

Abstract
p97, an essential chaperone in endoplasmic reticulum-associated degradation and organelle biogenesis, contains two AAA domains (D1 and D2) and assembles as a stable hexamer. We present a quantitative analysis of nucleotide binding to both D1 and D2 domains of p97, the first detailed study of nucleotide binding to both AAA domains for this type of AAA+ ATPase. We report that adenosine 5′-O-(thiotriphosphate) (ATPγS) binds with similar affinity to D1 and D2, but ADP binds with higher affinity to D1 than D2, offering an explanation for the higher ATPase activity in D2. Stoichiometric measurements suggest that although both ADP and ATPγS can saturate all 6 nucleotide binding sites in D1, only 3–4 of the 6 D2 sites can bind ATPγS simultaneously. ATPγS binding triggers a downstream cooperative conformational change of at least three monomers, which involves conserved arginine fingers and is necessary for ATP hydrolysis.
The “ATPases associated with various cellular activities” (AAA+)3 superfamily of proteins utilize ATP binding and hydrolysis to induce conformational change. This conformational change provides mechanical energy used in diverse functions to remodel substrate proteins and nucleic acids (1). AAA+ ATPases are defined by the presence of one or two AAA domains, and many form homohexameric rings. The enzymatic mechanisms of the more complex AAA+ ATPases have remained largely unclear, especially those like p97, which contain two AAA domains and thus 12 ATPase active sites within a functional hexamer.
p97 (also known as vasolin-containing protein (VCP) in mammals and Cdc48p in yeast) is an abundant and essential eukaryotic AAA-ATPase that remodels substrate protein complexes and is best characterized in endoplasmic reticulum-associated degradation and homotypic membrane fusion events of organelle biogenesis following mitosis (2, 3). A proposed Cdc48p segregase activity has been demonstrated in vitro for a membrane-tethered transcription factor (activated by a mechanism that co-opts the later stages of endoplasmic reticulum-associated degradation), which, in the presence of Cdc48p and Ufd1p-Npl4p adaptor, was separated from a polyubiquitinated membrane anchor (4). Although the detailed mechanism by which p97 carries out remodeling activity remains obscure, it involves the formation of multiple p97-adaptor complexes and conformational change during the ATPase hydrolysis cycle (2, 5–9).
p97 consists of three domains: an N domain that interacts with many proteins and two homologous AAA domains with ATPase activity, D1 and D2. The AAA domains contain conserved motifs necessary for nucleotide binding (Walker A) and hydrolysis (Walker B), and mutation of these motifs may be used to specifically decrease these functions (10). p97 has a stable homohexameric structure with the D1 and the D2 domains forming two stacked rings around a central pore and N domains protruding away from the D1 domains (6). The N domain position varies depending upon bound proteins (such as p47) and nucleotides, suggesting that the ATP hydrolysis cycle propels changes of position of bound adaptors (7). Communication of conformational changes resulting from ATP binding and hydrolysis has been proposed through two routes. Firstly, within a monomer, flexible linkers between the N, D1, and D2 domains could propagate conformational change between domains (11). Secondly, AAA domains within a AAA ring are potentially functionally linked by two arginine residues, contained within the second region of homology motif (SRH), that may protrude into the neighboring active site (12). These residues are conserved in p97 and are necessary for ATP hydrolysis (13).
Most knowledge of how the p97 hexamer functions through the ATP hydrolysis cycle has been gained from structural and biochemical studies. Strikingly, all crystal structures of p97 (1e32; 1YQ0; 1YQI; 1YPW) show D1 bound to ADP, whereas D2 has been crystallized bound to an ATP analog (AMPPNP), a transition state analog (ADP-AlF3), ADP, and with no nucleotide (14–18). Non-quantitative biochemical studies support these nucleotide-bound states; ADP has been identified already bound to recombinant p97 (19), and ADP and ATP have been shown to bind to D2 (20). Interestingly, there is preliminary evidence that ATP may not be bound by all sites in the D2 ring simultaneously as just 2.23 modified ATP molecules cross-linked to a p97 hexamer (21) and, furthermore, the structure of p97 with ADP-AlF3 bound to D2 shows full occupancy of the AlF3 moiety in only one of three monomers in the asymmetric unit (14).
To further investigate the mechanism of p97, we have carried out a systematic characterization of the nucleotide binding to p97 D1 and D2 domains. By exploiting mutations of the Walker A motif to limit nucleotide binding to D1 and D2 selectively, we have determined the dissociation constants and stoichiometry of ADP and ATP analog (ATPγS) binding to D1 and D2 individually. Additionally, detailed tryptophan fluorescence measurements have revealed a conformational change of p97 upon ATPγS binding to D2. Taken together, these results provide new insight into the molecular mechanism of p97.
EXPERIMENTAL PROCEDURES
Mutagenesis and Protein Preparation—An expression vector containing full-length rat wild-type p97 (pTrc His B p97) with an N-terminal His6 tag was constructed, and point mutations to the Walker A motif (K251A and K524A) and the SRH motif (R359A and R635A) were introduced by the QuikChange® method (Stratagene). All constructs were expressed at 37 °C in the Escherichia coli Rosetta 2 (DE3) strain (Novagen). Clarified lysates were loaded onto a HiTrap chelating column (GE Healthcare) charged with Ni2+ and eluted by imidazole gradient.
The purity and oligomeric state of preparations was confirmed by SDS and non-denaturing PAGE. The concentration of p97 monomers was quantified by absorbance at 280 nm. Samples, where required, were digested with apyrase enzyme (Potato apyrase, Grade VII, Sigma-Aldrich) as described previously (19). Apyrase was physically separated from p97 by a dialysis membrane during the digest to prevent contamination of p97. All binding experiments were carried out in the following buffer: 50 mm Tris (pH 8), 20 mm MgCl2, 2 mm EDTA, and all concentrations of p97 are of monomers.
