittel et al., 1999; Sabelko-Downes et al., 1999). One such population of cells is γδ T cells. Although present in small numbers in the CNS during EAE, their absence results in chronic disease likely due to an extension of inflammation leading to permanent tissue damage (Ponomarev and Dittel, 2005). γδ T cell regulation of inflammation and EAE disease severity was found to be dependent upon their expression of FasL, which is likely used to eliminate the encephalitogenic T cell population (Ponomarev and Dittel, 2005).
Other TNF-family members expressed by activated CD4 T cells implicated in death of oligodendrocytes and perhaps neurons are TNF-α and lymphotoxin (LT)-α, the production of which has been associated with the encephalitogenic potential of Th1 clones (Powell et al., 1990). TNF-α and LT bind to two receptors TNFR1 and TNFR2, which can induce apoptosis. Confirmation for a role in Fas and TNFR1 in oligodendrocyte cell death was obtained using mice that lacked oligodendrocyte expression of either Fas or TNFR1, or both. Only mice lacking both molecules were completely protected from EAE and did not show signs of demyelination (Hovelmeyer et al., 2005).
8. Neuronal Dysfunction, Transection and Death
It is becoming increasingly clear that neuronal damage or even death are key events in the pathogenesis of MS (Bjartmar et al., 2003; Medana and Esiri, 2003). Even early MS lesions have been shown to have signs of neuronal damage (Kornek et al., 2000; Kuhlmann et al., 2002). Although the mechanisms involved in neuronal damage are not clear in MS, possibilities include glutamate exocitotoxicity, transection of axons, TNF-family-induced cell death, stress due to loss of oligodendrocytes and induction of a transport defect. T cells can produce glutamate, although whether they contribute to neuronal death by this mechanism has not been carefully studied. Proapoptotic TNF-family members were discussed above. The loss of oligdendrocytes is very likely a contributing factor to neuronal stress and loss of function. Oligodendrocytes support both the health and activity of neurons (Anderson et al., 1998; Edgar et al., 2004; Linker et al., 2005). Although not the focus of this review, CD8 T cells possess the same mechanisms of cytotoxicity as Th1 T cells and produce many of the same cytokines. In an in vitro study, CD8 T cells were shown to transect axons (Edgar et al., 2004). The contribution of CD8 T cells in the pathogenesis and regulation of inflammation associated with MS has not been extensively studied. However, evidence exists that they can be an important player in the disease process.
Recently, we have shown that activated CD4 T cells produce a factor that directly results in the destabilization of microtubules in neuronal axons (Shriver and Dittel, 2006). Specifically, we showed that β3-tubulin becomes disassembled, a process that results in a transport defect (Shriver and Dittel, 2006). Neuronal function is dependent upon both retrograde and anterograde transport along the axon. Cytokines are transported up to the cell body and neurotransmitters are transported to the synapse. Without the transport of neurotransmitters the neuron cannot function properly, and the lack of cytokines puts the neuron at risk for death (Guzik and Goldstein, 2004). We found that the process was reversible upon removal of the activated T cells (Shriver and Dittel, 2006). Thus control of the pathogenic T cell population is essential for the regulation of inflammation, which is followed by resolution of disease symptoms.
9. Summary
The control of the pathogenic T cell population by a variety of mechanisms is essential to control the extent of inflammation. If the T cell population is not controlled within the CNS, a potential consequence is enhanced inflammation that is sustained for an extended period of time increasing the chance of oligodendrocyte and neuron cell loss. Since neurons cannot be replaced, this scenario would results in permanent tissue damage and debilitating MS. Although this review has focused on immune-mediated mechanisms that CD4 T cells employ to feedback and control the extent of their effector functions, the CNS also has the capacity to regulate the T cell response. One such mechanism is the cannabinoid system. CD4 T cells upregulate the cannabinoid receptor 2 (CNR2) upon activation and this receptor is an important modulator of T cell effector function. CNR2 can negatively regulate T cell effector functions by downmodulating cell proliferation and cytokine production and promoting apoptosis (Klein et al., 2003). The CNR2 ligands 2-archidonoylglycerol and anandamide are constitutively produced in the CNS (Salzet et al., 2000), and thus when the T cells enter the CNS they are immediately subjected to regulation. This regulation was evident when EAE was induced with CNR2-deficient T cells resulting in a very severe EAE disease course that was associated with increased numbers of encephalitogenic T cells in the CNS (Maresz et al., 2007). Thus a balance between the immune system and the CNS microenvironment also exists, and is required to prevent uncontrolled inflammation in the CNS.
