

Abstract
Activation-induced DNA cytosine deaminase (AID) is required for somatic hypermutation (SHM) and efficient class switch recombination (CSR) of immunoglobulin (Ig) genes. We created AID-transgenic mice that express AID ubiquitously under the control of a β–actin promoter. When crossed with AID−/− mice, the AID-transgenic, AID−/− mice carried out SHM and CSR, showing that the AID transgenes were functional. However, the frequencies of SHM in V- and switch-regions, and CSR were reduced compared to those in a wildtype AID background. Several criteria suggested that the inefficiency of SHM was due to reduced AID activity, rather than lack of recruiting error-prone DNA repair. High levels of AID mRNA were produced in resting B cells and kidney, cells that do not express AID in wildtype mice. Compared with these cells, activated B cells expressed about an order of magnitude less AID mRNA suggesting that there may be a post-transcriptional mechanism that regulates AID mRNA levels in professional AID producers but not other cells. The AID protein expressed in resting B cells and kidney was phosphorylated at serine-38. Despite this modification, known to enhance AID activity, resting B cells did not undergo SHM. Apparently, the large amounts of AID in resting B cells are not targeted to Ig genes in vivo, in contrast to findings in vitro.
Keywords: Activation-induced DNA cytosine deaminase, Somatic hypermutation, Class switch recombination, Immunoglobulin, Transgenic mice
1. Introduction
Naïve B lymphocytes are triggered to express activation-induced DNA cytosine deaminase (AID)3 and undergo somatic hypermutation (SHM) and class switch recombination (CSR) during T cell-stimulated immune responses (reviewed in (Longerich et al., 2006)). The detailed molecular mechanisms leading from AID expression to mutations in the variable regions of Ig light and heavy chain genes and heavy chain CSR are not understood. Especially intriguing are the facts that AID deaminates cytosine to uracil in Ig genes, but not most other genes in activated B cells, and that error-prone DNA repair mechanisms are recruited to sites where AID-created deoxy-uracils exist (reviewed in (Longerich et al., 2006)). Deoxy-uracils arising in other genes and outside the variable regions and their vicinity in Ig genes are eliminated and cytosines are reinstalled by a relatively error-free copying of the guanine on the opposite DNA strand (Friedberg et al., 2006).
We produced transgenic mice expressing AID transgenes at high levels in all cells in order to evaluate whether allowing excess AID to be produced would change certain characteristics of SHM. For example, we had previously shown that the presence of two copies of the CAGGTG hexamer in an Ig transgene leads to an over 4-fold increase in SHM that was completely reversed when the hexamer was changed to AAGGTG (Michael et al., 2003). CAGGTG is a good binding site for the E-box protein E47, AAGGTG is not. The SHM increase by CAGGTG was not accompanied by increased transcription of the Ig transgene, suggesting that CAGGTG, a component of all Ig enhancers, is an enhancer of somatic hypermutation in activated B cells. We postulated that the CAGGTG sequence was involved in attracting AID to Ig genes via transacting-factor binding to this site and inducing a conformational change of the transcription complex that permitted binding of the limited amounts of AID present in mutating B cells. We wanted to test whether ectopically expressed AID would obviate the effect of extra SHM enhancers.
Tansfection of AID into cell lines in culture induced cytosine deamination in a number of genes leading to C/G to T/A transitions without C/G transversions or mutations at A and T (Martin et al., 2002; Yoshikawa et al., 2002). This finding suggested that error-prone repair of the AID-induced uracil and neighboring nucleotides may require the in vivo maturation of B cells to the SHM state. It was therefore of interest whether AID expression under the control of a β-actin promoter would induce normal SHM patterns in activated B cells and whether it would cause mutations in resting B cells.
Finally, AID phosphorylated at serine38 was shown to be more active (Basu et al., 2005; McBride et al., 2006; Pasqualucci et al., 2006). Conflicting results were obtained with AID transfected into an embryonic kidney cell line, 293: it did not become phosphorylated nor did it induce SHM in one report (Basu et al., 2005), in another report it did become S38 phosphorylated in 293 cells as well as in fibroblasts (McBride et al., 2006). It was of interest to determine whether transgenic AID would undergo this phosphorylation and, if so, whether the phosphorylation was restricted to activated B cells.
AID-transgenic mice were studied by others and found to undergo SHM and CSR at reduced levels, despite higher amounts of AID protein in total AID-transgenic B cells than in wild type B cells (Muto et al., 2006). Also, PNA-lo B cells were found to undergo little if any SHM. We have made similar observations and expanded the analysis of such mice by comparing levels of AID mRNA and protein in resting and activated B cells in AID-transgenic versus wildtype mice. We also investigated the phosphorylation status of serine38 of AID in B cells and kidney. Further, we analyzed the 3’ decline rate of mutations during SHM and compared the mutability of high- and low-SHM targets in AID-transgenic and wildtype mice. It was of interest to determine in our study whether activated and resting B cells differentially regulated AID expression, to further consider why resting AID-transgenic B cells did not mutate their Ig genes, and whether the reduced SHM and CSR were due to reduced activity of transgenic AID, or reduced error-prone repair mechanisms.
2. Material and methods
2.1. Miice
The plasmid mAID-pCXN2 was a gift of T. Honjo (Kyoto University, Japan) (Okazaki et al., 2003) and was digested with Sal I /Hind III to remove most of the plasmid sequence. The purified AID-transgene DNA fragment was used to generate AID-transgenic (Tg) mice on a C57BL/6NTac background. Two lines of mice, #14 and #18, with about twenty copies of the AID transgene each (not shown) were generated. The transgenic mice were bred to C57BL/6 mice, keeping the transgene hemizygous. For certain experiments, the transgenic mice were further crossed with AID−/− mice at The University of Illinois at Chicago Animal Facilities. The Animal Care and Institutional Biosafety Committees at the University of Chicago and the University of Illinois gave approvals for the animal protocols used. Primers specific for the endogenous AID are located in the 5’ untranslated region and exon 1 (Muramatsu et al., 2000). The 5’ untranslated region of the endogenous AID is absent in the AID transgene. Primers specific for both the endogenous and transgenic AID genes were sense 5’ CCATTTCAAAAATGTCCGCT 3’ and anti-sense 5’ CAGGTGACGCGGTAACACC 3’ and were located in exon 2 and 3, respectively.
2.2. Mouse immunization and cell purification
Mice were immunized by i.p. injection of 108 sheep red blood cells (SRBC) on day 1, inoculated with the same amount of SRBC on day 8, and sacrificed on day 11. Spleens were removed and minced. Red blood cells were lysed with red blood cell lysing buffer (Sigma, St. Louis, MO). The remaining white cells were sorted using a Mo-Flo sorter (BD Biosciences) at the University of Chicago Immunology Core Facility after the cells were stained with fluorescein-peanut lectin agglutinin (PNA) and PE-anti-B220 (BD Pharmingen). The B220+PNAhigh and B220+PNAlow B cells were collected for DNA, RNA, and protein isolations.
2.3. In vitro stimulation of B cells
Mouse CD19+CD43− B cells were purified according to Miltenyi Biotec’s instructions (Auburn, CA). Briefly, mouse spleen cells were attached to magnetic beads conjugated with anti-mouse-CD43 monoclonal antibody (Miltenyi Biotec, Auburn, CA). The CD43− cells were further attached to anti-mouse-CD19 beads. The CD43−CD19+ B cells were cultured for 5 days in RPMI1640 containing 10% FBS in the presence or absence of 50 ug/ml of LPS and 25 ng/ml of IL-4. DNA, RNA and protein were collected from naïve and activated B cells.
