

Abstract
The aim of this study was to characterize the interaction between mTOR and ERK in primary endothelial cells (EC) following MHC class I and integrin ligation. Ligation of MHC class I molecules or integrins on the surface of EC leads to phosphorylation of ERK at Thr202/Tyr204. We utilized small interfering RNA (siRNA) blockade of mTOR and proteins involved in mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) to define a relationship between mTOR and ERK following MHC class I signaling. We found mTORC2 was responsible for MHC class I and integrin induced phosphorylation of ERK at Thr202/Tyr204. We corroborated these results demonstrating that long-term exposure to rapamycin also inhibited ERK pathway activation in response to MHC class I signaling. Our results demonstrate, for the first time, that engagement of either MHC class I or integrin on the surface of EC leads to ERK activation through an mTORC2-dependent pathway.
Keywords: MHC, HLA class I, ERK, mTORC2, siRNA, Endothelial cells, Signal transduction
mTOR plays an essential role in integrin and MHC class I-induced cell proliferation and survival through formation of two distinct multi-protein molecular complexes, mTORC1 and mTORC2 [1–4]. mTORC1 consists of mTOR in association with regulatory associated protein of mTOR (Raptor) and GβL [5]. mTORC1 phosphorylates p70 S6 Kinase and eukaryotic initiation factor 4E binding protein 1 which activate translational machinery to increase cell proliferation [6, 7]. mTORC2 is composed of mTOR binding to rapamycin insensitive companion of mTOR (Rictor), GβL, stress-activated protein kinase-interacting protein 1 (Sin1) and the newly discovered associations with PRoline-Rich protein 5 (PRR5) or the synonymous protein observed with Rictor 1 (Protor-1), and P-REX1 [8–12]. mTORC2 activates Akt at Ser473, Rac GTPase, cell migration and effects reorganization of the actin cytoskeleton [11–14]. Recent work utilizing mouse knockout models emphasizes the importance of mTOR for growth and proliferation in development [15, 16]. Homozygous mTOR (−/−) mice have arrested embryonic development at E5.5 [17]. Similarly, Raptor null mice die around E5.5 and exhibit an aberrant cellular phenotype. Rictor and GβL knockout mice live longer (E10.5), yet display defects in fetal vascular development [17, 18]. These data show both mTOR complexes are involved in signal transduction leading to cell proliferation, however, the mechanism(s) by which mTORC2 participates in mitogenic signaling remains less well defined.
ERK1/2 are members of a family of serine/threonine kinases activated by a variety of growth factors, cell surface receptors, integrins and MHC class I, all of which play a critical role in cell proliferation, differentiation, survival and reorganization of the actin cytoskeleton [19]. In this study, we identified a novel role for mTORC2 in MHC class I and integrin mediated activation of the ERK pathway. We show that MHC class I or integrin stimulation, but not growth factor, induces phosphorylation of ERK at Thr202/Tyr204 through mTORC2. These studies identify MHC class I molecules and integrins as upstream signaling pathways that position the ERK/mitogen-activated protein kinase (MAPK) pathway downstream of mTORC2. mTORC2 regulation of the ERK/MAPK pathway may play an important role in MHC class I and integrin mediated cell proliferation.
MATERIAL AND METHODS
Antibodies and Chemicals
Mirus TransIT-TKO® transfection reagent was purchased from Mirus Corporation (Madison, WI). mAB (HB-95™) W6/32 was purchased from the American Type Culture Collection (Manassas, VA) and purified by protein A-agarose affinity chromatography. mAb against Vinculin (V9131), rapamycin, protein A-agarose, bovine fibronectin and poly-L-lysine were purchased from Sigma-Aldrich (St. Louis, MO). Recombinant human basic fibroblast frowth factor (bFGF) was purchased from R&D systems (Minneapolis, MN). Soybean trypsin inhibitor was purchased from Invitrogen (Carlsbad, CA). Rabbit polyclonal antibody against phospho-ERK Thr202/Tyr204, ERK, mTOR and Raptor were purchased from Cell Signaling Technology (Beverly, MA). Rabbit polyclonal anti-Rictor antibody was purchased from Novus Biologicals (Littleton, CO). Goat anti-rabbit HRP antibody was purchased from Santa Cruz Biotechnologies (Santa Cruz, CA)
Cell Culture
Primary human aortic EC were isolated and cultured from the aortic rings of explanted donor hearts, as previously described [1, 20].
