

Abstract
The extracellular matrix (ECM) of the trabecular meshwork (TM) is thought to be important in regulating intraocular pressure (IOP) in both normal and glaucomatous eyes. IOP is regulated primarily by a fluid resistance to aqueous humor outflow. However, neither the exact site nor the identity of the normal resistance to aqueous humor outflow has been established. Whether the site and nature of the increased outflow resistance, which is associated with open-angle glaucoma, is the same or different from the normal resistance is also unclear.
The ECMs of the TM beams, juxtacanalicular region (JCT) and Schlemm’s canal (SC) inner wall are comprised of fibrillar and non-fibrillar collagens, elastin-containing microfibrils, matricellular and structural organizing proteins, glycosaminoglycans (GAGs) and proteoglycans. Both basement membranes and stromal ECM are present in the TM beams and JCT region. Cell adhesion proteins, cell surface ECM receptors and associated binding proteins are also present in the beams, JCT and SC inner wall region.
The outflow pathway ECM is relatively dynamic, undergoing constant turnover and remodeling. Regulated changes in enzymes responsible for ECM degradation and biosynthetic replacement are observed. IOP homeostasis, triggered by pressure changes or mechanical stretching of the TM, appears to involve ECM turnover. Several cytokines, growth factors and drugs, which affect the outflow resistance, change ECM component expression, mRNA alternative splicing, cellular cytoskeletal organization or all of these. Changes in ECM associated with open-angle glaucoma have been identified.
Keywords: Aqueous humor outflow resistance, proteoglycans, extracellular matrix proteins, extracellular matrix turnover
Introduction
The trabecular meshwork (TM) is thought to provide most of the flow resistance to aqueous humor outflow, although the alternative or uveoscleral outflow pathway does contribute to total aqueous humor drainage. (Bill and Maepea 1994; Schachtschabel et al. 2000; Weinreb 2000; Weinreb et al. 2002) Modulation of the trabecular outflow resistance is responsible for most of the regulation of intraocular pressure (IOP). (Brubaker 1970; Brubaker 1991) In open-angle glaucoma, sustained increases in the trabecular outflow resistance commonly result in elevated intraocular pressure (IOP). Elevated IOP is the primary risk factor for glaucomatous optic neuropathy, which is associated with progressive vision loss. Since glaucoma is responsible for around 25% of new blindness in developed nations, understanding the source, nature, and regulation of the normal and the glaucomatous trabecular outflow resistances is of considerable medical importance. (Quigley 1993,1996; Quigley and Vitale 1997)
The outer uveal and corneoscleral portions of the TM are highly fenestrated and composed of several irregular layers of ECM covered by TM beam cells (Fig. 1). The trabecular beams become more flattened and sheet-like in the deeper portions of this region. The beams and sheets are normally covered and maintained by continuous monolayers of TM cells, which reside on a typical basal lamina with a lamina rara and lamina densa. (Bhatt et al. 1995; Lütjen-Drecoll and Rohen 1996; Gong et al. 2002) The center or stroma of the beams exhibits typical collagen fibrils, elastic fibers and microfibril sheath-derived (SD) material. (Lütjen-Drecoll and Rohen 1996; Gong et al. 2002; Morrison and Acott 2003) Discontinuous tight junctional strands, which do not form continuous zonula occludens, and gap junctions are common between TM beam cells. (Grierson and Lee 1974; Raviola and Raviola 1981; Bhatt et al. 1995) Between the beams and sheets, the irregular intertrabecular spaces form torturous flow channels leading to the cribriform or juxtacanalicular (JCT) region, which lies adjacent to Schlemm’s canal. The size of the intratrabecular spaces is sufficiently large that it is hard to imagine them contributing to the outflow resistance. The cells on the outer layers of the TM are actively phagocytic and are thought to act primarily as pre-filters, removing cellular debris from the aqueous humor prior to its passage through the less porous inner JCT and SC regions.
Fig. 1.