2.4. DNA, RNA and protein
The genomic DNA from B220+PNAhigh, B220+PNAlow, and CD43−CD19+ cells was purified using either phenol/chloroform or the DNeasy tissue kit (Qiagen, Valencia, CA), while total RNA was isolated with the RNAqueous-4PCR kit (Ambion, Austin, TX). For Western blots, total protein was obtained by lysing the purified B cells in hypotonic buffer [10 mM Tris-Hcl (pH 7.4), 4mM EDTA, 30 mM Kcl, 1% NP40, 1mM NaF, and proteinase inhibitors (Roche, Germany), or loading buffer. For AID phospho-ser38 detection the protocol, anti-AID and antipS38 antibodies were previously described (McBride et al., 2006). In brief AID protein was partially purified from kidney, CD43− or CD43+ splenocyte whole cell extracts (20mM Tris pH 8, 400 mM NaCl, 1% NP40, 0.5mM EDTA, 25mM NaF, 1mM DTT) by anti-AID immunoprecipitation before Western blots were performed. For Western blots to determine PCNA in PNA-hi and PNA-lo B cells, anti-PCNA-HRP (PC10, Santa Cruz Biotech, CA) was used. The blot was reprobed, after stripping, with anti-actin antibodies (A2066, Sigma, MO).
2.5. Mutation analysis
The immunoglobulin heavy chain gene was amplified with Pfu Turbo using touchdown PCR as described (Longerich et al., 2005). The primers were specific to the J558 V region and the middle of the JC intron so that an ~ 1.2 kb DNA fragment containing the JH4 region and a part of the JC intron was amplified. The DNA fragment was purified from an agarose gel using the QIAquick Gel Extraction kit (Qiagen), inserted into the pCR-Blunt II-TOPO vector (Invitrogen, Carlsbad, CA), and amplified in the TOP10 E.coli (Invitrogen). Plasmid DNAs were sequenced at the University of Chicago Sequencing Center. Mutations were analyzed using the Sequencher 4.1. To investigate SHM, a 1.1 kb DNA fragment starting at the first nucleotide of the JC intron was analyzed. For mutation analysis in the Sμ region, two primers (5’Sμ/3’Sμ: TTA GAT AAA ATG GAT ACC TCA GTG G/CTC ATT CCA GTT CAT TAC AGT CTA C) were used to amplify an ~500 bp DNA fragment in the 5’ end of the Sμ region.
2.6. RT-PCR
RT was carried out according to the instructions of SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen). A semi-quantative PCR was carried out to determine relative levels of AID mRNA in different samples. AID cDNA was amplified in the presence of primers mAID126/mAID649 (5’-CTT TAC CAT TTC AAA AAT GTC CGC and 5’-ACT TCG TAC AAG GGC AAA AGG) (this pair of primers was also used for detecting the AID transgene). β-actin cDNA was amplified using the primers (5’-GTG GGC CGC TCT AGG CAC CAA and 5’-CTCTTT GAT GTC ACG CAC GAT TTC). The PCR conditions for AID were 94°C 90s; 94°C 30s, 57°C 30s, 72°C 50s, 32X or 33X; 72°C 370s; those for β-actin were 94°C 90s; 94°C 30s, 56°C 30s, 72°C 50s, 29X or 30X; 72°C 370s.
2.7. Switch recombination
All mice were used at 8 to 10 weeks of age. Purified splenic B cells were obtained and stimulated as described previously (Shanmugam et al., 2000). Semi-quantitative (q) RT-PCR was carried out as described (Wuerffel et al., 2001). Primers for GLTs (μγ3γ1), AID, Gapdh and postswitch transcripts (for μ → γ3 and μ → γ1 switching) were described (Ma et al., 2002; Muramatsu et al., 2000). FITC labeled rat anti-mouse IgG3 and rat anti-mouse IgG1 (BD Pharmingen, catalogue numbers 553403 and 553443) were used according to the manufacturer’s instructions. Cells were stained with propidium iodide and dead cells were excluded in the FACS analysis.
3. Results
3.1. AID transgenic mRNA
Our AID transgenic mice are similar to those reported by others, including relatively normal B cell development and predisposition to T cell lymphomas (Muto et al., 2006; Okazaki et al., 2003) (not shown). In two independent transgenic lines we studied, AID mRNA was found in all tissues tested. AID wildtype mice immunized with sheep red blood cells produced AID only in germinal center, PNA-hi B cells, but not in PNA-lo B cells (Fig.1B). However, in AID-transgenic mice AID mRNA was present at about 7-fold higher amounts in PNA-lo B cells compared with PNA-hi B cells. This is shown in Fig. 1 for line #14, relative to β-actin mRNA. In line #18 as well, PNA-lo B cells produced more AID mRNA than PNA-hi B cells (not shown). Surprisingly, kidney produced even higher levels of AID mRNA (10 fold higher than PNA-hi B cells) (Fig.1). It appears thus that the half-life of AID mRNA is controlled in mature B cells which normally are the only cells that produce AID, but is not controlled in other cell types.
Fig. 1.
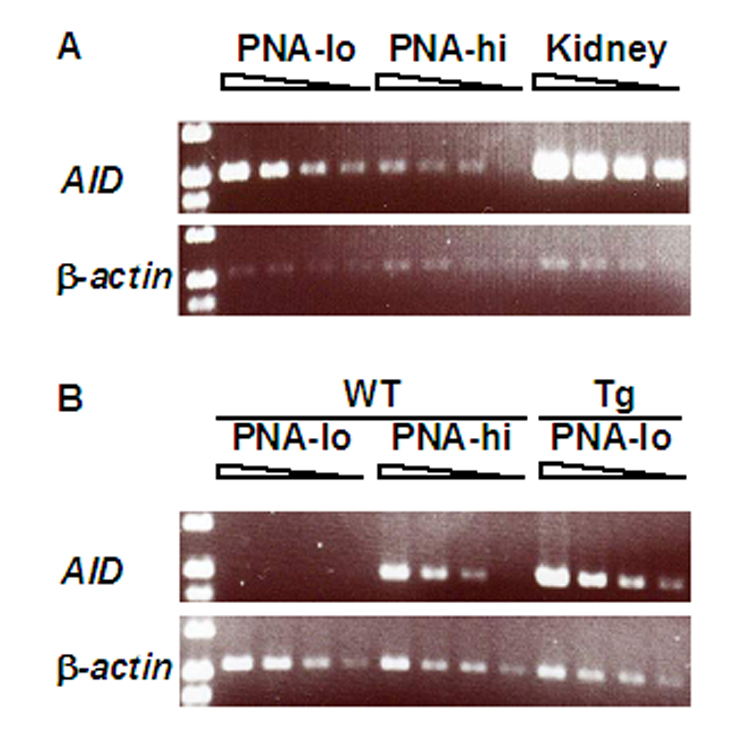
AID mRNA levels are higher in PNA-lo B cells and kidney than in PNA-hi B cells of AID-transgenic mice. A, PNA-lo and PNA-hi B cells and kidney from AID-transgenic mice of line #14. B, PNA-lo and PNA-hi B cells from wildtype mice and PNA-lo B cells from AID-transgenic mice of line #14. The total cycles of PCR for β-actin were 29 and for AID were 32. There was no PCR product in the samples without RT (no DNA contamination) (data not shown).
3.2. Ectopic expression of AID does not cause somatic hypermutation in naïve B cells
AID-transgenic PNA-lo or CD43- B cells express AID mRNA (Fig.1) and protein (Fig. 2) at a high level and if AID was sufficient for SHM, the IgH gene in these cells should be mutated. However, AID-transgenic PNA-lo B cells showed essentially no mutations (Table 1 A). On the other hand, PNA-hi B cells of these mice had normal levels of mutations (Table 1A) compared with non-transgenic mice (Longerich et al., 2005; Shen et al., 2006).
Fig. 2.