siRNA Design and Transfection
We designed an mTOR siRNA duplex corresponding to bases 5309–5327 from the open reading frame of Human FRAP1 mRNA 5′-CCA AAG UGC UGC AGU ACU AUU-3′. The RNA sequence used as a negative control (GL-2) for siRNA activity was 5′ CGU ACG CGG AAU ACU UCG A-dT.dT-3′. Human Rictor 5′ ACU UGU GAA GAA UCG UAU C-dT.dT -3′; Human Raptor 5′ GGA CAA CGG CCA CAA GUA C.dT.dT-3′; siRNA duplexes were purchased from Dharmacon Inc. (Lafayette, CO). The conditions for transfection were optimized for the efficient and specific transfection of HAEC using siGLO Lamin A/C siRNA as previously described (Dharmacon Inc, Lafayette, CO) [1].
Western Blot analysis
Serum-starved cultures of EC were treated as described in the individual experiments. Western blot studies were performed as previously described [1].
Fibronectin Stimulation
EC were transfected with control GL-2 or Rictor siRNA for 48 h then harvested by brief trypsin/EDTA treatment (0.05% trypsin, 2mM EDTA) (Invitrogen, Carlsbad, CA) and stimulated with fibronectin or poly-L-lysine as previously described [21].
RESULTS
mTOR is required for class I-induced phosphorylation of ERK at Thr202/Tyr204
To examine the relationship between mTOR and ERK following MHC class I ligation, EC were transfected with mTOR or control siRNA for 48 h and treated with anti-MHC class I antibody for various times. Ligation of MHC class I molecules induced a marked increase in the phosphorylation of ERK at Thr202/Tyr204 in EC transfected with control siRNA. In contrast, knockdown of mTOR completely blocked MHC class I-induced ERK activation (Fig. 1A). As a control, mTOR siRNA specifically inhibited mTOR total protein expression whereas; nontargeting GL-2 siRNA had no effect (Fig. 1B). These results indicate that class I mediated ERK activation is dependent upon mTOR.
Fig. 1. Ligation of MHC class I molecules leads to the phosphorylation of ERK at Thr202/Tyr204 through mTOR.

A, EC were transfected with 100nM mTOR siRNA or GL-2 control siRNA. After 48 h the EC were stimulated with 1 μg/mL anti-MHC class I mAb W6/32 for various time points. EC were lysed and immunoblotted with antibody to ERK Thr202/Tyr204 and ERK total protein. B, To confirm mTOR knockdown efficiency and equal loading, lysates were immunoblotted with antibodies to total mTOR and Vinculin. Data represent at least three independent experiments.
mTORC2 regulates class I-induced phosphorylation of ERK at Thr202/Tyr204
We next examined whether mTOR complexed with Rictor (mTORC2) or Raptor (mTORC1) was required for MHC class I-induced phosphorylation of ERK. siRNA was used to specifically knock down either Rictor or Raptor expression in EC to identify which of these mTOR complexes is responsible for MHC class I-induced ERK activation. siRNA knockdown of Rictor inhibited class I-induced phosphorylation of ERK at Thr202/Tyr204 compared to control siRNA (Fig. 2A). In contrast, knockdown of Raptor failed to block MHC class I-induced phosphorylation of ERK at Thr202/Tyr204 (Fig. 2B). These results show that class I mediated ERK activation is dependent on mTORC2.
Fig. 2. Knockdown of Rictor blocks MHC class I-induced phosphorylation of ERK at Thr202/Tyr204.