Much work has gone into the study of how inflammation is controlled within the CNS. The CD4 T cell is uniquely equipped to regulate the balance between pro- and anti-inflammatory responses. They perform these functions by being a heterogeneous population of cells capable of regulation through both soluble factors and cell-cell contact. This delicate balance can be easily disrupted as any number of immune perturbations can result in more severe disease in EAE models. Thus every case of MS can be due to unique genetic manifestations, making blanket treatment options challenging. Targeting of the almighty CD4 population for MS therapies will continue to be a focus and as we learn more about their function, we become increasingly closer to learning their secrets.
Acknowledgments
Because parts of this review was presented in a generalized format, the author apologizes for not specifically referencing many of the original articles in which novel and important immunological findings were first described.
Abbreviations
- APC
antigen presenting cells
- BM
bone marrow
- CNR2
cannabinoid receptor 2
- CNS
central nervous system
- CD
cluster of differentiation
- EAE
experimental autoimmune encephalomyelitis
- GM-CSF
granulocyte macrophage-colony stimulating factor
- IL-2R
IL-2 receptor
- IPEX
immunodysregulation, polyendocrinopathy, enteropathy, X-linked
- IFN
interferon
- IL
interleukin
- MHC
Major Histocompatibility Complex
- mAb
monoclonal antibodies
- MS
multiple sclerosis
- MBP
myelin basic protein
- NO
nitric oxide
- TCR
T cell receptor
- Th1
T helper 1
- TGF
transforming growth factor
- Treg
T regulatory
- TNF
tumor necrosis factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- Anderson TJ, Schneider A, Barrie JA, Klugmann M, McCulloch MC, Kirkham D, Kyriakides E, Nave KA, Griffiths IR. Late-onset neurodegeneration in mice with increased dosage of the proteolipid protein gene. J Comp Neurol. 1998;394:506–519. doi: 10.1002/(sici)1096-9861(19980518)394:4<506::aid-cne8>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Becher B, Durell BG, Miga AV, Hickey WF, Noelle RJ. The clinical course of experimental autoimmune encephalomyelitis and inflammation is controlled by the expression of CD40 within the central nervous system. J Exp Med. 2001;193:967–974. doi: 10.1084/jem.193.8.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Wujek JR, Trapp BD. Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J Neurol Sci. 2003;206:165–171. doi: 10.1016/s0022-510x(02)00069-2. [DOI] [PubMed] [Google Scholar]
- Carson MJ. Microglia as liaisons between the immune and central nervous systems: functional implications for multiple sclerosis. Glia. 2002;40:218–231. doi: 10.1002/glia.10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TT, Jabs C, Sobel RA, Kuchroo VK, Sharpe AH. Studies in B7-deficient mice reveal a critical role for B7 costimulation in both induction and effector phases of experimental autoimmune encephalomyelitis. J Exp Med. 1999;190:733–740. doi: 10.1084/jem.190.5.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittel BN. Evidence that Fas and FasL contribute to the pathogenesis of experimental autoimmune encephalomyelitis. Arch Immunol Ther Exp (Warsz) 2000;48:381–388. [PubMed] [Google Scholar]