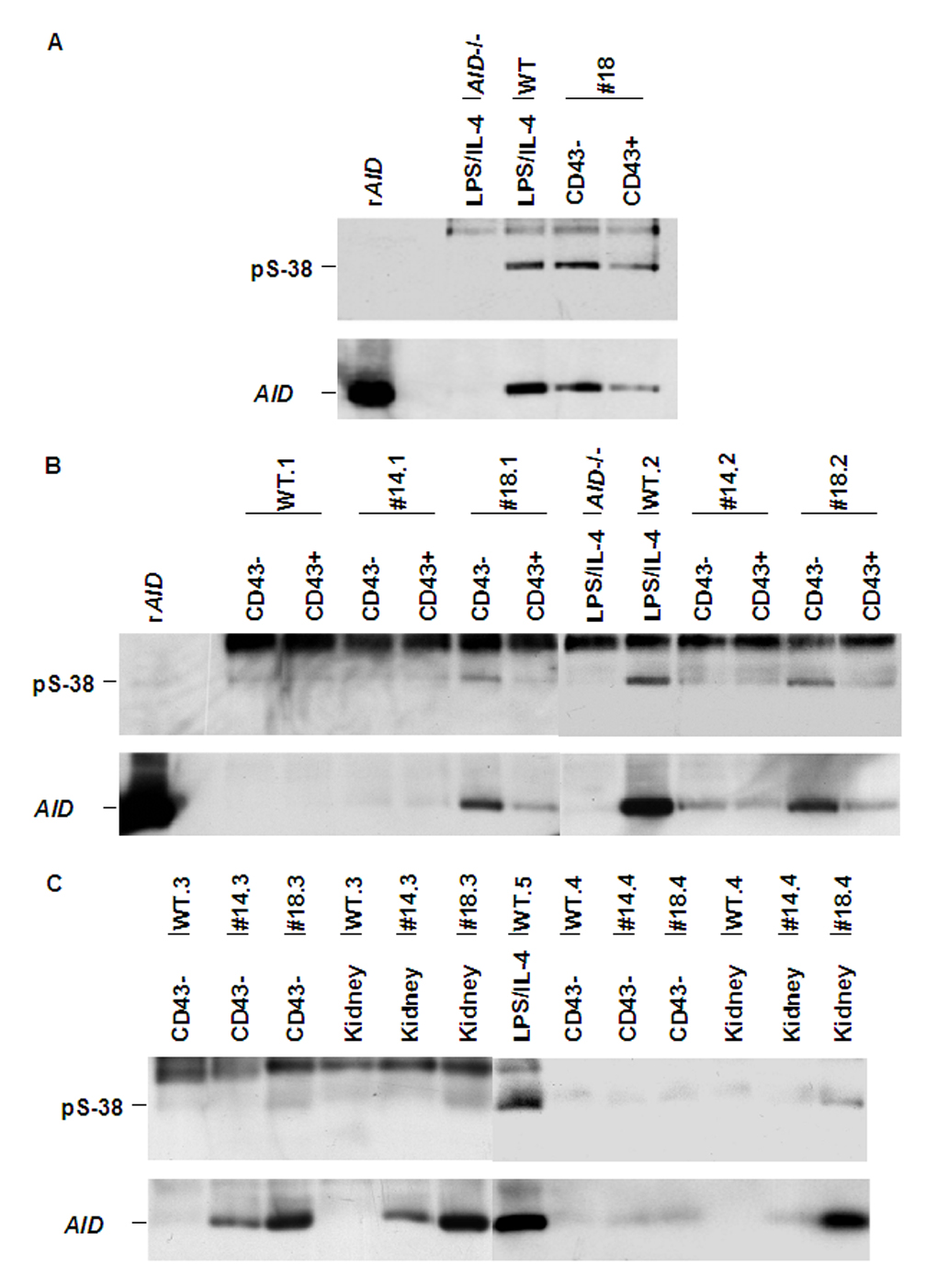
Phosphorylated serine 38 of AID in AID-transgenic mice. AID was immuno-precipitated from cell lysates with anti-AID antibodies and the immunoprecipitates were revealed in Western blots with either anti-pS38 (top) or anti-AID (bottom). The positions of AID and S38-phosphorylated AID are indicated. The band at the top of the blots probed for pS-38 is nonspecific (it also appears in AID −/− cells).
A, rAID, recombinant AID produced in E. coli; the four other lanes contain IPs from 50 million cells: AID−/− and wt, CD43- B cells were stimulated in culture with LPS and IL-4 for 3 days; #18, CD43− or CD43+ cells from AID transgenic mice line #18 (the cells were not activated in vitro).
B and C, Immuno-precipitation and Western blots as in A. Five wt (.1 to .5) and four different AID transgenic mice each of lines #14 and #18 (.1 to .4) are indicated. The AID−/− and wt.2 CD43- B cells were stimulated for 3 days with LPS and IL4.
B, Left six lanes: 40 million sorted, CD43− or CD43+ cells/lane. Right six lanes: 60 million cells per lane.
C, Left six lanes: 40 million CD43− cells or kidney. Right seven lanes: 20 million cells pre lane.
| Table 1A. Ectopic expression of AID does not target the IgH gene in naïve B cells | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lines | Micea | Ages | Titers | PNA | Total clones | Total bases | Mutant clones | Mutant bases | Mutations in hotspots | Mutation frequency | |
| 1 | 14 | 7396 | 5 months | 1:1024 | low | 20 | 22000 | 0 | 0 | 0 | |
| 2 | high | 50 | 55550 | 25 | 94 | 24 (26)c | 16.9×10−4 | ||||
| 3 | C892b | 12 months | No response | low | 36 | 39600 | 3 | 6 | 1.5×10−4 | ||
| 4 | high | 22 | 24200 | 11 | 73 | 18 (25) | 30.2×10−4 | ||||
| 5 | 18 | 1 | 3 months | Not done | low | 20 | 22000 | 0 | 0 | 0 | |
| 6 | high | 20 | 22000 | 13 | 67 | 15 (22) | 30.5×10−4 | ||||
| Table 1B. Overexpression of AID can restore SHM with a low mutation frequency | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lines | Micea | Ages | Titers | PNA | Total clones | Total bases | Mutant clones | Mutant bases | Mutations in hotspots | Mutation frequency | |
| 1 | 14 | T078 | 4.5months | 1:512 | low | 28 | 31350 | 1 | 2 | 0.6×10−4 | |
| 2 | high | 69 | 75900 | 29 | 58 | 21 (36)c | 7.6×10−4 | ||||
| 3 | 92 | 7.5months | 1:512 | high | 15 | 16500 | 2 | 6 | 0 (0) | 3.6×10−4 | |
| 4 | 908 | 9 months | 1:1024 | high | 24 | 26400 | 12 | 27 | 5(19) | 10×10−4 | |
| 5 | 18 | T040 | 2 months | 1:256 | low | 23 | 25300 | 0 | 0 | 0 | |
| 6 | high | 13 | 14850 | 0 | 0 | 0 | |||||
| 7 | T017b | 9 months | 1:512 | low | 32 | 35200 | 2 | 2 | 0.6×10−4 | ||
| 8 | high | 31 | 34100 | 5 | 13 | 2(15) | 3.8×10−4 | ||||
| 9 | 68 | 11 months | 1:512 | high | 28 | 30800 | 5 | 16 | 7(43) | 5.2×10−4 | |
| 10 | 72 | 11 months | 1:1024 | high | 28 | 30800 | 8 | 16 | 3(19) | 5.2×10−4 | |
| Table 1C. Mutations in the switch region of activated B cells | |||||||
|---|---|---|---|---|---|---|---|
| Mouse | IL-4, LPS | Clones | Bases | Mutant clones | Mutant bases | Mut. freq. (clones) | Mut. freq. (bases) |
| AID transgenic, AID−/− | no | 39 | 24297 | 2 | 2 | 0.05 | 0.8×10−4 |
| yesa | 37 | 23051 | 6 | 8 | 0.16 | 3.5×10−4 | |
| Table 1D. Transgenic AID does not equalize mutation frequencies in different Igκ transgenes | ||||||||
|---|---|---|---|---|---|---|---|---|
| Mousea | Age | Titer | PNA | Clones | Bases | Mutant clones | Mutant bases | Mutation frequency |
| RS | 16 months | low | 32 | 20992 | 0 | 0 | 0 | |
| high | 47 | 30832 | 15 | 232 | 75.2×10−4 | |||
| RSA | 15 months | low | 32 | 20992 | 1 | 2 | 1.0×10−4 | |
| high | 47 | 30832 | 11 | 44 | 14.2×10−4 | |||
Mice were AID transgenic, AID+/+.