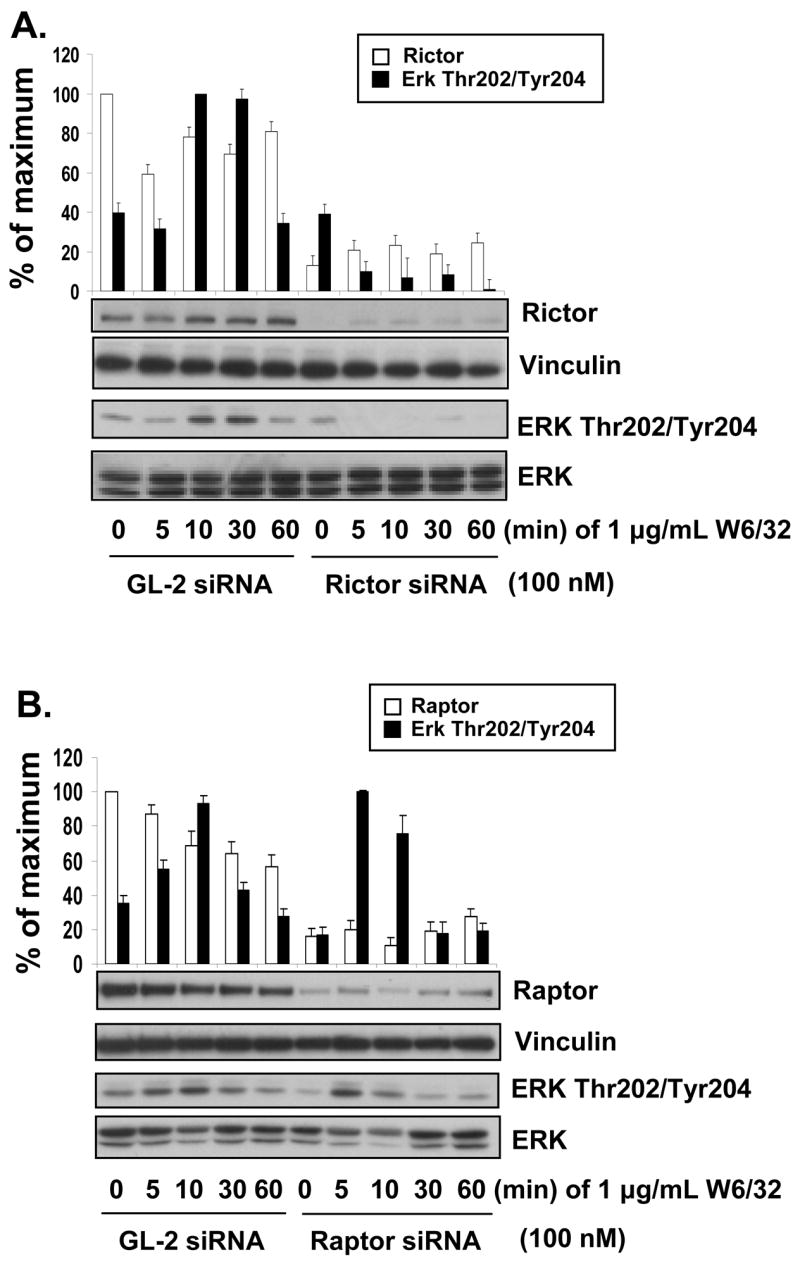
A, EC were transfected with 100 nM Rictor siRNA or GL-2 control siRNA. After 48 h the EC were stimulated with 1 μg/mL anti-MHC class I mAb W6/32 for various time points. EC were lysed and immunoblotted with antibodies to Rictor, Vinculin, ERK Thr202/Tyr204 and ERK. B, EC were transfected with 100 nM Raptor siRNA or GL-2 control siRNA. After 48 h the EC were stimulated with anti-MHC class I mA at various time points. EC were lysed and immunoblotted with antibodies to Raptor, Vinculin, ERK Thr202/Tyr204 and ERK. Densitometry results are expressed as the percentage maximal increase in phosphorylation above control values (mean ±S.E.). Data is representative of three independent experiments.
mTORC2 regulates ERK activation following integrin stimulation
Integrin signaling activates both the mTOR and MAPK pathways in a variety of cell types increasing cell proliferation, migration and survival through activation of focal adhesion kinase (FAK), the mTOR/p70S6Kinase and ERK pathways [2–4]. To determine if mTORC2 is required for integrin activation of ERK, EC were transfected with Rictor or GL-2 control siRNA for 48 h, placed in suspension for 30 min and then plated onto poly-L-lysine or fibronectin coated plates to initiate ligation of integrin molecules. In the presence of GL-2 control siRNA, integrin engagement with fibronectin stimulated ERK phosphorylation at Thr202/Tyr204 at 10 min compared to poly-L-lysine negative controls. In contrast, EC transfected with Rictor siRNA and plated onto fibronectin did not show any detectable increase in the phosphoryation of ERK at Thr202/Tyr204 (Fig. 3A). These results demonstrate, for the first time, that integrin-induced ERK phosphorylation at Thr202/Tyr204 in EC is dependent on mTORC2, similar to MHC class I ligation.