Quantification of Bound Nucleotide by Heat Denaturation— Bound nucleotide was released by incubating 75 μl of 100 μm p97 at 100 °C. The precipitated protein was removed by centrifugation, the supernatant was decanted, and the volume was measured. The concentration of adenine nucleotide was calculated from absorbance at 260 nm (ε = 15 400 m-1 cm-1), and the amount of nucleotide released was expressed as the ratio of nucleotide to p97 monomer. Error bars in the legends for Figs. 2 and 4 represent the standard deviation of greater than six measurements.
FIGURE 2.

Affinity of ADP for D1 and D2. A, heat denaturation measurements of the amount of ADP prebound to wild type and Walker A mutants show that ADP is prebound to D1. B, fluorescence anisotropy measurements of B-ADP binding to wild type and Walker A mutants confirm that B-ADP binds selectively to D1. C, linear regression of the apparent Kd of B-ADP at different concentrations of ADP allows calculation of the actual Kd of B-ADP and ADP. D, ITC shows that ADP binds to two types of sites with distinct affinities (fitted to a two-site model). E, ADP binding to p97 pretreated with apyrase (to remove prebound ADP) was measured by ITC and fitted to a two-site model (as in panel D). The calculated Kd (○, ▵) and ΔH (▪, ♦) of D1 and D2 do not vary significantly from untreated p97 (shown in purple). App. stoichiometry, apparent stoichiometry.
FIGURE 4.

Stoichiometry and cooperativity associated with ATPγS binding p97. A, ITC profile of wild-type p97 binding to ATPγS. The data are fitted to a competition model calculating that p97 is saturated at 1.6 ATPγS per monomer. B, modified heat denaturation experiments in which ATPγS was added to p97 (with ADP prebound or removed by apyrase digest). Results show that ATPγS binding to p97 is saturated at 1.5–1.6 molecules of nucleotide per monomer (9–10 per hexamer) and is independent of ADP prebound to D1. C, tryptophan fluorescence changes of wild type and Walker A mutants in response to ATPγS binding show that the fluorescence change results from ATPγS binding to D2 and is not altered by nucleotide bound to D1. Fits to a cooperative binding equation calculate greater than 2.8 monomers positively cooperate. D, tryptophan fluorescence of SRH mutant (R359A,R635A) shows no positive cooperativity. Fitting to a single site model shows that ATPγS is bound with similar affinity to ITC measurements of wild-type p97. E, ITC data of R359A,R635A binding to ATPγS indicate that the stoichiometry of ATPγS binding is 1.66 molecules of ATPγS per monomer (10 per hexamer), similar to wild-type p97.
The stoichiometry of ATPγS bound by p97 was estimated by mixing 1 ml of 10 μm p97 with a moderate excess of ATPγS (Sigma-Aldrich) and spin-concentrating to 10% of the original volume. Protein concentration and volume were recalculated, and the total amount of nucleotide in the sample was determined. The concentration of unbound ATPγS (assumed to be equal to the starting concentration of ATPγS) was subtracted from the total nucleotide concentration, giving the amount of nucleotide bound to p97. Molar excesses greater than 4-fold were not used as the total nucleotide concentration measurements were dominated by unbound nucleotide.
Fluorescence Spectroscopy—Fluorescence measurements were made at 25 °C with a Fluoromax-3 spectrofluorimeter (Jobin Yvon Horiba), and data were fitted using GraFit (Erithacus Software). Fluorescence anisotropy experiments used 50 nm BODIPY-labeled ADP (B-ADP) (Invitrogen) in a glass cuvette (λex: 595 nm, slits 3 nm; λem: 620 nm, slits 10 nm). Each ADP/B-ADP competition experiment used a constant concentration of ADP (1, 5, 10, or 20 μm) and titrated p97, mixing fresh p97, ADP, and B-ADP for each point to correct for prebound ADP. The apparent Kd (App.K Ld) at each of the four ADP concentrations ([Itot]) was plotted, and linear regression calculated the Kd of B-ADP (K Ld) and ADP (K Id) for p97 by the following equation (adapted from Ref. 22).
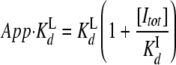 |
Tryptophan fluorescence intensity measurements were made by adding ATPγS to 500 μl of 1 μm p97 in a quartz cuvette (λex: 295 nm, slits 2 nm; λem: 345 nm, slits 5 nm). To minimize photobleaching, fresh p97 was used for each concentration of ATPγS. Fluorescence intensity changes as ATPγS increased were fitted to a single site or cooperative binding equation (23) (mean and standard deviation of multiple titrations are listed in supplemental Table 2).
Isothermal Titration Calorimetry—Isothermal titration calorimetry (ITC) measurements were made with a Microcal VP-ITC microcalorimeter (Microcal) equilibrated at 25 °C, typically titrating nucleotide against 50–300 μm p97. Data were analyzed using Origin (OriginLab) and fitted to models describing one and two types of binding site and competition between two ligands (24, 25). (Competition models were fitted using the concentration of prebound ADP determined by heat denaturation experiments.) As data did not show features suggesting more complex binding modes and fitted well to two-site and competition binding models giving results consistent with prior knowledge, higher dimensional models were not fitted. Fitting results of at least three repetitions of each experiment were averaged, and the mean and standard deviations are listed in supplemental Table 1).