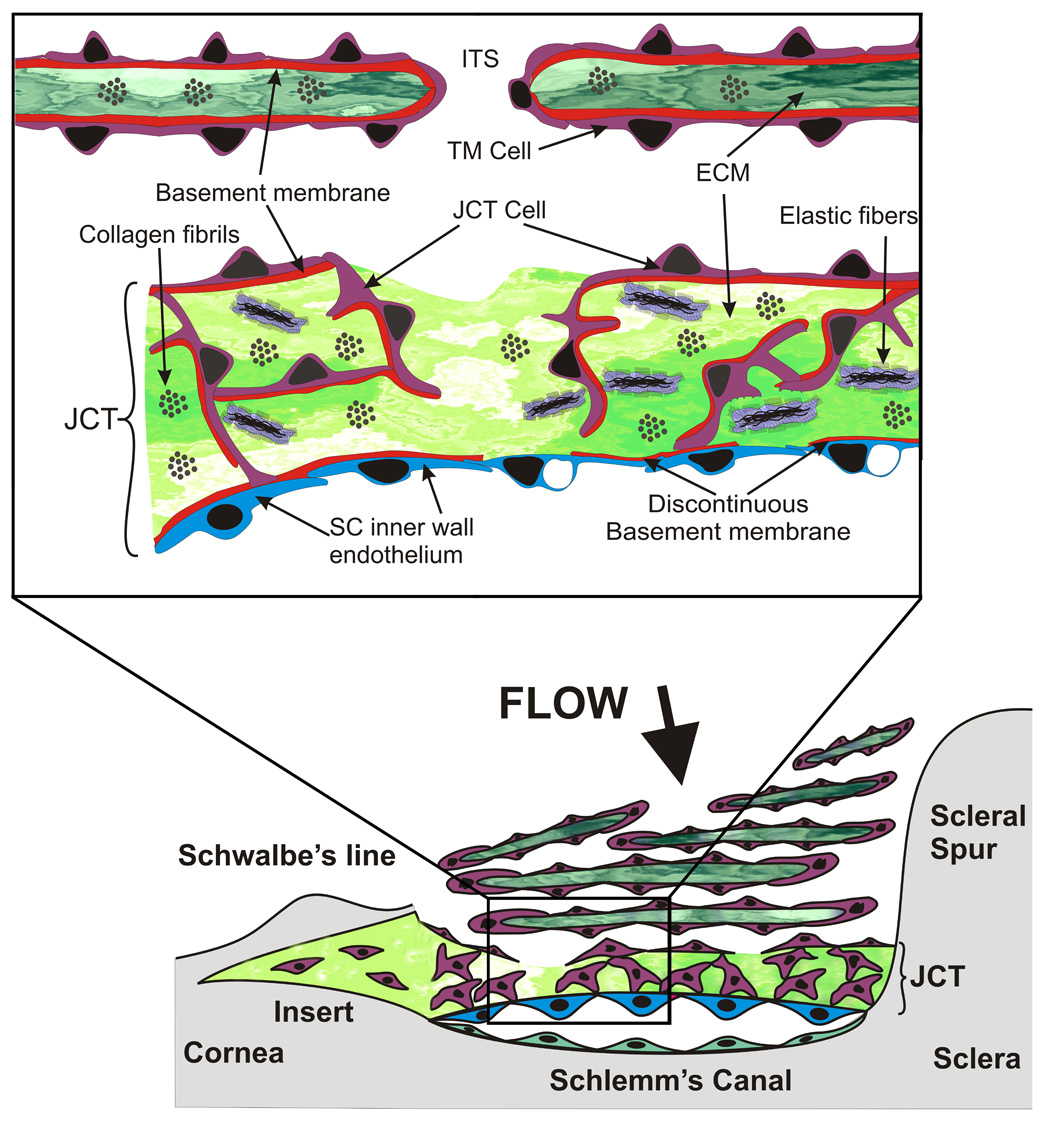
Diagram of the outflow pathway and juxtacanalicular or cribriform region. The lower portion of the figure shows a stylized view of the TM and the upper inset shows an expanded view of the JCT region. The figure is modified from several sources.(Tripathi 1971, 1977; Grierson et al. 1978; Rohen et al. 1981; Rohen 1983; Grierson and Calthorpe 1989; Epstein and Rohen 1991; Maepea and Bill 1992; Lutjen-Drecoll 1999; Parc et al. 2000)
The JCT region is composed of an amorphous ECM with a discontinuous scattering of several layers of cribriform or JCT cells on the trabecular surface and embedded within the ECM (Fig. 1). (Tripathi 1971; Inomata et al. 1972; Tripathi 1977; Grierson et al. 1978; Rohen et al. 1981; Rohen 1983; Grierson and Calthorpe 1989; Epstein and Rohen 1991; Maepea and Bill 1992; Lutjen-Drecoll and Rohen 1994; Lütjen-Drecoll and Rohen 1996; Lutjen-Drecoll 1999; Parc et al. 2000) These JCT cells form occasional contacts with each other, often via extended processes using gap and adherens junctions.(Inomata et al. 1972; Grierson et al. 1978) They also form similar junctions with SC inner wall endothelial cells. (Inomata et al. 1972; Grierson et al. 1978) No continuous occludens junctions are apparent between JCT cells or between JCT and SC cells. SC inner wall cells form a continuous monolayer and exhibit occasional gap junctions and extensive continuous tight occludens junctions. (Inomata et al. 1972; Grierson et al. 1978; Raviola and Raviola 1981; Bhatt et al. 1995) The SC inner wall cells reside on a discontinuous basal lamina that may be affected by the movement of aqueous humor through these cells. (Hogan et al. 1971; Inomata et al. 1972; Grierson et al. 1978; Bhatt et al. 1995; Lutjen-Drecoll 1999; Gong et al. 2002; Fuchshofer et al. 2006) Whether JCT cells have basal lamina appears to be controversial. (Marshall et al. 1990; Tawara and Inomata 1994; Hernandez and Gong 1996; Lütjen-Drecoll and Rohen 1996; Fuchshofer et al. 2006) These cells have been considered to be fibroblast-like and thus without any basal lamina. However, basement membrane proteins, type IV collagen and laminin, and basement membrane proteoglycans have been identified in the JCT region and associated with the JCT cells. (Marshall et al. 1990; Tawara and Inomata 1994; Fuchshofer et al. 2006) One study found basement membrane-like material adjacent to all JCT cells. (Fuchshofer et al. 2006) This varied in amount and location with individual cells and was observed associated with at least half of the cell surface in some cases. It was generally not observed of the face of these cells where they abut the “open spaces” found scattered throughout the JCT region. (Fuchshofer et al. 2006)
Possible sites of the outflow resistance
Sequential serial dissection and microcapillary studies with perfused eyes suggest that the trabecular outflow resistance resides somewhere within the deepest 1/4 to 1/3 of the TM. (Grant 1958; Grant 1963; Ellingsen and Grant 1972; Rosenquist et al. 1989; Maepea and Bill 1992; Bill and Maepea 1994) When a microcapillary was inserted from Schlemm’s canal toward the anterior chamber, the resistance was bypassed at 7–14 µm from the inner wall of Schlemm’s canal. This is approximately the thickness of the juxtacanalicular region (Fig. 1). (Gong et al. 2002; Johnson 2006) This observation appears to eliminate the SC inner wall endothelium as a source of resistance, while positively implicating the remainder of the JCT. However, it has been argued that the microcapillary used in these studies was sufficiently large in bore that it may have deformed the inner wall a portion of this distance prior to breaching it. (Johnson 2006) Thus, the precise location of the resistance has not been definitively established. Most of the resistance does appear to reside within the JCT or SC inner wall endothelium, or both. (Lutjen-Drecoll 1999; Ethier 2002; Johnson 2006)