The mouse developed a T cell lymphoma with enlarged spleen and completely down-regulated CD3 marker and did not respond to SRBC immunization.
() Percentage of total mutations at C and G in AID hotspots (WRC, GYW); 10% of C and G are in hotspots in the 1,100 bp JC intron DNA fragment.
Mice were AID transgenic, AID−/−.
The mouse developed a T cell lymphoma with enlarged spleen, but it responded to SRBC immunization.
See table IA.
CD43- B cells were activated in vitro with LPS and IL-4 for 5 days. The 5’ Sμ region was sequenced in cells without and with activation.
Line 18 AID-TG, AID+/+ mice were immunized with SRBC. 656 base pairs in the center of the VJ region of the RS or RSA transgenes, respectively, were sequenced in PNAhigh or PNAlow B cells.
To test the possibility that the transgenic AID protein did not function properly, we generated AID-transgenic, AID−/− mice by breedings with AID−/− mice. SHM of IgH genes was readily seen in activated B cells in both lines #14 and #18 (Table 1B). Thus, the transgenic AID is functional and can sustain SHM. As in the AID-transgenic mice with endogenous wildtype AID, also in the absence of endogenous AID few mutations were observed in PNA-lo B cells. The few mutations found in cells sorted by FACS to be PNA-lo may have come from B cells that originally were PNA-hi and had incorporated the PNA-lectin during the experiment. Thus, despite strong expression, the AID transgene does not override the inability of PNA-lo B cells to undergo SHM.
The mutation frequencies in the absence of endogenous AID were lower than those in the AID-transgenic mice with endogenous AID (Table 1A), similar to the findings by others (Muto et al., 2006). The mutation pattern however was not changed in the AID-transgenic, AID−/− mice compared with wild type mice (Longerich et al., 2005; Rada et al., 2002; Shen et al., 2006) (Fig. 3). The usual A>T bias was observed in the AID−/− mice with the AID-transgene (Fig. 3). As in wild type mice (Longerich et al., 2005; Shen et al., 2006), most mutations accumulated in the 5’ region of the JCH intron, while in the 3’ end of the sequenced 1.1 kb region, mutations were scarce (Fig. 4). This suggests that the decline of mutation frequencies always observed beyond about 2 kb from the promoter is not due to lack of AID. Furthermore, the WRC/GYW hotspot motif was preferentially mutated (Table 1A and B): on average, the proportion of hotspot-mutations at C and G were 24% in the AID-wt background and 22% in the AID−/− background, while the proportion of hotspot C and G was only 10% in the sequenced region. Thus, ectopically expressed AID recapitulates the mutation preferences of native AID.
Fig. 3.

Normal mutation pattern in IgH gene of AID-transgenic mice. The mutations were collected from PNA-hi B cells of AID-transgenic/AID +/+ (endogenous) (top) and AID-transgenic/AID−/− (bottom) mice (see Table 1 A and B). The adjusted percentage of mutations in A, C, G, and T, respectively was based on the nucleotide composition in the 1.1 kb sequenced DNA fragment. No identical sequences in the same PCR reaction were observed in this study.
Fig. 4.
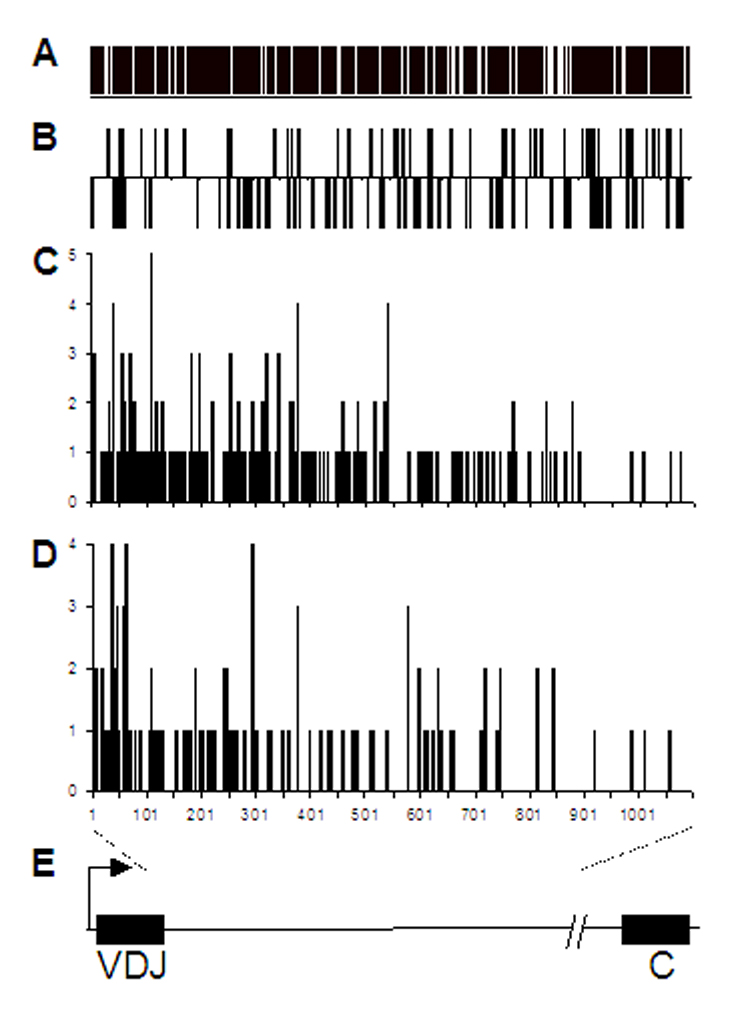
Gradual decline of mutations 3’ of VDJH also in AID-transgenic mice. A, Occurrence of cytidine in the 1.1 kb region at the start of the Ig heavy chain JC intron. The bars are Cs on both DNA strands. B, Positions of WRC hotspots in both DNA strands. C, Mutation pattern in the AID-trangenic, AID+/+ mice. D, Mutation pattern in the AID-transgenic, AID−/− mice. E, The Ig heavy chain gene (not to scale). Bent arrow, transcription initiation site; variable region (VDJ), JC intron (the line between VDJ and C), and constant region (C) are indicated. The sequenced region extends from 1 to 1,100 bp of the JC intron.
3.3. Serine-38 of the AID transgene is phosphorylated in naïve B cells
AID phosphorylation at Serine-38 enhances SHM (Basu et al., 2005; McBride et al., 2006). To determine whether the lack of SHM in AID-transgenic B cells was due to altered phosphorylation we analyzed AID immunoprecipitates by Western blotting with anti-phospho-Ser38 AID antibodies. We found that AID protein was phosphorylated in CD43− (naïve) B cells in AID-transgenic mice and the percentage of phosphorylated AID in these cells was at least as high as that in wild-type B cells that had been activated with LPS and IL-4 (Fig. 2A). AID was phosphorylated at Ser-38 in CD43− B cells of both transgenic lines, #14 and #18 (Fig. 2B). Thus, phosphorylation of AID is not sufficient for SHM in naïve B cells. Furthermore, AID protein in kidney was also S38 phosphorylated, especially in line #18 (Fig.2C).