RESULTS
Mutagenesis of Walker A and SRH Motifs—We sought to explore nucleotide binding to recombinant rat p97 by studying the D1 and D2 domains separately. To selectively reduce nucleotide binding affinity of individual AAA domains, we mutated the Walker A motif (GXXXGKT) that interacts with bound nucleotide (17) (Fig. 1). Lysine to alanine mutation of the Walker A motif was made in D1 (K251A) and D2 (K524A) both individually and together (K251A,K524A). To also probe potential communication around the hexamer ring, an arginine finger in the SRH motif was mutated to alanine in both D1 and D2 (R359A,R635A). In D1 at least, crystal structures (e.g. PDB code 1e32) show that this arginine residue protrudes into the active site of the neighboring monomer (Fig. 1, see also supplemental Fig. 1).
FIGURE 1.

The active site of p97 D1 domain. The figure shows the crystal structure (PDB code 1e32) of the active site of p97 D1 domain (17) showing ADP bound at the interface of two monomers (blue and gray). Conserved residues crucial for nucleotide binding (Walker A Lys-251) and hydrolysis (Walker B Glu-305) are shown, as are two arginine residues (Arg-359 and Arg-362) contributed by the SRH motif of the neighboring monomer.
Introduction of these mutations to recombinant p97 did not alter the oligomeric structure (see supplemental Fig. 2A); however, these mutations did affect ATPase activity (see supplemental Fig. 2B). Similar to findings of other groups, p97 carrying the Walker A mutation to the D1 domain alone (K251A) showed a moderate decrease in ATPase activity, whereas p97 carrying mutations in the D2 domain (K524A, K251A,K524A, R359A,R635A) showed little ATPase activity when compared with wild type (13, 26, 27). This is consistent with the previously suggested hypothesis that the D2 domain is the major site of ATPase activity (27, 28).
Characterization of ADP Binding to p97—Other studies found that ADP is prebound to recombinant preparations of p97 (14–19). Therefore, prior to carrying out ligand binding experiments, we made a detailed characterization of nucleotide already bound to p97. Samples of p97 were heat-denatured, the precipitated protein was removed by centrifugation, and the resulting supernatant was examined spectroscopically. The supernatant had an absorption maximum at 260 nm, consistent with adenine-based nucleotides. Further analysis by HPLC showed co-elution with ADP but not with ATP controls (data not shown), indicating that ADP is prebound to our p97 preparations. We quantified the amount of ADP prebound to p97 and found that typically 0.9 ± 0.2 molecules of ADP were present per p97 monomer (Fig. 2A).
To determine which AAA domains ADP was bound to, heat denaturation experiments were repeated using Walker A mutants designed to specifically reduce the affinity of nucleotide in D1 and D2. HPLC confirmed that all mutants contained ADP (data not shown). However, p97 mutated in D2 alone (K524A) contained a similar amount of prebound ADP as wild type, whereas p97 mutated in D1, either alone (K251A) or in combination with D2 (K251A,K524A), contained little prebound ADP (Fig. 2A). This indicates that ADP is prebound solely to D1 as Walker A mutation of this domain is sufficient to reduce prebound ADP. Mutations of the SRH motif of D1 and D2 (R359A,R635A) did not alter the amount of ADP prebound to D1 consistent with structural predictions that Arg-359 does not interact with ADP (17). These experiments define the initial nucleotide-bound state of our recombinant p97 preparations, showing that ADP is prebound to 90% of wild-type D1 domains (0.9 ADP molecules per monomer).
From these results, we hypothesized that ADP binds with highest affinity to D1 since it is retained throughout the purification and normal dialysis of recombinant p97. To determine the affinity of ADP for D1, we carried out binding experiments using fluorescent BODIPY-ADP (B-ADP) as a probe. Wild-type p97 was added incrementally to B-ADP, and binding was detected by the resulting increase of fluorescence anisotropy (Fig. 2B). Binding experiments were then performed with D1 and D2 Walker A mutants to identify which domain B-ADP binds to. The D2 mutant exhibited similar B-ADP binding to the wild-type, suggesting that B-ADP does not bind to D2. Mutation of D1 (K251A and K251A,K524A), however, disrupted normal B-ADP binding (7-fold greater concentration necessary for half-maximal saturation), thus confirming that B-ADP binds to D1 (Fig. 2B). The dissociation constant (Kd) of ADP binding to D1 was determined by measuring the apparent Kd of B-ADP at four concentrations of competing unlabeled ADP. Plotting the apparent Kd of B-ADP against the competing ADP concentration allowed the Kd of ADP to be estimated at 1.4 μm (Fig. 2C).
ADP binding to wild-type p97 was also investigated by ITC. We titrated an excess of ADP against wild-type p97, yielding an exothermic binding profile (Fig. 2D). Fits of a single site binding model were inconsistent with the binding profile at higher ADP concentrations, indicating that ADP also binds to a second site. Other studies have demonstrated that D2 can also bind ADP (14, 20) and, in agreement with this, a two-site binding model fitted the experimental data well. The two-site model predicts a higher affinity site with a Kd of 1.1 ± 0.8 μm and apparent stoichiometry of 0.2 ± 0.05 and a low affinity site with a calculated Kd of 86 ± 51 μm but poorly defined stoichiometry due to the low affinity of this site (stoichiometry was set to 1 for fitting; see supplemental Table 1). The apparent stoichiometry of the high affinity site (0.2 ± 0.05) is consistent with the saturation of a small population of vacant D1 domains, as predicted by Fig. 2A, and supporting this, the Kd is the same as measured previously for D1 (Fig. 2C). Combining these results, we interpret that ADP binds with significantly higher affinity to D1 (Kd ∼ 1 μm) than D2 (Kd ∼ 90 μm).