Fig. 3. Integrin stimulates ERK phosphorylation through Rictor.

A, EC were transfected with 100 nM Rictor siRNA or GL-2 control siRNA. After 48 h EC were trypsinized, kept in suspension (Suspend) for 30 min and then replated on poly-L-lysine (Lys) or fibronectin (FN) coated dishes for 10 min. EC were lysed and immunoblotted with antibody to ERK Thr202/Tyr204, ERK and Rictor. Data is representative of three independent experiments. B, EC were transfected with 100 nM Rictor siRNA or GL-2 control siRNA for 48 h. EC were stimulated with 1 ng/mL or 20 ng/mL bFGF for 10 or 30 min. EC were lysed and immunoblotted with antibodies to ERK Thr202/Tyr204, ERK, Rictor and Vinculin. Data is representative of three independent experiments.
Growth factor stimulation of ERK does not require mTORC2
Next we determined if mTORC2-dependent ERK activation is a pathway selectively induced by MHC class I and integrin ligation or is also triggered by growth factor stimulation. For this, EC were treated with bFGF to stimulate ERK phosphorylation in the absence of Rictor. EC were transfected with Rictor siRNA or control siRNA for 48 h and treated with bFGF for 10 or 30 min. Knockdown of Rictor did not inhibit bFGF induced phosphorylation of ERK at Thr202/Tyr204 (Fig. 3B). These data support the hypothesis that ERK activation through mTORC2 is selective for MHC class I and integrin signaling.
Long-term rapamycin treatment blocks MHC class I-induced activation of ERK
Exposure of cells to the drug rapamycin blocks activation of the mTOR pathway. Short-term (5–30 min) exposure to rapamycin selectively blocks mTORC1 signaling [5, 6]. In contrast, long-term treatment with rapamycin (>2h) inhibits mTOR association with components of the mTORC2 complex, Rictor and Sin1 [13, 22]. To corroborate the results obtained with mTOR, Rictor and Raptor siRNA knockdown, we determined the effect of short and long-term exposure to rapamycin on MHC class I-induced phosphorylation of ERK. We observed a dramatic decrease in MHC class I-induced ERK activation after long-term treatment with rapamycin which was not apparent after short term exposure (Fig. 4). This result provides an independent line of evidence supporting the hypothesis that MHC class I activation of the ERK pathway is regulated via an mTORC2-dependent pathway.
Fig. 4. Long-term rapamycin treatment blocks MHC class I-induced phosphorylation of ERK at Thr202/Tyr204.

EC were treated with 30nM of rapamycin for various time points and stimulated with 1μg/mL anti-MHC class I mAb W6/32 for 30 min. EC were lysed and immunoblotted with antibodies to ERK Thr202/Tyr204 and ERK total protein. Data represent at least three independent experiments.
DISCUSSION
Our results reveal for the first time, that ligation of MHC class I molecules on the surface of EC leads to activation of ERK1/2 through an mTORC2-dependent pathway. siRNA-mediated knockdown of either mTOR or Rictor, but not Raptor, inhibited MHC class I-induced phosphorylation of ERK at Thr202/Tyr204. These data provide further evidence for distinct functions of mTORC1 and mTORC2 in cell signaling.
Recent studies have shown that short-term treatment with rapamycin inhibits mTORC1 function whereas, long-term treatment prevents the association between mTOR and Rictor and thereby abrogates mTORC2 function [22]. To provide an independent line of evidence linking ERK activation to mTORC2 following MHC class I ligation, we treated EC with rapamycin for various times and found that phosphorylation of ERK at Thr202/Tyr204 in response to MHC class I ligation was inhibited following long-term (>2h) exposure to rapamycin. These results substantiate our findings with siRNAs and support the hypothesis that MHC class I ligation leads to ERK activation through mTORC2. Interestingly, recent data in fission yeast shows that TOR2 controls entry into mitosis through the MAPK pathway providing a molecular mechanism for the control of cell size [23]. Thus, the link between mTORC2 and ERK found in our study might have precedent in lower organisms and represent an evolutionarily conserved regulatory pathway.