Segmental flow
One caveat that may add complexity is the segmental flow pattern observed particularly in older human eyes. By all appearances, SC in humans and higher primates is completely open and unobstructed at normal IOP levels. Since there are approximately 25–30 collector channels distributed around a human eye, (Hogan et al. 1971) it seems that fluid within SC should exhibit relatively free circumferential flow. This would allow drainage via adjacent or even other more distant collector channels. However, a number of studies have documented segmental flow differences around the circumference of the eye. (Grant 1956; Grant 1958; Grant 1963; Buller and Johnson 1994; Parc et al. 2000; Ethier and Chan 2001; Gottanka et al. 2001; Hann et al. 2005; Hann and Johnson 2007) TM dissections studies and other observations suggest that fluid may not be free to move around the circumference of SC. (Hogan et al. 1971; Rosenquist et al. 1989; Lutjen-Drecoll 1999; Ethier 2002; Ethier et al. 2004; Johnstone 2004; Johnson 2006) Clinical observations that multiple site trabeculectomy or stent placement improves drainage, further highlight this phenomenon. Evaluation of F-actin cytoskeletal alignments within SC inner and outer wall endothelial cells and shear stress modeling of radial flow in SC, particularly relative to collector channels, suggests that this is more complicated than originally perceived. (Ethier et al. 2004)
Possible sources of outflow resistance
Glycosaminoglycans (GAGs) within the TM have long been a favorite hypothetical source of the outflow resistance. (Bárány 1953; Bárány, E.H. and Scotchbrook, S. 1954; Pedler 1956; Francois 1975) These long repeat disaccharide chains with a high density of carboxyl and sulfate groups, which are negatively charged at physiologic pH, would be the ideal material to form the outflow resistance. (Francois 1975; Rohen and Lütjen-Drecoll 1981; Rohen and Lutjen-Drecoll 1982; Acott 1992, 1994a; Acott and Wirtz 1996; Hernandez and Gong 1996; Yue 1996) Initial perfusion studies with enzymes, which selectively degrade GAGs, were found to reduce the outflow resistance by approximately 50% in bovine eyes. (Bárány 1953; Bárány, E. H. and Scotchbrook, S. 1954; Bárány and Woodin 1954; Pedler 1956) Since then, variations of these experiments using eyes from different species perfused with several different GAG-degrading enzymes and conditions have given somewhat conflicting results. (Peterson and Jocson 1974; Knepper et al. 1984; Sawaguchi et al. 1992; Sawaguchi et al. 1993; Hubbard et al. 1997) In dog (Van Buskirk and Brett 1978) and rabbit (Knepper et al. 1984) eyes, in vivo, hyaluronidase treatment increased outflow facility. In bovine perfused organ culture, chondroitinase ABC increased outflow facility, while hyaluronidase and heparatinase did not have significant effects. (Sawaguchi et al. 1993) In porcine perfused anterior segment organ culture, chondroitinase ABC increased outflow facility significantly and a combination of chondroitinase ABC, heparatinase and hyaluronidase was even more effective. (Keller et al. 2007a) In primates, one study found that intracameral injection of chondroitinase ABC increased outflow (Sawaguchi et al. 1992), one study found that hyaluronidase increased outflow (Peterson and Jocson 1974), and one study found that neither chondroitinase ABC nor hyaluronidase had significant effects on outflow. (Hubbard et al. 1997) In human perfused anterior segment organ culture, heparatinase was shown to increase outflow (Johnson and Bahler 1999) and inhibition of GAG biosynthesis or GAG sulfation increased outflow. (Keller et al. 2007a) Thus, a contribution of GAGs to the outflow resistance seems highly likely, but a direct role in providing the resistance, particularly in humans and higher primates, has not been unequivocally established.