We next asked whether ADP prebound to D1 influenced the binding of ADP to D2. p97 was therefore treated with apyrase for varying times to remove differing amounts of ADP from D1 (19), and ADP binding was then measured by ITC. Longer apyrase treatments led to higher measured stoichiometries of ADP binding to D1; however, D1 and D2 ADP binding parameters (Kd and ΔH) remained similar to untreated p97 (Fig. 2E). Thus, under these conditions, ADP binding to D2 is not influenced by ADP bound to D1. Supporting this, the Kd of ADP binding to D2 when D1 ADP binding was disrupted (K251A) was in the same range as D2 in wild-type p97 as determined by ITC (data not shown).
In summary, we have identified ADP prebound to the D1 domain of our p97 preparations (consistent with other studies (14–19)) and have quantified the amount ADP bound to wild type and mutants. Measurements of the affinity of ADP binding to D1 have revealed an apparent Kd of 1 μm. Given that ADP is found in all D1 domains of p97 crystal structures (PDB code 1e32; 1YQ0; 1YQI; 1YPW), this affinity is lower than predicted. However, ADP is bound in a deep pocket in the crystal structure, in between two monomers. This suggests a slow off rate and a possibly even slower on rate, explaining the low Kd. Furthermore, different buffer conditions used for crystallization versus enzymatic assays could also potentially contribute to this (14–19). In contrast to D1, the D2 ring has comparatively low affinity for ADP, predicting a role as the major site of ATP hydrolysis.
Affinity of ATPγS Binding to D1 and D2—We wanted to examine ATP binding to p97. However, as we detected residual ATPase activity in p97 mutants designed to knock out catalysis of ATP hydrolysis (data not shown), we instead used a hydrolysis-resistant ATP analog. ATP analog, AMPPNP, has much lower affinity for p97 than ATP (21), indicating that for the purposes of binding experiments, it does not accurately represent ATP. Our preliminary experiments showed that ATPγS bound to p97 with considerably higher affinity than AMPPNP (data not shown), and so we therefore used ATPγS to mimic ATP. For these and subsequent ITC experiments, we did not remove ADP from D1 by apyrase digest as the treatment did not provide the large amounts of ADP-free p97 necessary. However, we have carefully characterized ADP already bound to p97, allowing it to be accounted for in analysis.
We first measured ATPγS binding to D2 Walker A mutant, K524A, which is deficient in D2 nucleotide binding and has ADP prebound to D1 (Fig. 2A). The mid-point of the binding curve estimates that a small population of D1 sites (0.16 ± 0.03 D1 sites per monomer) is unoccupied with prebound ADP and binds to ATPγS early in the titration. The data do not fit well to a single site model, implying that competition with prebound ADP must also be modeled. A competition model assuming that 0.9 molecules of ADP are present per monomer (from Fig. 2A) fitted closely to the experimental data. This fit predicts that ATPγS binds to D1 with a Kd of 2.1 ± 0.6 μm and an overall stoichiometry of 1.05 ± 0.04 molecules of ATPγS per monomer (6.3 ATPγS per hexamer) (Fig. 3A). The Kd of ADP for D1 calculated by the fit (0.6 ± 0.15 μm) is in reasonably good agreement with independent ITC measurements of ADP binding to wild type (Fig. 2D) and K524A (data not shown).
FIGURE 3.

Affinity of ATPγS for p97 Walker A mutants. A, ITC data of ATPγS binding to D2 Walker A mutant (K524A). Data are fitted to a competition model to account for prebound ADP. B, ITC data of ATPγS binding to p97 mutated in the D1 Walker A motif (K251A, fitted to a two-site model). Note the jagged profiles of raw data results from titration of different volumes of ligand.
We next tested ATPγS binding to D2 using the D1 Walker A mutant, K251A, which has reduced affinity for nucleotide in D1 and so contains little prebound ADP (Fig. 2A). Fits of a two-site model to ITC binding profiles predicted a high affinity site with a Kd of 3 ± 2 μm and a low affinity site with a Kd of 74 ± 34 μm (Fig. 3B, and see supplemental Table 1). (The model fitted best when the stoichiometry of ATPγS binding the high affinity site was 1; however, modified heat denaturation experiments (described later) did not confirm this, indicating a stoichiometry of 0.75 (see supplemental Fig. 3A).) We assigned the high affinity site as ATPγS binding to D2 because additional mutation of D2 (K251A,K524A) disrupted high affinity ATPγS binding (see supplemental Fig. 3B). The low affinity site could be a subset of low affinity D2 domains, or more likely, residual binding of ATPγS to the mutated D1 domain.
In contrast to the different affinities of ADP binding to D1 and D2, ATPγS binding to Walker A mutants suggests that D1 and D2 have similar affinities for ATPγS (D1 Kd = 2 μm and D2 Kd = 3 μm). Additionally, our data are consistent with measurements of the affinity of ADP binding (Fig. 2, C and D), providing evidence that ATPγS binding to D1 does not alter the affinity of ADP bound to other D1 domains.
Stoichiometry of ATPγS Binding to Wild-type p97—To investigate ATPγS binding to wild-type p97, we carried out further ITC experiments. The ITC binding profile of ATPγS titrated against wild-type p97 showed that ATPγS binding was exothermic at lower ATPγS concentrations but became endothermic at higher ATPγS concentrations (Fig. 4A, top panel). The mid-point of the exothermic portion indicated a stoichiometry of 0.66 ± 0.125 ATPγS molecules binding per p97 monomer. This is consistent with ATPγS initially binding to four vacant sites per hexamer, presumably from the empty D2 ring and the small population of ADP-free D1 domains. The later endothermic phase corresponds to ATPγS competing with ADP for binding sites in the D1 ring and represents the difference in enthalpy of ADP dissociation and ATPγS association.