3.4. PCNA protein is elevated in PNA-hi B cells
One possibility for the lack of mutations in PNA-lo transgenic B cells was that error-prone repair was not recruited. Since mono-ubiquitinated PCNA appears to be required for loading error-prone DNA polymerases on DNA, we compared PCNA protein in PNA-lo and PNA-hi B cells (Fig. 5). Overall PCNA levels, as well as mono-ubiquitinated PCNA were several-fold higher in PNA-hi than PNA-lo B cells of wild type and AID-transgenic mice, in agreement with their likely recruitment of lesion-bypass polymerases during DNA replication and SHM. PNA-lo B cells, that are not replicating their DNA and presumably employ PCNA for DNA repair, did not have detectable mono-ubiquitinated PCNA.
Fig. 5.
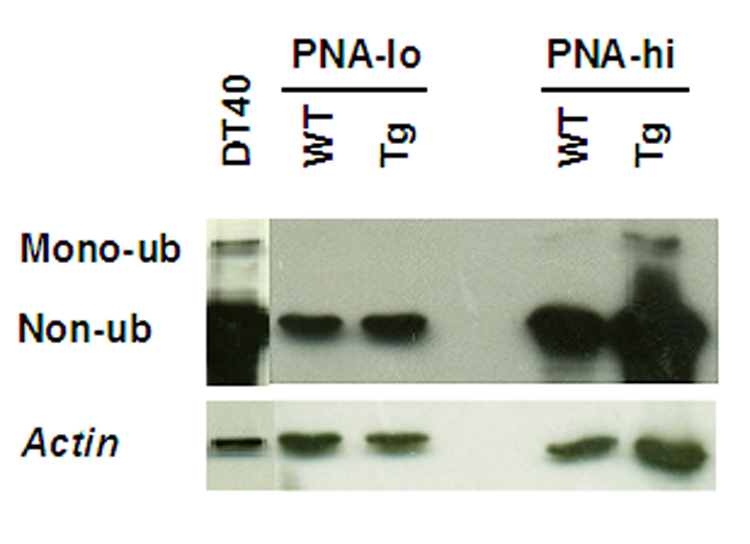
PCNA in B cells. Western blot of PNA-lo and PNA-hi B cells from wildtype and AID-transgenic mice. Top, anti-PCNA; bottom, anti-actin. DT40, chicken B cell line as a positive control; in this cell mono-ubiquitination has been shown essential for efficient SHM (Arakawa et al., 2006).
3.5. The differential mutability of two SHM test genes is not equalized in AID transgenic mice
It is still a mystery how AID gets targeted to Ig genes, but not most other genes expressed in germinal center B cells reviewed in (Longerich et al., 2006). We found previously that an Ig-κ SHM target transgene, RS, was mutated at least four times more than several other related transgenes (Michael et al., 2002; Michael et al., 2003). The transgenes with lower mutability included the RSA variant of the RS transgene in which two CAGGTG sites within the V region were altered to AAGGTG (Michael et al., 2003). We had concluded that AID was more efficiently attracted to the RS transgene due to greater binding of postulated SHM enhancing factors to CAGGTG cis-elements enriched in the RS transgene (Michael et al., 2003). Since AID-transgenic mice of the #18 line expressed more AID protein in CD43+ B cells (Fig. 2B) than control wild type mice, it was possible that a requirement for special targeting of AID to Ig genes was dispensed with in the AID-transgenic mice. The RS or RSA transgenes were therefore combined with the AID transgenes by crossing the respective mice. The mice were immunized with SRBC and PNA-hi and PNA-lo B cells were isolated from spleen and tested for SHM in the κ-transgenes. Table 1 D shows the same differential mutability in the RS versus RSA transgenes in PNA-hi B cells as was observed with wild type AID (Michael et al., 2003). RS-transgenic, AID-transgenic mice mutated the RS transgene at a frequency of 75×10−4 bp, 5.4 times more than the 14×10−4 frequency in the RSA mice. As observed for the endogenous heavy chain genes, also the RS and RSA transgenes were not significantly mutated in PNA-lo B cells of AID-transgenic mice (Table 1D). Thus, the differential mutability of the RS and RSA transgenes is unlikely due to limiting AID levels and is therefore likely caused by reduced access of AID to the RSA transgene.
3.6. Switch recombination
To examine the capacity of the AID-transgene to support CSR we activated splenic B cells from WT and AID-transgenic, AID−/− (line # 14) mice with LPS and LPS+IL4. Germline transcripts (GLTs) of μ and γ3 and AID transcripts were assessed by semi-quantitative RT-PCR and compared to the GAPDH loading control following 48 hours of stimulation (Fig. 6A). The μ and γ3 GLTs are expressed following LPS activation and are not affected by IL4 treatment whereas γ1 and ε GLT expression is IL4 inducible in both WT and AID-transgenic, AID−/− B cells. It should be noted that although IL4 does not repress the γ3 GLT in LPS activated B cells, it does suppress μ–>γ3 CSR. Using an RT-PCR assay specific for the endogenous AID gene, we found that AID transcription was evident in the WT but not AID-transgenic, AID−/− B cells following LPS and LPS+IL4 activation, whereas AID expression was detected in WT and AID-transgenic, AID−/− B cells using an RT-PCR assay that will detect both endogenous and transgenic AID. The post-switch transcript (PST) expression profile, taken after 5 days of activation, confirms successful CSR in the B cell cultures, whereas no PSTs were found for AID−/− B cells similarly activated (Fig. 6B). A representative FACS analysis of B cells stimulated with LPS and LPS+IL4 for 5 days, confirms the PST findings and indicates that these stimulation conditions specifically induce μ–>γ3 and μ–>γ1 CSR, respectively (Fig.7). Notably, CSR from AID-transgenic, AID−/− B cells was reproducibly, about 70% that of WT for both induced IgG3 and IgG1 expression. This level of CSR in the AID-transgenic, AID−/− B cells is higher than that found for a previously described AID-transgenic, AID−/− mouse (Muto et al., 2006).
Fig. 6.