- Dittel BN, Merchant RM, Janeway CA., Jr Evidence for Fas-dependent and Fas-independent mechanisms in the pathogenesis of experimental autoimmune encephalomyelitis. J Immunol. 1999;162:6392–6400. [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Yool D, Zhang SC, Fowler JH, Montague P, Barrie JA, McCulloch MC, Duncan ID, Garbern J, Nave KA, Griffiths IR. Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J Cell Biol. 2004;166:121–131. doi: 10.1083/jcb.200312012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- Fleetwood AJ, Cook AD, Hamilton JA. Functions of granulocyte-macrophage colony-stimulating factor. Crit Rev Immunol. 2005;25:405–428. doi: 10.1615/critrevimmunol.v25.i5.50. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- Gerritse K, Laman JD, Noelle RJ, Aruffo A, Ledbetter JA, Boersma WJ, Claassen E. CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci U S A. 1996;93:2499–2504. doi: 10.1073/pnas.93.6.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ, Becher B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. 2005;11:328–334. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16:111–135. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- Guzik BW, Goldstein LS. Microtubule-dependent transport in neurons: steps towards an understanding of regulation, function and dysfunction. Curr Opin Cell Biol. 2004;16:443–450. doi: 10.1016/j.ceb.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Haines JL, Terwedow HA, Burgess K, Pericak-Vance MA, Rimmler JB, Martin ER, Oksenberg JR, Lincoln R, Zhang DY, Banatao DR, Gatto N, Goodkin DE, Hauser SL. Linkage of the MHC to familial multiple sclerosis suggests genetic heterogeneity. The Multiple Sclerosis Genetics Group. Hum Mol Genet. 1998;7:1229–1234. doi: 10.1093/hmg/7.8.1229. [DOI] [PubMed] [Google Scholar]
- Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hovelmeyer N, Waisman A, Rulicke T, Prinz M, Priller J, Becher B, Aguzzi A. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. 2005;11:146–152. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- Hovelmeyer N, Hao Z, Kranidioti K, Kassiotis G, Buch T, Frommer F, von Hoch L, Kramer D, Minichiello L, Kollias G, Lassmann H, Waisman A. Apoptosis of oligodendrocytes via Fas and TNF-R1 is a key event in the induction of experimental autoimmune encephalomyelitis. J Immunol. 2005;175:5875–5884. doi: 10.4049/jimmunol.175.9.5875. [DOI] [PubMed] [Google Scholar]
- Infante-Duarte C, Kamradt T. Th1/Th2 balance in infection. Springer Semin Immunopathol. 1999;21:317–338. doi: 10.1007/BF00812260. [DOI] [PubMed] [Google Scholar]
- Kishimoto T. Interleukin-6: from basic science to medicine--40 years in immunology. Annu Rev Immunol. 2005;23:1–21. doi: 10.1146/annurev.immunol.23.021704.115806. [DOI] [PubMed] [Google Scholar]
- Klein TW, Newton C, Larsen K, Lu L, Perkins I, Nong L, Friedman H. The cannabinoid system and immune modulation. J Leukoc Biol. 2003;74:486–496. doi: 10.1189/jlb.0303101. [DOI] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T, Linington C, Schmidbauer M, Lassmann H. Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol. 2000;157:267–276. doi: 10.1016/S0002-9440(10)64537-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakowski M, Owens T. Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur J Immunol. 1996;26:1641–1646. doi: 10.1002/eji.1830260735. [DOI] [PubMed] [Google Scholar]
- Kuhlmann T, Lingfeld G, Bitsch A, Schuchardt J, Bruck W. Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain. 2002;125:2202–2212. doi: 10.1093/brain/awf235. [DOI] [PubMed] [Google Scholar]
- Le Bras S, Geha RS. IPEX and the role of Foxp3 in the development and function of human Tregs. J Clin Invest. 2006;116:1473–1475. doi: 10.1172/JCI28880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levings MK, Allan S, d'Hennezel E, Piccirillo CA. Functional dynamics of naturally occurring regulatory T cells in health and autoimmunity. Adv Immunol. 2006;92:119–155. doi: 10.1016/S0065-2776(06)92003-3. [DOI] [PubMed] [Google Scholar]
- Ligers A, Xu C, Saarinen S, Hillert J, Olerup O. The CTLA-4 gene is associated with multiple sclerosis. J Neuroimmunol. 1999;97:182–190. doi: 10.1016/s0165-5728(99)00072-7. [DOI] [PubMed] [Google Scholar]