Since prebound ADP was also present, we fitted a competition model that accounts for 0.9 molecules of prebound ADP per monomer (from Fig. 2A) but assumes that D1 and D2 have the same affinity for ATPγS (Fig. 3). In addition, we fixed the Kd of ADP at 0.85 μm and assumed that low affinity ADP binding to D2 was negligible. The fit estimated the average Kd of ATPγS to be 1.7 μm (Fig. 4A), and notably, the total stoichiometry of ATPγS was 1.6 ± 0.16 molecules per monomer (9.6 molecules ATPγS per hexamer).
We confirmed this predicted stoichiometry by measuring the maximal amount of ATPγS bound by p97 through a modified heat denaturation protocol. An excess of ATPγS was added to wild-type p97, and the majority of unbound ATPγS was removed before the sample was heat-denatured. (The excess of ATPγS was limited to 4-fold as experimental error increased substantially at higher concentrations.) Binding to p97 was close to saturation when ∼1.5 molecules of nucleotide were bound per p97 monomer (9 molecules per hexamer) (Fig. 4B). Repeating this experiment using apyrase-treated p97 gave a similar result (Fig. 4B), demonstrating that the maximal amount of ATPγS bound is not influenced by the presence of ADP in D1.
Together these results indicate that within a p97 hexamer, only 9–10 nucleotides fill the 12 possible binding sites. Given that the D1 and D2 rings have differing structural properties (6) and that crystallographic structures show full occupancy of D1 (14–18) but partial occupancy of D2 in the case of ADP-AlF3 (14), we speculate that one ring is fully occupied by ATPγS, whereas the other ring is partially occupied. The balance of evidence suggests that the D1 ring is fully occupied with 6 molecules of ATPγS, whereas the D2 ring can only bind 3–4 molecules of ATPγS.
Cooperativity Associated with ATPγS Binding to D2—We next probed the mechanism of ATPγS binding to the D2 ring by exploiting a Trp residue positioned on the linker between D1 and D2 and close to the D2 active site (Trp-476). The fluorescence of this residue is reported to increase with D2 nucleotide binding (20) due to a change in the environment of the Trp residue resulting from either the proximity of nucleotide to Trp-476 or a conformational change in p97. Titration of ATPγS into wild-type p97 produced a sigmoidal increase in fluorescence that fitted to a cooperative binding equation with a Hill coefficient (nH) of 2.8 ± 0.3 (Fig. 4C and supplemental Table 2) (23). We repeated the titration with the D1 and D2 Walker A mutants to confirm that this fluorescence change was a consequence of ATPγS binding to D2. In the same ATPγS concentration range, mutation of D2 (K524A and K251A,K524A) was sufficient to prevent any increase in Trp fluorescence (Fig. 4C, see supplemental Table 2). This clearly demonstrates that changes in Trp fluorescence are exclusively associated with ATPγS binding to the D2 ring alone.
The sigmoidal increase in Trp fluorescence indicates positive cooperativity within the hexamer upon ATPγS binding to D2. The Hill coefficient of 2.8 implies that three or more p97 monomers positively cooperate upon ATPγS binding to the D2 ring. A similar effect was observed in p97 purified from rat liver cytosol (nH = 4 ± 0.1), in this case indicating cooperation of at least four p97 monomers (see supplemental Fig. 4 and Table 2). Although the significance in the slight difference in Hill coefficient between recombinant and endogenous p97 is unclear, both results suggest a cooperative change involving at least 3–4 monomers.
We asked whether the nucleotide state of D1 alters the cooperative fluorescence changes by comparing ATPγS titrations when D1 is in differing nucleotide-bound states. We compared Trp fluorescence profiles of wild-type p97 (with ADP prebound to D1; Fig. 2A) to the D1 K251A mutant (D1 empty and reduced affinity for ATPγS; Figs. 2A and 3B) and apyrase-treated p97 (D1 empty and ATPγS binding to both domains; Fig. 4B). The three profiles were very similar (Fig. 4C), demonstrating that the nucleotide-bound state of D1 does not influence the cooperative Trp-476 fluorescence changes associated with ATPγS binding to D2.
Cooperative Conformational Change Downstream of ATPγS Binding—Since it has been proposed that the activity of individual monomers in a hexameric ring are coordinated by arginine fingers (from the SRH motif) protruding from one monomer to the next (13), we asked whether these arginine residues in p97 (Arg-359, Arg-635) are involved in the cooperative fluorescence changes we observed on ATPγS binding. Significantly, ATPγS binding to the double arginine mutant (R359A,R635A) resulted in a decrease in the magnitude of the fluorescence change (reduced from 1–1.5 in wild type to 1–1.3 in R359A,R635A) and a loss of the sigmoidal shape of the binding curve indicating no positive cooperativity (Fig. 4D, see supplemental Table 2). Fitting to a single site model predicted that ATPγS binds with a Kd of 1.9 ± 0.6 μm to R359A,R635A. This is comparable with previous measurements for wild-type p97 and confirms that these mutations do not disrupt the affinity of ATPγS binding (Fig. 4A, see also supplemental Table 2). The smaller increase in Trp-476 fluorescence of R359A,R635A (from 1 to 1.3) can be assigned directly to nucleotide binding alone, whereas the larger cooperative increase (from 1 to 1.5) observed with wild-type p97 is consistent with nucleotide binding and an additional cooperative conformational change.