An important question to be investigated is how MHC class I molecules transduce their signals. Because MHC class I molecules express no signaling motifs in their cytoplasmic domain it is hypothesized that MHC class I molecules interact with an accessory protein(s) in order to activate downstream signal transduction. Our data raise the possibility that members of the integrin family may cooperate with MHC class I molecules to elicit activation of proteins involved in the MHC class I signaling pathway. Our results show that ligation of integrin molecules with fibronectin in EC induced mTORC2 dependent phosphorylation of ERK similar to MHC class I molecules. Numerous reports have shown ligation of integrin molecules leads to the phosphorylation of ERK in many cell types, including EC [24–26]. However, fibronectin stimulation of beta-1 or beta-3 integrins in cytotoxic T cells fails to activate ERK [27]. Rather, integrin ligation facilitated strong ERK activation only in the presence of TCR/CD3 co-stimulation suggesting that cooperation between the TCR/CD3 and integrins is required to activate ERK in cytotoxic T lymphocytes [27]. In view of our results and these considerations, it will be of interest to determine whether class I molecules and integrins cooperate to transduce signals in response to extracellular matrix components.
In contrast to MHC class I and integrin ligation, EC treated with bFGF stimulates ERK activation independently of mTORC2. These results are consistent with recent studies showing that siRNA knockdown of Sin1 failed to inhibit the phosphorylation of ERK in response to a variety of growth factors including bFGF [28]. In addition, phosphorylation of ERK induced by insulin growth factor-1 or platelet derived growth factor did not differ between Rictor null and control mouse embryonic fibroblasts [18], consistent with the notion that polypeptide growth factors that act via tyrosine kinase receptors promote ERK activation through an mTORC2-independent pathway. Collectively, these data underscore a differential role of mTORC2 in mediating ERK activation in response to integrins, MHC class I ligation and polypeptide growth factors.
Numerous studies have shown that development of post-transplant anti-donor HLA antibodies are a contributing factor to the progression of transplant vasculopathy (TV) [29, 30]. Furthermore, passive transfer of anti-donor MHC class I antibodies to experimental heart transplant recipients lacking T and B lymphocytes induces TV [31]. We reported activation of the mTOR pathway in capillary and intramyocardial artery and vein endothelium of cardiac allografts from T and B cell deficient RAG1.KO hosts that were passively transfused with anti-donor MHC class I antibody [32]. The mTOR pathway has been shown to play a critical role in the process of vascular intimal proliferation. A 12 month clinical trial study found that cardiac transplant recipients treated with the mTOR inhibitor everolimus had significantly reduced intimal thickness compared to the azathioprine treated group [33]. Similarly, a 2-year study found everolimus treated patients had significantly reduced acute rejection and limited progression of TV [34]. Our results identify ERK as a new target of mTOR following activation by anti-MHC antibody and suggest that blocking of both mTORC1 and mTORC2 may be necessary to inhibit MHC class I mediated cell proliferation.
In summary, our findings reveal that mTORC2 is required for integrin and MHC class I-induced ERK activation, but not for growth factor induced ERK activation. This cross-talk between the mTOR and MAPK pathways emphasizes the importance of mTOR to regulate both transcriptional and translational initiation machinery. In addition, our data demonstrates signal transduction pathway similarities between MHC class I and integrin molecules. Determining the membrane proximal signaling events initiated after class I crosslinking should permit the identification of molecules such as integrins that interact with MHC to propagate intracellular signals.