When combined with hydrodynamic modeling, initial morphometric analysis of transmission electron micrographs of the TM, suggested that the visible ECM within the JCT did not contain sufficient electron dense material to account for the outflow resistance. (McEwen 1958; Ethier et al. 1986) It was postulated that some “invisible” GAG-like substance filled the “empty spaces.” GAG chains would clearly not retain their native conformations through the harsh processing steps and are unlikely to be stained sufficiently to be visible in the micrographs. Recent quick-freeze deep-etch and electron microscopy studies show a much more extensive ECM in the JCT. (Gong et al. 2001; Gong et al. 2002) However, the actual visualized material is still insufficient to account for the outflow resistance. Although this method shows dramatically increased amounts of ECM, the highly-charged GAG side chains would still be expected to collapse completely in response to the processing necessary for even this approach. Thus, even this much-improved electron micrographic method highlights the presence of numerous micron-sized “open spaces” within the JCT that can easily account for more fluid conductivity than is observed experimentally. (Gong et al. 2001; Gong et al. 2002; Johnson 2006)
Cells or ECM or both provide the outflow resistance
Studies comparing the outflow facility of in vivo to in vitro human eyes and evaluating the effects of temperature and of metabolic poisons on outflow facility all suggest that aqueous humor is not transported by an active cellular metabolic process. (Bárány 1953; Van Buskirk and Brant 1974; Epstein et al. 1981; Epstein and Anderson 1984; Suzuki and Anderson 1993) A number of agents that primarily act on cytoskeletal organization have been shown to modulate outflow facility. (Kaufman and Erickson 1982; Johnson 1997; Peterson et al. 1997; Tian et al. 1998; Epstein et al. 1999; Sabanay et al. 2000; Rao et al. 2001; Vittitow et al. 2002; Rao et al. 2005) These studies suggest that cells somewhere in the TM or the SC inner wall, or both, are directly involved in forming the outflow resistance. Perfusion of a fibronectin fragment that disrupts integrin-ECM interactions and could affect either ECM or cytoskeleton also increases outflow facility. (Santas et al. 2003) A recent study showing effects of cytoskeletal modifying agents on matrix metalloproteinases (MMPs) further adds to the complexity of assigning direct roles for the cells or ECM. (Sanka et al. 2007) The various GAG-modifying studies mentioned above suggest a direct ECM contribution to the outflow resistance. The initiation of ECM turnover in the TM, mediated by members of the MMP family, also increases outflow facility. (Bradley et al. 1998) Perhaps more importantly, the inhibition of endogenous trabecular MMP activity reduces outflow facility. Thus ongoing ECM turnover is required to maintain the appropriate outflow resistance. (Bradley et al. 1998) The microcapillary observations mentioned earlier also argue for the JCT ECM and against SC inner wall cells. (Maepea and Bill 1992)