We also used ITC experiments to measure the stoichiometry of ATPγS binding to R359A,R635A, fitting a competition model to the data to account for prebound ADP (Fig. 2A). The total stoichiometry of ATPγS binding derived from this fit was 1.66 ± 0.07 (Fig. 4E), similar to that obtained for wild-type p97 (1.6; Fig. 4A). The Kd for ATPγS binding to D1 and D2 (7.8 ± 1.6 μm) was slightly increased but of a similar magnitude to wild-type p97, and the competition phase of the data was slightly more endothermic, which we attribute to altered conformational flexibility within the system.
These results show that mutation of this SRH arginine finger does not significantly affect the affinity or stoichiometry of ATPγS binding, but does lead to a loss of cooperativity. The cooperativity observed for wild-type p97 therefore appears to be associated with a conformational change that occurs downstream of ATPγS binding, and is mediated by the SRH arginine finger. Since mutations of arginine fingers of the SRH motif are also associated with loss of ATPase activity (see supplemental Fig. 2B) (13), we conclude that this cooperative conformational change is required for ATP hydrolysis.
DISCUSSION
This work presents the first quantitative study of the nucleotide binding properties of both D1 and D2 rings of a Type II AAA ATPase and reveals striking differences between the homologous AAA domains of p97. Despite sharing over 40% sequence identity, we have determined markedly different dissociation constants of ADP binding to the D1 and D2 rings: around 1 μm for D1 and 90 μm for the D2 ring (summarized in supplemental Table 3). In contrast, the affinity of ATPγS binding to D1 and D2 is similar for both rings, with a dissociation constant of ∼2 μm. Other work has suggested that the D2 ring is the major site of ATP hydrolysis (27), a finding supported by crystal structures that capture D2 (PDB code 1e32; 1YQ0; 1YQI; 1YPW) in multiple nucleotide-bound states, but D1 bound only to ADP (14–16). Our results support these data and suggest that the difference in affinity of ADP for D1 and D2 provides a plausible explanation for these observations.
Although the affinity of ATPγS for D1 and D2 is similar, our results show that the stoichiometry of binding is distinct; ATPγS binding is limited to a subset of D2 domains (3–4 out of 6 possible sites), whereas the D1 ring binds ATPγS to full occupancy. The stoichiometry of ATPγS binding to D2 predicts that ATPγS binding to the first 3–4 D2 sites leads to changes in the final 2–3 D2 sites to prevent ATPγS binding. Fig. 5 summarizes possible symmetrical and random arrangements of ATPγS binding to D2 consistent with our results. The partial occupancy of ATPγS in D2 is in agreement with earlier studies. i) Only 2.23 molecules of photoactivated ATP derivative were cross-linked to endogenous p97 (21), and ii) only one D2 domain of the three monomers in the asymmetric unit of the p97-ADP-AlF3 crystal structure had full occupancy of AlF3 (14). In the AAA+ ATPase superfamily, there is a precedent for partial occupancy of ATP in homohexameric AAA proteins with single rings such as ClpX, RuvB, and Hs1U (29–31), but this is the first report for a tandem AAA protein and suggests a potential conservation of mechanism despite the wide variety of AAA+ ATPase functions.
FIGURE 5.

Models of ATP binding to p97 D2 domain. The complete D2 ring is shown uppermost with the D1 ring and N domains below (partially obscured). Four of the possible arrangements of ATP binding consistent with our measurements of stoichiometry and cooperativity of ATPγS binding are shown. 3 or 4 molecules of ATP could bind symmetrically as a trimer of dimers (A) or a dimer of trimers (B), or alternatively, in an asymmetric organization (C and D). Additionally, ATP could bind to three or four neighboring D2 domains (not shown).
We have additionally tested in a limited set of situations whether the nucleotide-bound state of one ring alters the affinity of the other. Our results show that ADP and ATPγS binding in D2 is unchanged by the presence of ADP bound in D1 and does not alter the affinity of ADP bound to D1. Furthermore, the stoichiometry of ATPγS binding to D2 is unaffected by ADP bound in D1, and conformational changes in D2 are the same, regardless of the nucleotide-bound state of the D1 ring. Our results provide the first evidence that the nucleotide binding properties of D1 and D2 rings are independent. This does not rule out an interdependence of D1 and D2 in other aspects of p97s mechanism, such as the detailed kinetics of ATP hydrolysis or in the transfer of conformational energy to potential adaptors. However, under the conditions of the experiments carried out here, both rings can act independently in terms of nucleotide binding.
In this study, we carried out a detailed analysis of tryptophan fluorescence changes previously shown to arise from nucleotide binding (20) and have identified a positive cooperative conformational change associated with ATPγS binding to D2 in both recombinant and endogenous p97. The Hill coefficient of the conformational change predicts that this involves at least three monomers, and we have shown that the conserved arginine residues of the SRH motif (most likely Arg-635) are key to mediating this change. Interestingly, this positive cooperative conformational change does not regulate ATPγS binding because we find that mutation of the key SRH residues (R359A,R635A) prevents cooperativity but does not significantly alter ATPγS binding affinity or stoichiometry. However, this mutation does decrease ATP hydrolysis to low levels, in agreement with previous studies (13), suggesting that the positive cooperative conformational change could be an essential component of the ATP hydrolysis pathway. Studies of mixed mutant hexamers have also indicated that the potential intermonomer contacts made by these residues contribute to the ATP hydrolysis pathway (13). Interestingly, crystal structures of the D1 domain (e.g. PDB code 1e32) unambiguously show Arg-359 protruding into the active site of the neighboring monomer (17), potentially giving directionality to any conformational change.