Acknowledgments
The authors thank Dr. Judith A. Berliner for helpful discussions and critical reading of the manuscript. This research was supported by NIH grant RO1AI42819 to E.F.R. R.J. and E.R. were supported by DK55003 and DK56930.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jindra PT, Jin YP, Rozengurt E, Reed EF. HLA Class I Antibody-Mediated Endothelial Cell Prolilferation via the mTOR Pathway. J Immunol. 2008;180:2357–2366. doi: 10.4049/jimmunol.180.4.2357. [DOI] [PubMed] [Google Scholar]
- 2.Han S, Khuri FR, Roman J. Fibronectin stimulates non-small cell lung carcinoma cell growth through activation of Akt/mammalian target of rapamycin/S6 kinase and inactivation of LKB1/AMP-activated protein kinase signal pathways. Cancer Res. 2006;66:315–323. doi: 10.1158/0008-5472.CAN-05-2367. [DOI] [PubMed] [Google Scholar]
- 3.Sakakibara K, Liu B, Hollenbeck S, Kent KC. Rapamycin inhibits fibronectin-induced migration of the human arterial smooth muscle line (E47) through the mammalian target of rapamycin. Am J Physiol Heart Circ Physiol. 2005;288:H2861–2868. doi: 10.1152/ajpheart.00561.2004. [DOI] [PubMed] [Google Scholar]
- 4.Kornberg L, Earp HS, Parsons JT, Schaller M, Juliano RL. Cell adhesion or integrin clustering increases phosphorylation of a focal adhesion-associated tyrosine kinase. J Biol Chem. 1992;267:23439–23442. [PubMed] [Google Scholar]
- 5.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177–189. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 6.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 7.Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S, Yoshino K, Hara K, Tanaka N, Avruch J, Yonezawa K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem. 2003;278:15461–15464. doi: 10.1074/jbc.C200665200. [DOI] [PubMed] [Google Scholar]
- 8.Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006;20:2820–2832. doi: 10.1101/gad.1461206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 10.Pearce LR, Huang X, Boudeau J, Pawlowski R, Wullschleger S, Deak M, Ibrahim A, Gourlay R, Magnuson MA, Alessi DR. Identification of Protor as a novel Rictor-binding component of mTOR-complex-2. Biochem J. 2007;405:513–522. doi: 10.1042/BJ20070540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hernandez-Negrete I, Carretero-Ortega J, Rosenfeldt H, Hernandez-Garcia R, Calderon-Salinas JV, Reyes-Cruz G, Gutkind JS, Vazquez-Prado J. P-Rex1 links mTOR signaling to Rac activation and cell migration. J Biol Chem. 2007;282:23708–23715. doi: 10.1074/jbc.M703771200. [DOI] [PubMed] [Google Scholar]
- 12.Woo SY, Kim DH, Jun CB, Kim YM, Haar EV, Lee SI, Hegg JW, Bandhakavi S, Griffin TJ. PRR5, a novel component of mTOR complex 2, regulates platelet-derived growth factor receptor beta expression and signaling. J Biol Chem. 2007;282:25604–25612. doi: 10.1074/jbc.M704343200. [DOI] [PubMed] [Google Scholar]
- 13.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 14.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 15.Gangloff YG, Mueller M, Dann SG, Svoboda P, Sticker M, Spetz JF, Um SH, Brown EJ, Cereghini S, Thomas G, Kozma SC. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol Cell Biol. 2004;24:9508–9516. doi: 10.1128/MCB.24.21.9508-9516.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murakami M, Ichisaka T, Maeda M, Oshiro N, Hara K, Edenhofer F, Kiyama H, Yonezawa K, Yamanaka S. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol Cell Biol. 2004;24:6710–6718. doi: 10.1128/MCB.24.15.6710-6718.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 18.Shiota C, Woo JT, Lindner J, Shelton KD, Magnuson MA. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev Cell. 2006;11:583–589. doi: 10.1016/j.devcel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 19.Chang F, Steelman LS, Lee JT, Shelton JG, Navolanic PM, Blalock WL, Franklin RA, McCubrey JA. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia. 2003;17:1263–1293. doi: 10.1038/sj.leu.2402945. [DOI] [PubMed] [Google Scholar]