However, due to the highly interdependent nature of the bidirectional interactions between cells and their ECMs, an approach which can clearly differentiate between the actual direct contribution of either the cells or the ECM to the aqueous outflow resistance has not yet been devised. Consequently, although both the cells and the ECM are clearly critical to maintaining the outflow resistance, their respective direct contributions remain unclear. Aspects of this uncertainty and a variety of other relevant studies have been discussed in detail. (Johnson and Erickson 2000; Ethier 2002; Fautsch and Johnson 2006; Johnson 2006)
A model reflecting a combined contribution of cells and ECM has considerable appeal, particularly in light of the inconclusive evidence discussed above. (Johnson et al. 1992; Ethier 2002; Overby et al. 2002; Johnson 2006) If flow through the inner wall cells of Schlemm’s canal is constrained to a subpopulation of the inner wall “pores” that are not fixation artifacts, then the idea of “funneling” becomes tenable. With a limited number of real pores serving as the necks of funnels, fluid movement through the ECM of the JCT would be constrained to regions near and upstream from the funnel openings. These portions of the ECM and the SC inner wall cells would together provide the outflow resistance. Unfortunately, although this model helps to resolve the apparent conflicting observations, a clear method to test this combined resistance model has also yet to be devised.
ECM components identified within the TM
A variety of ECM components have been identified within the TM. Most of these are similar to those found in other tissues, although a few apparently unique isoforms have been identified. Only limited information is available about the detailed molecular organization, exact composition and relative abundance of these components. Presumably, these nuances will be very important in defining the unique function(s) and properties of the outflow pathway. Studies with general microscopic stains and electron microscopy identified GAGs, fibrillar collagens, unevenly-spaced or curly collagen, elastic fibrils with associated sheath-derived plaque material, basement membrane and amorphous basement membrane-like materials and other non-specific ECM components. (Hogan et al. 1971; Francois 1975; Grierson and Lee 1975; Lütjen-Drecoll et al. 1981; Richardson 1982; Lütjen-Drecoll et al. 1986; Lütjen-Drecoll and Tamm 1987; Lütjen-Drecoll and Rohen 1996; Lutjen-Drecoll 1999; Gong et al. 2002)
GAGs
The GAG composition in TM extracts was determined by sequential enzymatic degradation with various GAG-degrading enzymes. The sources included TM from a few hours postmortem human eyes, stationary human anterior segment organ cultures, perfused human anterior segment organ cultures and densely-confluent early-passage human TM cell cultures. (Grierson and Lee 1975; Schachtschabel et al. 1977; Knepper et al. 1981; Schachtschabel et al. 1982; Rohen et al. 1984; Acott et al. 1985; Yue and Elvart 1987; Acott et al. 1988; Tschumper et al. 1990; Acott 1994a; Knepper et al. 1996b) Relatively good agreement was found in the amount of each type of GAG in the TM using these models and all of the normal GAGs were identified. Of the total trabecular GAGS, approximately 20–25% is hyaluronic acid or hyaluronan (HA), 40–60% is chondroitin and dermatan sulfates (CS and DS), 5–10% is keratan sulfate (KS) and 15–20% is heparan sulfate (HS). (Francois 1975; Rohen and Lütjen-Drecoll 1981; Rohen and Lutjen-Drecoll 1982; Acott 1992, 1994a; Acott and Wirtz 1996; Yue 1996) These percentages are based on Alcian blue or similar GAG staining or 3H- or 35S-radiolabel incorporation and are not strictly calibrated molar amounts. When normalized to a relative ng amount based on GAG standards, HA was 15 %, CS/DS was 38%, KS was 6.5% and HS was 40% of the total. (Knepper et al. 1996a)