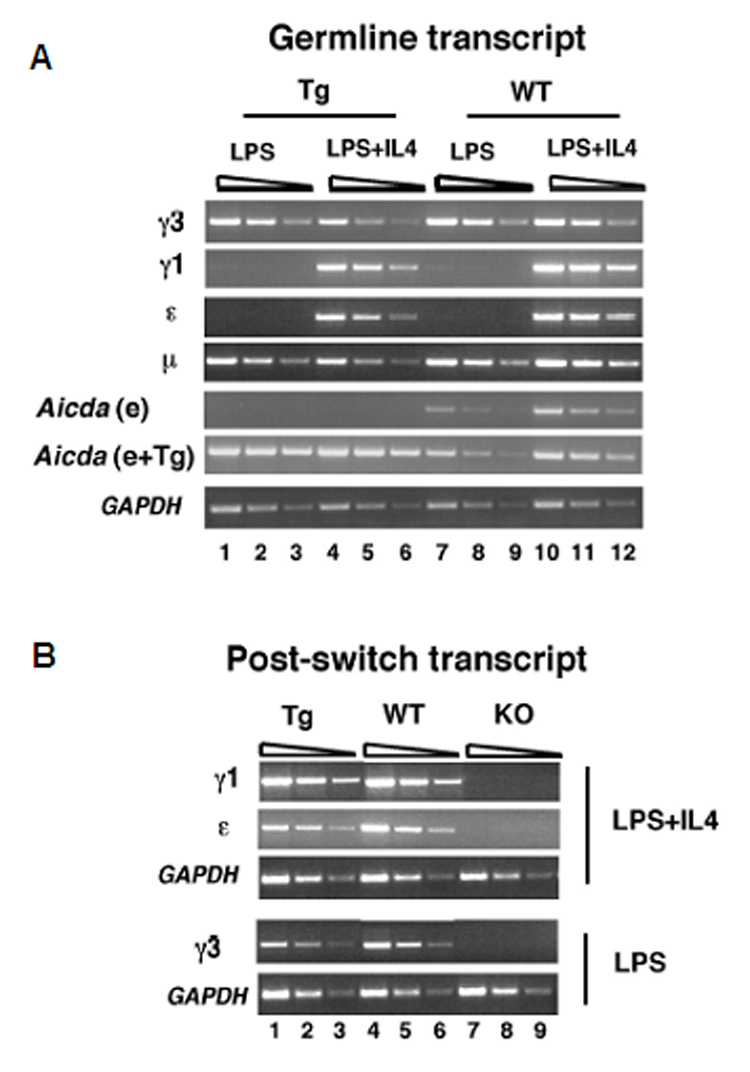
CSR occurs in AID-transgenic/AID−/− mice. A, Germline transcripts (GLTs) were analyzed by semi-quantitative RT-PCR using cDNAs derived from splenic B cells from AID-transgenic/AID−/− and WT mice that were activated with LPS or LPS +IL4 for 48 hours. Gapdh RT-PCR products were harvested after 29 cycles (lanes 1, 4, 7,10), 27 cycles (lanes 2, 5, 8, 11), 25 cycles (lanes 3, 6, 9, 12). GLT and AID RT-PCR products were harvested after 33 cycles (lanes 1, 4, 7, 10), 31 cycles (lanes 2, 5, 8, 11) and 29 cycles (lanes 3, 6, 9, 12). AID originating from the endogenous (e) alone (Muramatsu et al., 2000) or from a combination of endogenous genes and transgenes (e+Tg) (see Materials and Methods) are shown. B, Post-switch transcripts (PSTs) were analyzed by semi-quantitative RT-PCR using cDNAs derived from splenic B cells from AID-transgenic/AID−/−, AID+/+ and AID−/− mice that were activated with LPS or LPS+IL4 for 5 days. Gapdh PCR products were harvested after 29 cycles (lanes 1, 4, 7), 27 cycles (lanes 2, 5, 8) and 25 cycles (lanes 3, 6, 9). PST PCR products were harvested after 33 cycles (lanes 1, 4, 7), 31 cycles (lanes 2, 5, 8), and 29 cycles (lanes 3, 6, 9).
Fig. 7.
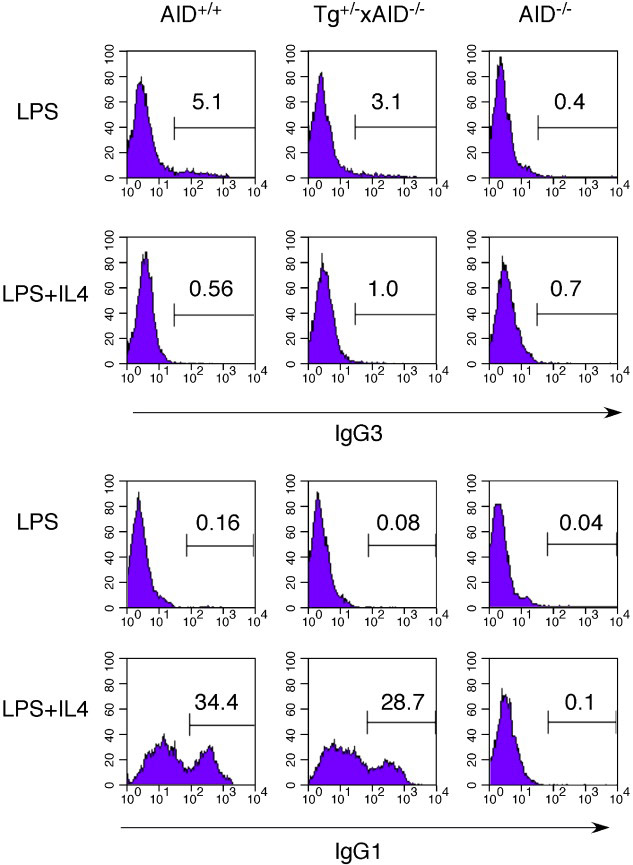
FACS analysis of μ → γ3 and μ → γ1 CSR in AID+/+, AID-transgenic/AID−/−, and AID−/− mice. Splenic B cells were isolated from age-matched mice and activated with LPS or LPS+IL4 for 5 days then analyzed by FACs for CSR. Results are representative of three independent experiments. Flow cytometric analyses of IgG3- (top) or IgG1- (bottom) expressing B cells and the percentages of positive cells are indicated in each diagram.
To investigate whether ectopically expressed AID allows mutations in the Ig heavy chain switch regions, CD43− cells of AID-transgenic, AID−/− mice were activated with LPS and IL-4 for 5 days. An insignificant level of mutations were seen in the 5’ Sμ region in CD43− naïve B cells, despite the high levels of AID mRNA (Fig. 1) and AID protein (Fig. 2) in these cells. In the activated B cells, the 5’ Sμ region showed about four times more mutations compared with the naïve B cells (Table 1C). However, the mutation frequency was several-fold lower than in wild type B cells (Petersen et al., 2001; Schrader et al., 2003). Thus, both for V region SHM and switch region SHM, the AID transgene does not provide the same level of activity as does the endogenous AID gene.
4. Discussion
4.1. AID mRNA and protein in AID transgenic mice
The AID transgene is expressed as mRNA in all tissues tested (spleen, heart, liver, kidney, B cells; Fig. 1 and not shown). Strikingly, the levels of AID-transgenic mRNA in PNA-lo B cells are several times higher than in PNA-hi B cells. Even larger amounts of AID mRNA are seen in kidney. It is no surprise that transgenic AID is expressed in all cells, since the transgene is under the control of a β-actin promoter. However, it appears that in PNA-hi B cells, the only cells that normally express AID, the mRNA level may be regulated, resulting in the similar levels in AID-transgenic and wild type mice (Fig.1). Given the high amounts of AID mRNA in the PNA-lo B cells and kidney, one may assume that the transcription rate is equally high in PNA-hi B cells. Perhaps there exists a post-transcriptional regulation of the mRNA levels, e.g. by an miRNA process (Kloosterman and Plasterk, 2006), that is specific for PNA-hi (mutating) B cells.
All cells from AID-transgenic mice that we tested (spleen, kidney, B cells; Fig. 2 and not shown) also produced AID protein. Concordant with their higher mRNA levels (Fig.1), resting B cells (CD43−) of AID-transgenic mice contain almost as much AID protein as LPS+IL-4 activated B cells from wild type mice (Fig.2). The AID protein is phosphorylated at serine-38 in CD43− B cells (Fig. 2). The proportion of AID that is S38 phosphorylated can be higher in resting, CD43− B cells of AID-transgenic mice than in activated B cells of wild type mice (Fig. 2A). Serine-38 had been reported to increase the efficiency of AID in cytosine deamination (Basu et al., 2005; McBride et al., 2006; Pasqualucci et al., 2006).
AID expressed in 293 cells (embryonic kidney) was found not highly phosphorylated and inactive (Basu et al., 2005). However, when phosphorylated at serine-38 by protein kinase A, AID could deaminate cytosine in a cell-free, transcription-dependent assay and induce CSR when transfected into B cells (Basu et al., 2005; Pasqualucci et al., 2006). Others have found that transfected AID is phosphorylated in cells other than lymphocytes (McBride et al., 2006) and that transfected AID is active for SHM and/or CSR in fibroblasts (Martin and Scharff, 2002; McBride et al., 2006; Okazaki et al., 2002; Yoshikawa et al., 2002) and SHM in B cell lymphomas, hybridomas, and Chinese hamster cells (Martin and Scharff, 2002). Our data support the notion that S38 phosphorylation of AID is not a specialty of activated B cells. Our data also show that, unlike AID in a cell-free assay and in transfected cells, S38-phosphorylated transgenic AID does not induce SHM in resting B cells in vivo.