In vivo, p97 is thought to separate or translocate protein complexes by using ATP binding and hydrolysis to change conformation. Changes in conformation upon nucleotide binding (6–8, 14, 19, 20, 32) and also the nucleotide dependence of assembly of some p97-adaptor complexes has already been demonstrated (33, 34); however, there is lack of detailed knowledge of the steps involved. Our identification of a 3–4 ATP binding stoichiometry and cooperativity of at least three monomers suggests that the D2 ring of the p97 hexamer does not operate a concerted mechanism in which all D2 sites simultaneously bind ATP, hydrolyze ATP, and change conformation (10). Instead, the stoichiometry and cooperativity are more consistent with a processive mechanism where multiple rounds of ATP hydrolysis and conformational change form a full reaction cycle. Further experiments utilizing linked oligomers are required to investigate whether ATP binding and hydrolysis follow a symmetrical and rotational mechanism or are a stochastic process as has been shown for ClpX (31).
In comparison with other AAA+ enzymes, our results suggest that the relationship between D1 and D2 is similar to that of close homologue, N-ethylmaleimide-sensitive factor (NSF). The stability of ADP bound to the D1 domain of p97 resembles one AAA ring of NSF (NSF D2), which binds ATP with high affinity (Kd = 30 nm) and has only been crystallized bound to ATP or ATP analogs (35–37). In common with the D2 ring of p97, the other AAA ring of NSF (NSF D1) shows higher ATPase activity (38, 39). In another tandem AAA+ ATPase, Hsp104, ATP hydrolysis in one ring (NBD1) is regulated by the nucleotide-bound state of the other (NBD2) (40), and substrate remodeling activity is linked to impaired hydrolysis at individual AAA domains (41). Although our results suggest that nucleotide binding to the D1 and D2 rings of p97 is independent, we do not rule out more complex interdependence of ATP hydrolysis in the presence of adaptor/substrate proteins similar to Hsp104. Intriguingly, the p97 D2 ring shares some resemblance to a distant relative with a single AAA ring, ClpX, which also binds 3–4 ATP molecules per hexamer in a similar range of affinity (Kd = 0.5 μm compared with 2 μm for D2 binding ATPγS) and moreover displays a positive cooperative change accompanying ATP binding (31). Taken together, these similarities suggest that despite the presence of an additional AAA ring in p97, combined with evolutionary and functional divergence, elements of the ATP hydrolysis mechanism of homohexameric AAA+ proteins may be conserved.
Supplementary Material
Acknowledgments
We are very grateful to Ingrid Dreveny and Valerie Pye for fruitful discussions and John Eccleston for critically reading the manuscript. We would thank Colin Davis and Ciaran McKeown for technical assistance and Suhail Islam for preparation of structural figures.
This work was supported by a Welcome Trust grant (to P. S. F. and X. Z.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains four supplemental figures, three supplemental tables, and supplemental methods.
Author's Choice—Final version full access.
Footnotes
The abbreviations used are: AAA, ATPases associated with various cellular activities; ITC, isothermal titration calorimetry; B-ADP, BODIPY ADP; NSF, N-ethylmaleimide-sensitive factor; SRH, second region of homology; HPLC, high pressure liquid chromatography; PDB, Protein Data Bank; ATPγS, adenosine 5′-O-(thiotriphosphate); AMPPNP, 5′-adenylyl-β,γ-imidodiphosphate; ADP-AIF3, adenosine 5′-diphosphate-aluminum fluoride.
References
- 1.Hanson, P. I., and Whiteheart, S. W. (2005) Nat. Rev. Mol. Cell Biol. 6 519-529 [DOI] [PubMed] [Google Scholar]
- 2.Ye, Y. (2006) J. Struct. Biol. 156 29-40 [DOI] [PubMed] [Google Scholar]
- 3.Uchiyama, K., and Kondo, H. (2005) J Biochem. (Tokyo) 137 115-119 [DOI] [PubMed] [Google Scholar]
- 4.Shcherbik, N., and Haines, D. S. (2007) Mol. Cell 25 385-397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dreveny, I., Pye, V. E., Beuron, F., Briggs, L. C., Isaacson, R. L., Matthews, S. J., McKeown, C., Yuan, X., Zhang, X., and Freemont, P. S. (2004) Biochem. Soc. Trans. 32 715-720 [DOI] [PubMed] [Google Scholar]
- 6.Pye, V. E., Dreveny, I., Briggs, L. C., Sands, C., Beuron, F., Zhang, X., and Freemont, P. S. (2006) J. Struct. Biol. 156 12-28 [DOI] [PubMed] [Google Scholar]
- 7.Beuron, F., Dreveny, I., Yuan, X., Pye, V. E., McKeown, C., Briggs, L. C., Cliff, M. J., Kaneko, Y., Wallis, R., Isaacson, R. L., Ladbury, J. E., Matthews, S. J., Kondo, H., Zhang, X., and Freemont, P. S. (2006) EMBO J. 25 1967-1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rouiller, I., DeLaBarre, B., May, A. P., Weis, W. I., Brunger, A. T., Milligan, R. A., and Wilson-Kubalek, E. M. (2002) Nat. Struct. Biol. 9 950-957 [DOI] [PubMed] [Google Scholar]