Localizations of the GAGs within the TM have been determined by similar enzymatic degradation methods using various GAG stains and light or transmission electron microscopy (TEM). Hyaluronan binding protein and GAG-specific antibodies have also been used to localize GAGs within the TM. Most studies found HA primarily in the JCT region with less in the remainder of the TM using HA binding protein as a probe (Keller et al, manuscript in review) (Lütjen-Drecoll et al. 1990; Hernandez and Gong 1996; Lerner et al. 1997) or quantitative hyaluronidase removal of Alcian blue staining. (Knepper et al. 1996b) The other GAGs are sulfated, facilitating alternative staining methods. When GAGs were stratified within the JCT of normal human eyes, fg/µm2 values obtained were 7.78 for HA, 8.18 for CS, 0.29 for DS, and 2.48 for HS (where HS included a small contribution from undegraded material). (Knepper et al. 1996b) Using 4-layer stratification, HA and HS were found to be higher near SC and declined somewhat towards the trabecular side of the JCT, while CS and DS were found to increase modestly towards the trabecular side of the JCT. (Knepper et al. 1996b)
Using antibodies specific to the various sulfated GAG chains, we (Keller et al, manuscript in review) found for that all cell layers in the TM and JCT, CS, DS and HS were present at higher levels just beneath the cells. CS/DS GAGs were also found as small clumps in the center of TM beams, apparently associated with collagen fibrils and elastin microfibrils. The JCT showed relatively dense staining throughout for CS, DS and HS sulfated GAGs. Using Cuprolinic or Cupromeronic Blue to stain sulfated GAG chains in conjunction with GAGase treatments, sulfated proteoglycans have been localized at the ultrastructural level within the TM. (Tawara et al. 1989; Gong et al. 1992; Tawara and Inomata 1994; Hernandez and Gong 1996; Hubbard et al. 1997) Using Cuprolinic Blue, small thin structures were found to be closely associated with collagen fibrils. A large thick structure was associated more peripherally with and between collagen fibril bundles and associated with fine filaments. Both of these contained predominantly DS/CS GAGs. Basement membrane-associated structures contained HS GAGs. (Gong et al. 1992) Although they did not identify the proteoglycan core proteins in this study, the small thin structures were probably one or more small leucine-rich proteoglycans (SLRPs) such as decorin or biglycan; the large thick structure was probably versican; and the basement membrane-associated HS proteoglycan was probably perlecan. Using Cupromeronic Blue and a slightly different analysis, fairly similar distributions were obtained. (Tawara et al. 1989) In one study, these sulfated proteoglycans were found associated with JCT cell basement membrane-like material. (Tawara and Inomata 1994)
Proteoglycans
With the exception of hyaluronan, (Itano and Kimata 2002) GAGs are synthesized directly as covalent side-chains on serine/threonine or asparagine groups of specific proteoglycan core proteins. (Funderburgh 2002; Silbert and Sugumaran 2002; Sugahara and Kitagawa 2002) Studies employing several methods of identification showed that the TM expresses a number of proteoglycans (PGs) (Table 1). This includes a number of SLRPs, CSPGs, HSPGs, and other types of PGs (Table 1). (Murphy et al. 1987; Tanihara et al. 1995; Wirtz et al. 1997; Knepper, P et al. 1998; Friedman et al. 2000; Friedman et al. 2002; Ishibashi et al. 2002; Knepper, P et al. 2002; Borras 2003; Lo et al. 2003; Filla et al. 2004; Vittitow and Borras 2004; Zhao et al. 2004; Chen et al. 2005; Vittal et al. 2005; Keller et al. 2007b)
Table 1.