4.2. Somatic hypermutation and class switch recombination in AID-transgenic mice
The transgenic AID protein is functional as shown by the occurrence of somatic hypermutation (SHM) and class switch recombination (CSR) in mice that are null for endogenous AID (Fig. 3, Fig. 6, Fig. 7; Table 1). However, the frequencies of both SHM and CSR were reduced in AID-transgenic mice on an endogenous AID−/− background compared with the frequencies in a wildtype AID background, as was also found by others (Muto et al., 2006). SHM mutation frequencies were maximally 10×10−4 in older transgenic mice of the #14 line and 5.2×10−4 in the #18 line. In contrast, in the wildtype AID background, the SHM frequencies were up to three to six times higher, as high as 30×10−4 in both lines #14 and #18. Is this relative inefficiency of the transgenic AID gene due to decreased AID activity or diminished recruitment of error-prone DNA repair? The patterns of point mutations were essentially the same in AID-transgenic mice with an AID−/− backgound and with an AID+/+ background with respect to the proportion of mutations at A and T as well as transversions at C and G (Fig. 3). These patterns change when lesion bypass polymerases are diminished or when the uracil glycosylase Ung or both Ung and DNA mismatch repair are inactive (reviewed in (Longerich et al., 2006; Shen et al., 2006)). This suggests that post-AID functions are intact in the AID-transgenic mice, such as the recruitment of error-prone repair in SHM. Also, hallmarks of AID access to Ig genes are the same in the AID-transgenic mice on an endogenous AID−/− background compared with wildtype: AID hotspots are preferentially targeted (Table 1 A and B) and the track of SHM decreases at the same rate as in wild type mice toward the 3’ end of the mutated region of the heavy chain gene (Fig. 4) (Longerich et al., 2005; Shen et al., 2006). Furthermore, two Ig-κ̣ transgenes that differ in SHM susceptibility relative to the numbers of CAGGTG enhancers of SHM (Michael et al., 2003) showed the same differential mutability as in AID wild type mice (Table 1 D). Thus, it appears that the AID transgene may be less efficient in inducing SHM than the endogenous AID gene, but that the recruitment of error-prone repair mechanisms may be intact. Lower efficiency of the AID transgene is also supported by the diminished CSR activity since CSR depends little if at all on error-prone mechanisms which are recruited in the case of SHM (Storb and Stavnezer, 2002).
Why would transgenic AID induce SHM less efficiently than endogenous AID? We can only speculate but the findings may hint at some interesting control of endogenous AID expression. First, over-expression of AID may lead to a suppressive effect. Second, endogenous AID may be modified in as yet unknown ways during the cell cycle critical for a high level of mutations, but, based on its transcription from a non-AID promoter and posttranscriptional events, the ectopic AID may have a different cell cycle regulation and thus be less able to reach correct levels (or modifications) in a particular phase so that the protein cannot access DNA as frequently as in the physiological condition.
4.3. Why do naïve B cells not undergo SHM despite high levels of transgenic AID?
Naive B cells express transgenic AID that is phosphorylated at serine-38. Nevertheless, no significant mutations were observed in PNA-lo B cells of AID-transgenic mice whose PNA-hi B cells readily underwent SHM. Thus, Ser38 phosphorylated AID is not sufficient for SHM in vivo, while it causes cytosine deamination in a cell free system or in transfected cells (Basu et al., 2005; Pasqualucci et al., 2006). Possibly, error-prone repair mechanisms are not recruited in resting B cells. Mono-ubiquitinated PCNA appears to be required for targeting lesion-bypass polymerases (Arakawa et al., 2006; Lehmann, 2006) and PNA-lo B cells produced several-fold lower levels of PCNA and considerably less mono-ubiquitinated PCNA than activated B cells (Fig.5), perhaps suggesting lack of error-prone repair of AID-created uracils. However, if AID had acted in PNA-lo B cells in the absence of error-prone repair, AID activity should have resulted in C to T transitions (Longerich et al., 2005; Rada et al., 2002). Such AID footprints were not observed. It appears therefore that the large amounts of AID in AID-transgenic PNA-lo B cells are simply not targeted to the expressed Ig genes. It is not very likely that uracils are efficiently eliminated in PNA-lo B cells by error-free repair since Ung and Smug uracil glycosylases are very low or undetectable in these cells.
Since resting B cells show no mutations, apparently their precursors, preB cells, also do not mutate. PreB cells may lack unknown activators of SHM or express inhibitors. It is possible that activators can be induced, since bone marrow preB cells infected with the Abelson murine leukemia virus undergo SHM (Gourzi et al., 2006).
PreB cells share certain properties with PNA-hi B cells: cell replication is ongoing (Hoffmann and Melchers, 2003) and E47 protein is high (Quong et al., 2002) in both cell types. While both replication and E47 may be required for SHM, they are obviously not sufficient. PreB cells also express several factors (Hoffmann et al., 2002) that are present in replicating PNA-hi cells and known to be important for SHM: e.g. lesion bypass polymerases (Zeng et al., 2001), Ung (Di Noia et al., 2006; Rada et al., 2002) and Msh2 (Marra et al., 1996).
However, the frequency and number of cell cycles are different in preB cells and PNA-hi B cells. PreB cells undergo about 4 to 5 cell divisions with normal cell cycle times of about one per day before exiting the cell cycle (Hoffmann and Melchers, 2003). PNA-hi B cells participate in germinal center reactions that persist for about 2 to 3 weeks (Schwickert et al., 2007) and antigen-activated B cells migrate back and forth between the dark and light zones of the germinal center (Allen et al., 2007; Camacho et al., 1998; Schwickert et al., 2007). The cell cycle time for germinal center B cells has been estimated to be as short as 6 hrs (MacLennan, 1994), but may be 12 hrs or more (Allen et al., 2007) (and mutating DT40 cells cycle about every 8 hrs; S. Longerich and U.S., unpublished). Thus, potentially, PNA-hi B cells undergo about 28 to 56 cycles before exiting the germinal center. Possibly, collisions between replication and transcription complexes on Ig genes in mutating B cells may contribute to SHM (Shen, 2007). It remains a major challenge to investigate whether there are co-factors for AID in mutating B cells and/or potential inhibitors in non-mutating B cells, and whether DNA replication plays a role in the somatic hypermutation process.
Acknowledgements
We are grateful to Tasuku Honjo for the AID transgene. We thank Serhiy Pylawka for excellent experimental assistance, Ryan Duggan and James Marvin for help with flow cytometric cell sorting, William Buikema and Chris Hall for DNA sequencing, MARCUS PETER FOR ADVICE WITH PCNA UBIQUITINATION, and Michel Nussenzweig and Terry Martin for critical reading of the paper.
We gratefully acknowledge support by the University of Chicago Cancer Research Center for the use of the DNA Sequencing and Flow Cytometry Facilities.