- 20.Yeh M, Leitinger N, de Martin R, Onai N, Matsushima K, Vora DK, Berliner JA, Reddy ST. Increased transcription of IL-8 in endothelial cells is differentially regulated by TNF-alpha and oxidized phospholipids. Arterioscler Thromb Vasc Biol. 2001;21:1585–1591. doi: 10.1161/hq1001.097027. [DOI] [PubMed] [Google Scholar]
- 21.Jiang X, Jacamo R, Zhukova E, Sinnett-Smith J, Rozengurt E. RNA interference reveals a differential role of FAK and Pyk2 in cell migration, leading edge formation and increase in focal adhesions induced by LPA in intestinal epithelial cells. J Cell Physiol. 2006;207:816–828. doi: 10.1002/jcp.20629. [DOI] [PubMed] [Google Scholar]
- 22.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 23.Petersen J, Nurse P. TOR signalling regulates mitotic commitment through the stress MAP kinase pathway and the Polo and Cdc2 kinases. Nat Cell Biol. 2007;9:1263–1272. doi: 10.1038/ncb1646. [DOI] [PubMed] [Google Scholar]
- 24.Vomastek T, Iwanicki MP, Schaeffer HJ, Tarcsafalvi A, Parsons JT, Weber MJ. RACK1 targets the ERK/MAP Kinase pathway to link integrin engagement with focal adhesion disassembly and cell motility. Mol Cell Biol. 2007;27:8296–8305. doi: 10.1128/MCB.00598-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sawhney RS, Cookson MM, Omar Y, Hauser J, Brattain MG. Integrin alpha2-mediated ERK and calpain activation play a critical role in cell adhesion and motility via focal adhesion kinase signaling: identification of a novel signaling pathway. J Biol Chem. 2006;281:8497–8510. doi: 10.1074/jbc.M600787200. [DOI] [PubMed] [Google Scholar]
- 26.Wilson SH, Ljubimov AV, Morla AO, Caballero S, Shaw LC, Spoerri PE, Tarnuzzer RW, Grant MB. Fibronectin fragments promote human retinal endothelial cell adhesion and proliferation and ERK activation through alpha5beta1 integrin and PI 3-kinase. Invest Ophthalmol Vis Sci. 2003;44:1704–1715. doi: 10.1167/iovs.02-0773. [DOI] [PubMed] [Google Scholar]
- 27.Puente LG, Ostergaard HL. Beta 1/beta 3 integrin ligation is uncoupled from ERK1/ERK2 activation in cytotoxic T lymphocytes. J Leukoc Biol. 2003;73:391–398. doi: 10.1189/jlb.0402199. [DOI] [PubMed] [Google Scholar]
- 28.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 29.Reed EF, Demetris AJ, Hammond E, Itescu S, Kobashigawa JA, Reinsmoen NL, Rodriguez ER, Rose M, Stewart S, Suciu-Foca N, Zeevi A, Fishbein MC. Acute antibody-mediated rejection of cardiac transplants. J Heart Lung Transplant. 2006;25:153–159. doi: 10.1016/j.healun.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 30.Terasaki PI, Cai J. Humoral theory of transplantation: further evidence. Curr Opin Immunol. 2005;17:541–545. doi: 10.1016/j.coi.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 31.Russell PS, Chase CM, Colvin RB. Alloantibody- and T cell-mediated immunity in the pathogenesis of transplant arteriosclerosis: lack of progression to sclerotic lesions in B cell-deficient mice. Transplantation. 1997;64:1531–1536. doi: 10.1097/00007890-199712150-00005. [DOI] [PubMed] [Google Scholar]
- 32.Jindra PT, Hsueh A, Hong L, Gjertson DW, Shen XD, Gao F, Dang J, Mischel PS, Baldwin WM, 3rd, Fishbein MC, Kupiec-Weglinski JW, Reed EF. Anti-MHC Class I Antibody of Proliferation and Survival Signaling in Murine Cardiac Allografts. J Immunol. 2008;180:2214–2224. doi: 10.4049/jimmunol.180.4.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eisen HJ, Tuzcu EM, Dorent R, Kobashigawa J, Mancini D, Valantine-von Kaeppler HA, Starling RC, Sorensen K, Hummel M, Lind JM, Abeywickrama KH, Bernhardt P. Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients. N Engl J Med. 2003;349:847–858. doi: 10.1056/NEJMoa022171. [DOI] [PubMed] [Google Scholar]
- 34.Vigano M, Tuzcu M, Benza R, Boissonnat P, Haverich A, Hill J, Laufer G, Love R, Parameshwar J, Pulpon LA, Renlund D, Abeywickrama K, Cretin N, Starling RC, Eisen HJ. Prevention of acute rejection and allograft vasculopathy by everolimus in cardiac transplants recipients: a 24-month analysis. J Heart Lung Transplant. 2007;26:584–592. doi: 10.1016/j.healun.2007.03.005. [DOI] [PubMed] [Google Scholar]