- 9.Rouiller, I., Butel, V. M., Latterich, M., Milligan, R. A., and Wilson-Kubalek, E. M. (2000) Mol. Cell 6 1485-1490 [DOI] [PubMed] [Google Scholar]
- 10.Ogura, T., and Wilkinson, A. J. (2001) Genes Cells 6 575-597 [DOI] [PubMed] [Google Scholar]
- 11.Smith, G. R., Contreras-Moreira, B., Zhang, X., and Bates, P. A. (2004) J. Struct. Biol. 146 189-204 [DOI] [PubMed] [Google Scholar]
- 12.Ogura, T., Whiteheart, S. W., and Wilkinson, A. J. (2004) J. Struct. Biol. 146 106-112 [DOI] [PubMed] [Google Scholar]
- 13.Wang, Q., Song, C., Irizarry, L., Dai, R., Zhang, X., and Li, C. C. (2005) J. Biol. Chem. 280 40515-40523 [DOI] [PubMed] [Google Scholar]
- 14.DeLaBarre, B., and Brunger, A. T. (2005) J. Mol. Biol. 347 437-452 [DOI] [PubMed] [Google Scholar]
- 15.DeLaBarre, B., and Brunger, A. T. (2003) Nat. Struct. Biol. 10 856-863 [DOI] [PubMed] [Google Scholar]
- 16.Huyton, T., Pye, V. E., Briggs, L. C., Flynn, T. C., Beuron, F., Kondo, H., Ma, J., Zhang, X., and Freemont, P. S. (2003) J. Struct. Biol. 144 337-348 [DOI] [PubMed] [Google Scholar]
- 17.Zhang, X., Shaw, A., Bates, P. A., Newman, R. H., Gowen, B., Orlova, E., Gorman, M. A., Kondo, H., Dokurno, P., Lally, J., Leonard, G., Meyer, H., van Heel, M., and Freemont, P. S. (2000) Mol. Cell 6 1473-1484 [DOI] [PubMed] [Google Scholar]
- 18.Dreveny, I., Kondo, H., Uchiyama, K., Shaw, A., Zhang, X., and Freemont, P. S. (2004) EMBO J. 23 1030-1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davies, J. M., Tsuruta, H., May, A. P., and Weis, W. I. (2005) Structure (Camb.) 13 183-195 [DOI] [PubMed] [Google Scholar]
- 20.Wang, Q., Song, C., Yang, X., and Li, C. C. (2003) J. Biol. Chem. 278 32784-32793 [DOI] [PubMed] [Google Scholar]
- 21.Zalk, R., and Shoshan-Barmatz, V. (2003) Biochem. J. 374 473-480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng, Y., and Prusoff, W. H. (1973) Biochem. Pharmacol. 22 3099-3108 [DOI] [PubMed] [Google Scholar]
- 23.Hattendorf, D. A., and Lindquist, S. L. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 2732-2737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sigurskjold, B. W. (2000) Anal. Biochem. 277 260-266 [DOI] [PubMed] [Google Scholar]
- 25.Wiseman, T., Williston, S., Brandts, J. F., and Lin, L. N. (1989) Anal. Biochem. 179 131-137 [DOI] [PubMed] [Google Scholar]
- 26.Park, S., Rancour, D. M., and Bednarek, S. Y. (2007) J. Biol. Chem. 282 5217-5224 [DOI] [PubMed] [Google Scholar]
- 27.Song, C., Wang, Q., and Li, C. C. (2003) J. Biol. Chem. 278 3648-3655 [DOI] [PubMed] [Google Scholar]
- 28.Wang, Q., Song, C., and Li, C. C. (2003) Biochem. Biophys. Res. Commun. 300 253-260 [DOI] [PubMed] [Google Scholar]
- 29.Putnam, C. D., Clancy, S. B., Tsuruta, H., Gonzalez, S., Wetmur, J. G., and Tainer, J. A. (2001) J. Mol. Biol. 311 297-310 [DOI] [PubMed] [Google Scholar]
- 30.Bochtler, M., Hartmann, C., Song, H. K., Bourenkov, G. P., Bartunik, H. D., and Huber, R. (2000) Nature 403 800-805 [DOI] [PubMed] [Google Scholar]
- 31.Hersch, G. L., Burton, R. E., Bolon, D. N., Baker, T. A., and Sauer, R. T. (2005) Cell 121 1017-1027 [DOI] [PubMed] [Google Scholar]
- 32.Beuron, F., Flynn, T. C., Ma, J., Kondo, H., Zhang, X., and Freemont, P. S. (2003) J. Mol. Biol. 327 619-629 [DOI] [PubMed] [Google Scholar]
- 33.Boyault, C., Gilquin, B., Zhang, Y., Rybin, V., Garman, E., Meyer-Klaucke, W., Matthias, P., Muller, C. W., and Khochbin, S. (2006) EMBO J. 25 3357-3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uchiyama, K., Jokitalo, E., Kano, F., Murata, M., Zhang, X., Canas, B., Newman, R., Rabouille, C., Pappin, D., Freemont, P., and Kondo, H. (2002) J. Cell Biol. 159 855-866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matveeva, E. A., He, P., and Whiteheart, S. W. (1997) J. Biol. Chem. 272 26413-26418 [DOI] [PubMed] [Google Scholar]
- 36.Lenzen, C. U., Steinmann, D., Whiteheart, S. W., and Weis, W. I. (1998) Cell 94 525-536 [DOI] [PubMed] [Google Scholar]
- 37.Yu, R. C., Hanson, P. I., Jahn, R., and Brunger, A. T. (1998) Nat. Struct. Biol. 5 803-811 [DOI] [PubMed] [Google Scholar]
- 38.Sumida, M., Hong, R. M., and Tagaya, M. (1994) J. Biol. Chem. 269 20636-20641 [PubMed] [Google Scholar]
- 39.Whiteheart, S. W., Rossnagel, K., Buhrow, S. A., Brunner, M., Jaenicke, R., and Rothman, J. E. (1994) J. Cell Biol. 126 945-954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hattendorf, D. A., and Lindquist, S. L. (2002) EMBO J. 21 12-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doyle, S. M., Shorter, J., Zolkiewski, M., Hoskins, J. R., Lindquist, S., and Wickner, S. (2007) Nat. Struct. Mol. Biol. 14 114-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.