This work was supported by NIH grants AI47380 and AI053130 to U.S. and AI052400 to A.K.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen CD, Okada T, Tang HL, Cyster JG. Imaging of germinal center selection events during affinity maturation. Science. 2007;315:528–531. doi: 10.1126/science.1136736. [DOI] [PubMed] [Google Scholar]
- Arakawa H, Moldovan GL, Saribasak H, Saribasak NN, Jentsch S, Buerstedde JM. A role for PCNA ubiquitination in immunoglobulin hypermutation. PLoS Biol. 2006;4:e366. doi: 10.1371/journal.pbio.0040366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu U, Chaudhuri J, Alpert C, Dutt S, Ranganath S, Li G, Schrum J, Manis J, Alt F. The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature. 2005;438:508–511. doi: 10.1038/nature04255. [DOI] [PubMed] [Google Scholar]
- Camacho SA, Kosco-Vilbois MH, Berek C. The dynamic structure of the germinal center. Immunol Today. 1998;19:511–514. doi: 10.1016/s0167-5699(98)01327-9. [DOI] [PubMed] [Google Scholar]
- Di Noia JM, Rada C, Neuberger MS. SMUG1 is able to excise uracil from immunoglobulin genes: insight into mutation versus repair. Embo J. 2006;25:585–595. doi: 10.1038/sj.emboj.7600939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg E, Walker G, Siede W, Wood R, Schultz R, Ellenberger T. DNA Repair and Mutagenesis. Washington, D.C.: 2006. [Google Scholar]
- Gourzi P, Leonova T, Papavasiliou FN. A role for activation-induced cytidine deaminase in the host response against a transforming retrovirus. Immunity. 2006;24:779–786. doi: 10.1016/j.immuni.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Hoffmann R, Melchers F. A genomic view of lymphocyte development. Curr Opin Immunol. 2003;15:239–245. doi: 10.1016/s0952-7915(03)00047-5. [DOI] [PubMed] [Google Scholar]
- Hoffmann R, Seidl T, Neeb M, Rolink A, Melchers F. Changes in gene expression profiles in developing B cells of murine bone marrow. Genome Res. 2002;12:98–111. doi: 10.1101/gr.201501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman WP, Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11:441–450. doi: 10.1016/j.devcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Lehmann A. Translesion synthesis in mammalian cells. Exp Cell Res. 2006;312:2673–2676. doi: 10.1016/j.yexcr.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Longerich S, Basu U, Alt F, Storb U. AID in somatic hypermutation and class switch recombination. Curr Opin Immunol. 2006;18:164–174. doi: 10.1016/j.coi.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Longerich S, Tanaka A, Bozek G, Storb U. The very 5' end and the constant region of Ig genes are spared from somatic mutation because AID does not access these regions. J. Exp. Med. 2005;202:1443–1454. doi: 10.1084/jem.20051604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Wortis HH, Kenter AL. Two new isotype-specific switching activities detected for Ig class switching. J Immunol. 2002;168:2835–2846. doi: 10.4049/jimmunol.168.6.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLennan IC. Germinal centers. Annu Rev Immunol. 1994;12:117–139. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- Marra G, Chang CL, Laghi LA, Chauhan DP, Young D, Boland CR. Expression of human MutS homolog 2 (hMSH2) protein in resting and proliferating cells. Oncogene. 1996;13:2189–2196. [PubMed] [Google Scholar]
- Martin A, Bardwell PD, Woo CJ, Fan M, Shulman MJ, Scharff MD. Activation-induced cytidine deaminase turns on somatic hypermutation in hybridomas. Nature. 2002;415:802–806. doi: 10.1038/nature714. [DOI] [PubMed] [Google Scholar]
- Martin A, Scharff MD. Somatic hypermutation of the AID transgene in B and non-B cells. Proc Natl Acad Sci U S A. 2002;99:12304–12308. doi: 10.1073/pnas.192442899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride KM, Gazumyan A, Woo EM, Barreto VM, Robbiani DF, Chait BT, Nussenzweig MC. Regulation of hypermutation by activation-induced cytidine deaminase phosphorylation. Proc Natl Acad Sci U S A. 2006;103:8798–8803. doi: 10.1073/pnas.0603272103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael N, Martin TE, Nicolae D, Kim N, Padjen K, Zhan P, Nguyen H, Pinkert C, Storb U. Effects of sequence and structure on the hypermutability of immunoglobulin genes. Immunity. 2002;16:123–134. doi: 10.1016/s1074-7613(02)00261-3. [DOI] [PubMed] [Google Scholar]
- Michael N, Shen H, Longerich S, Kim N, Longacre A, Storb U. The E box motif CAGGTG enhances somatic hypermutation without enhancing transcription. Immunity. 2003;19:235–242. doi: 10.1016/s1074-7613(03)00204-8. [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and somatic hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- Muto T, Okazaki IM, Yamada S, Tanaka Y, Kinoshita K, Muramatsu M, Nagaoka H, Honjo T. Negative regulation of activation-induced cytidine deaminase in B cells. Proc Natl Acad Sci U S A. 2006;103:2752–2757. doi: 10.1073/pnas.0510970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki I, Hiai H, Kakazu N, Yamada S, Muramatsu M, Kinoshita K, Honjo T. Constitutive expression of AID leads to tumorigenesis. J. Exp. Med. 2003;197:1173–1181. doi: 10.1084/jem.20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki I, Kinoshita K, Muramatsu M, Yoshikawa K, Honjo T. The AID enzyme induces class switch recombination in fibroblasts. Nature. 2002;416:340–345. doi: 10.1038/nature727. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L, Kitaura Y, Gu H, Dalla-Favera R. PKA-mediated phosphorylation regulates the function of activation-induced feaminase (AID) in B cells. Proc Natl Acad Sci U S A. 2006;103:395–400. doi: 10.1073/pnas.0509969103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen S, Casellas R, Reina-San-Martin B, Chen H, Difilippantonio M, Wilson P, Hanitsch L, Celeste A, Muramatsu M, Pilch D, Redon C, Ried T, Bonner W, Honjo T, Nussenzweig M, Nussenzweig A. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quong MW, Romanow WJ, Murre C. E protein function in lymphocyte development. Annu Rev Immunol. 2002;20:301–322. doi: 10.1146/annurev.immunol.20.092501.162048. [DOI] [PubMed] [Google Scholar]
- Rada C, Williams G, Nilsen H, Barnes D, Lindahl T, Neuberger M. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr. Biol. 2002;12:1748–1755. doi: 10.1016/s0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- Schrader CE, Bradley SP, Vardo J, Mochegova SN, Flanagan E, Stavnezer J. Mutations occur in the Ig Smu region but rarely in Sgamma regions prior to class switch recombination. Embo J. 2003;22:5893–5903. doi: 10.1093/emboj/cdg550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwickert TA, Lindquist RL, Shakhar G, Livshits G, Skokos D, Kosco-Vilbois MH, Dustin ML, Nussenzweig MC. In vivo imaging of germinal centres reveals a dynamic open structure. Nature. 2007;446:83–87. doi: 10.1038/nature05573. [DOI] [PubMed] [Google Scholar]
- Shanmugam A, Shi MJ, Yauch L, Stavnezer J, Kenter AL. Evidence for class-specific factors in immunoglobulin isotype switching. J Exp Med. 2000;191:1365–1380. doi: 10.1084/jem.191.8.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HM. Activation-induced cytidine deaminase acts on double-strand breaks in vitro. Mol Immunol. 2007;44:974–983. doi: 10.1016/j.molimm.2006.03.015. [DOI] [PubMed] [Google Scholar]
- Shen HM, Tanaka A, Bozek G, Nicolae D, Storb U. Somatic hypermutation and class switch recombination in Msh6(−/−)Ung(−/−) double-knockout mice. J Immunol. 2006;177:5386–5392. doi: 10.4049/jimmunol.177.8.5386. [DOI] [PubMed] [Google Scholar]
- Storb U, Stavnezer J. Immunoglobulin genes: generating diversity with AID and UNG. Curr. Biol. 2002;12:R725–R727. doi: 10.1016/s0960-9822(02)01247-2. [DOI] [PubMed] [Google Scholar]
- Wuerffel RA, Ma L, Kenter AL. NF-kappa B p50-dependent in vivo footprints at Ig S gamma 3 DNA are correlated with mu-->gamma 3 switch recombination. J Immunol. 2001;166:4552–4559. doi: 10.4049/jimmunol.166.7.4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa K, Okazaki IM, Eto T, Kinoshita K, Muramatsu M, Nagaoka H, Honjo T. AID enzyme-induced hypermutation in an actively transcribed gene in fibroblasts. Science. 2002;296:2033–2036. doi: 10.1126/science.1071556. [DOI] [PubMed] [Google Scholar]
- Zeng X, Winter D, Kasmer C, Kraemer K, Lehmann A, Gearhart P. DNA polymerase eta is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nature Immunology. 2001;2:537–541. doi: 10.1038/88740. [DOI] [PubMed] [Google Scholar]