- Linker RA, Sendtner M, Gold R. Mechanisms of axonal degeneration in EAE--lessons from CNTF and MHC I knockout mice. J Neurol Sci. 2005;233:167–172. doi: 10.1016/j.jns.2005.03.021. [DOI] [PubMed] [Google Scholar]
- Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer-Gould A, Strober S, Cannella B, Allard J, Klonowski P, Austin A, Lad N, Kaminski N, Galli SJ, Oksenberg JR, Raine CS, Heller R, Steinman L. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–508. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- Mann MK, Maresz K, Shriver LP, Tan Y, Dittel BN. B cell regulation of CD4+CD25+ T regulatory cells and IL-10 via B7 is essential for recovery from experimental autoimmune encephalomyelitis. J Immunol. 2007;178:3447–3456. doi: 10.4049/jimmunol.178.6.3447. [DOI] [PubMed] [Google Scholar]
- Maresz K, Pryce G, Ponomarev ED, Marsicano G, Croxford JL, Shriver LP, Ledent C, Cheng X, Carrier EJ, Mann MK, Giovannoni G, Pertwee RG, Yamamura T, Buckley NE, Hillard CJ, Lutz B, Baker D, Dittel BN. Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB(1) on neurons and CB(2) on autoreactive T cells. Nat Med. 2007;13:492–497. doi: 10.1038/nm1561. [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Stephens LA, Anderton SM. Natural Recovery and Protection from Autoimmune Encephalomyelitis: Contribution of CD4+CD25+ Regulatory Cells within the Central Nervous System. J Immunol. 2005;175:3025–3032. doi: 10.4049/jimmunol.175.5.3025. [DOI] [PubMed] [Google Scholar]
- McQualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, Hamilton JA, Reid HH, Bernard CC. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp Med. 2001;194:873–882. doi: 10.1084/jem.194.7.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medana IM, Esiri MM. Axonal damage: a key predictor of outcome in human CNS diseases. Brain. 2003;126:515–530. doi: 10.1093/brain/awg061. [DOI] [PubMed] [Google Scholar]
- Miyara M, Sakaguchi S. Natural regulatory T cells: mechanisms of suppression. Trends Mol Med. 2007;13:108–116. doi: 10.1016/j.molmed.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- Nagata S, Suda T. Fas and Fas ligand: lpr and gld mutations. Immunol Today. 1995;16:39–43. doi: 10.1016/0167-5699(95)80069-7. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Dittel BN. {gamma}{delta} T Cells Regulate the Extent and Duration of Inflammation in the Central Nervous System by a Fas Ligand-Dependent Mechanism. J Immunol. 2005;174:4678–4687. doi: 10.4049/jimmunol.174.8.4678. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Maresz K, Yan T, Dittel BN. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activaton in microglial cells. J Neurosci. 2007a doi: 10.1523/JNEUROSCI.1922-07.2007. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev ED, Novikova M, Yassai M, Szczepanik M, Gorski J, Dittel BN. Gamma delta T cell regulation of IFN-gamma production by central nervous system-infiltrating encephalitogenic T cells: correlation with recovery from experimental autoimmune encephalomyelitis. J Immunol. 2004;173:1587–1595. doi: 10.4049/jimmunol.173.3.1587. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Shriver LP, Dittel BN. CD40 expression by microglial cells is required for their completion of a two-step activation process during central nervous system autoimmune inflammation. J Immunol. 2006;176:1402–1410. doi: 10.4049/jimmunol.176.3.1402. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Shriver LP, Maresz K, Dittel BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J Neurosci Res. 2005;81:374–389. doi: 10.1002/jnr.20488. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol. 2007b;178:39–48. doi: 10.4049/jimmunol.178.1.39. [DOI] [PubMed] [Google Scholar]
- Powell MB, Mitchell D, Lederman J, Buckmeier J, Zamvil SS, Graham M, Ruddle NH, Steinman L. Lymphotoxin and tumor necrosis factor-alpha production by myelin basic protein-specific T cell clones correlates with encephalitogenicity. Int Immunol. 1990;2:539–544. doi: 10.1093/intimm/2.6.539. [DOI] [PubMed] [Google Scholar]
- Sabelko KA, Kelly KA, Nahm MH, Cross AH, Russell JH. Fas and Fas ligand enhance the pathogenesis of experimental allergic encephalomyelitis, but are not essential for immune privilege in the central nervous system. J Immunol. 1997;159:3096–3099. [PubMed] [Google Scholar]
- Sabelko-Downes KA, Cross AH, Russell JH. Dual role for Fas ligand in the initiation of and recovery from experimental allergic encephalomyelitis. J Exp Med. 1999;189:1195–1205. doi: 10.1084/jem.189.8.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzet M, Breton C, Bisogno T, Di Marzo V. Comparative biology of the endocannabinoid system possible role in the immune response. Eur J Biochem. 2000;267:4917–4927. doi: 10.1046/j.1432-1327.2000.01550.x. [DOI] [PubMed] [Google Scholar]
- Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci U S A. 1991;88:7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriver LP, Dittel BN. T-cell-mediated disruption of the neuronal microtubule network: correlation with early reversible axonal dysfunction in acute experimental autoimmune encephalomyelitis. Am J Pathol. 2006;169:999–1011. doi: 10.2353/ajpath.2006.050791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176:287–292. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suppiah V, Alloza I, Heggarty S, Goris A, Dubois B, Carton H, Vandenbroeck K. The CTLA4 +49 A/G*G-CT60*G haplotype is associated with susceptibility to multiple sclerosis in Flanders. J Neuroimmunol. 2005;164:148–153. doi: 10.1016/j.jneuroim.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Suvannavejh GC, Dal Canto MC, Matis LA, Miller SD. Fas-mediated apoptosis in clinical remissions of relapsing experimental autoimmune encephalomyelitis. J Clin Invest. 2000;105:223–231. doi: 10.1172/JCI8561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A, Verhagen J, Blaser K, Akdis M, Akdis CA. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: the role of T regulatory cells. Immunology. 2006;117:433–442. doi: 10.1111/j.1365-2567.2006.02321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- Tran EH, Prince EN, Owens T. IFN-gamma shapes immune invasion of the central nervous system via regulation of chemokines. J Immunol. 2000;164:2759–2768. doi: 10.4049/jimmunol.164.5.2759. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Waldner H, Sobel RA, Howard E, Kuchroo VK. Fas- and FasL-deficient mice are resistant to induction of autoimmune encephalomyelitis. J Immunol. 1997;159:3100–3103. [PubMed] [Google Scholar]
- Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 Family Cytokines and the Expanding Diversity of Effector T Cell Lineages. Annu Rev Immunol. 2007 doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- Wei L, Laurence A, Elias KM, O'Shea JJ. IL-21 is produced by TH17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem. 2007 doi: 10.1074/jbc.M705100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildin RS, Freitas A. IPEX and FOXP3: clinical and research perspectives. J Autoimmun. 2005 25:56–62. doi: 10.1016/j.jaut.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol. 1996;157:3223–3227. [PubMed] [Google Scholar]
- Willenborg DO, Fordham SA, Staykova MA, Ramshaw IA, Cowden WB. IFN-gamma is critical to the control of murine autoimmune encephalomyelitis and regulates both in the periphery and in the target tissue: a possible role for nitric oxide. J Immunol. 1999;163:5278–5286. [PubMed] [Google Scholar]
- Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]