Proteoglycans found in the outflow pathway.
| Proteoglycan | Family | Main GAG(s) |
|---|---|---|
| Biglycan | SLRP | DS/CS |
| Decorin | SLRP | DS/CS |
| Fibromodulin | SLRP | KS |
| Lumican | SLRP | KS |
| Mimecan | SLRP | KS |
| Versican, CSPG 2 | CSPG | CS |
| Neurocan, CSPG 3 | CSPG | CS |
| Melanoma-assoc., CSPG 4 | CSPG | CS |
| Bamacan, CSPG 6 | CSPG | CS |
| Perlecan, HSPG 1 | HSPG | HS |
| LDL receptor, HSPG 2 | HSPG | HS |
| Syndecan 1 | Transmembrane HSPG | HS |
| Syndecan 2 | Transmembrane HSPG | HS |
| Syndecan 3 | Transmembrane HSPG | HS |
| Syndecan 4 | Transmembrane HSPG | HS |
| Glypican 3 | GPI anchored HSPG | HS |
| Glypican 4 | GPI anchored HSPG | HS |
| Glypican 5 | GPI anchored HSPG | HS |
| Testican 1 | SPARC/osteonectin | CS |
| Testican 3 | SPARC/osteonectin | CS |
| Secretory granule PG 1 | Serglycin | CS/HS |
| Collagen XII | Non-fibrillar collagen | CS |
| Collagen XV, endostatin | Non-fibrillar collagen | CS |
| CD44 | Cell surface HA receptor | CS/HS |
Some specific core proteins have been localized to regions of the TM by immunohistochemistry using light or transmission electron microscopy. Perlecan was localized to the basement membranes of the TM beams and SC inner wall cells. (Murphy et al. 1987) At the light level, syndecan 1 immunostaining in the TM/SC was light or absent. Syndecan 2, 3 and 4 immunostaining was observed through the TM/SC region with syndecan 3 immunostaining being somewhat stronger in the JCT/SC region. (Filla et al. 2004) As would be expected for a transmembrane protein, syndecan immunostaining was strongest at the cell edges. At the ultrastructural level, decorin and versican immunostaining was found within the electron dense elastic fiber cores and the surrounding microfibril sheath material, both within the TM beams and the JCT region. (Ueda et al. 2002; Ueda and Yue 2003) Both proteoglycans were also localized to the long-spaced (100–120 nm periodicity) collagen and the normal collagen fibrils. At the light level, we see similar versican and fibromodulin distributions with a significant amount also associated with the TM beam, JCT and SC cells. (Keller et al. 2007a)
Many of these proteoglycans have complex structures with multiple binding domains and exhibit alternative splicing. (Zhao and Russell 2005; Keller et al. 2007b) For example, versican (Fig. 2) is a large lectican, which can bind to hyaluronan chains and interact with various other ECM proteins or cellular receptors and has four alternative mRNA splicing variants. (Wight 2002; Wu et al. 2005; Kenagy et al. 2006) TM cells express these different splice forms and the ratios are changed with mechanical stretch, TGFβ, TNFα and IL-1α treatments (Keller, et. al. manuscript in preparation). (Zhao and Russell 2005; Keller et al. 2007b) As indicated, these splice variants change the number of potential GAG attachment sites from over 20 to 0, thus dramatically affecting the structure and functional properties of this proteoglycan. Versican is also cleaved by specific proteinases at various sites, which results in fragments with very different activities such as stimulation of cell division, disruption of chemokine function and apoptosis. (Kenagy et al. 2006) In the TM, the versican fragment produced by ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) 1 or 4, which cleave between Glu441 and Ala442 in the alpha GAG domain, is found in moderate abundance and may be higher in areas of high segmental outflow (Bradley, et. al., manuscript in preparation).
Fig. 2.
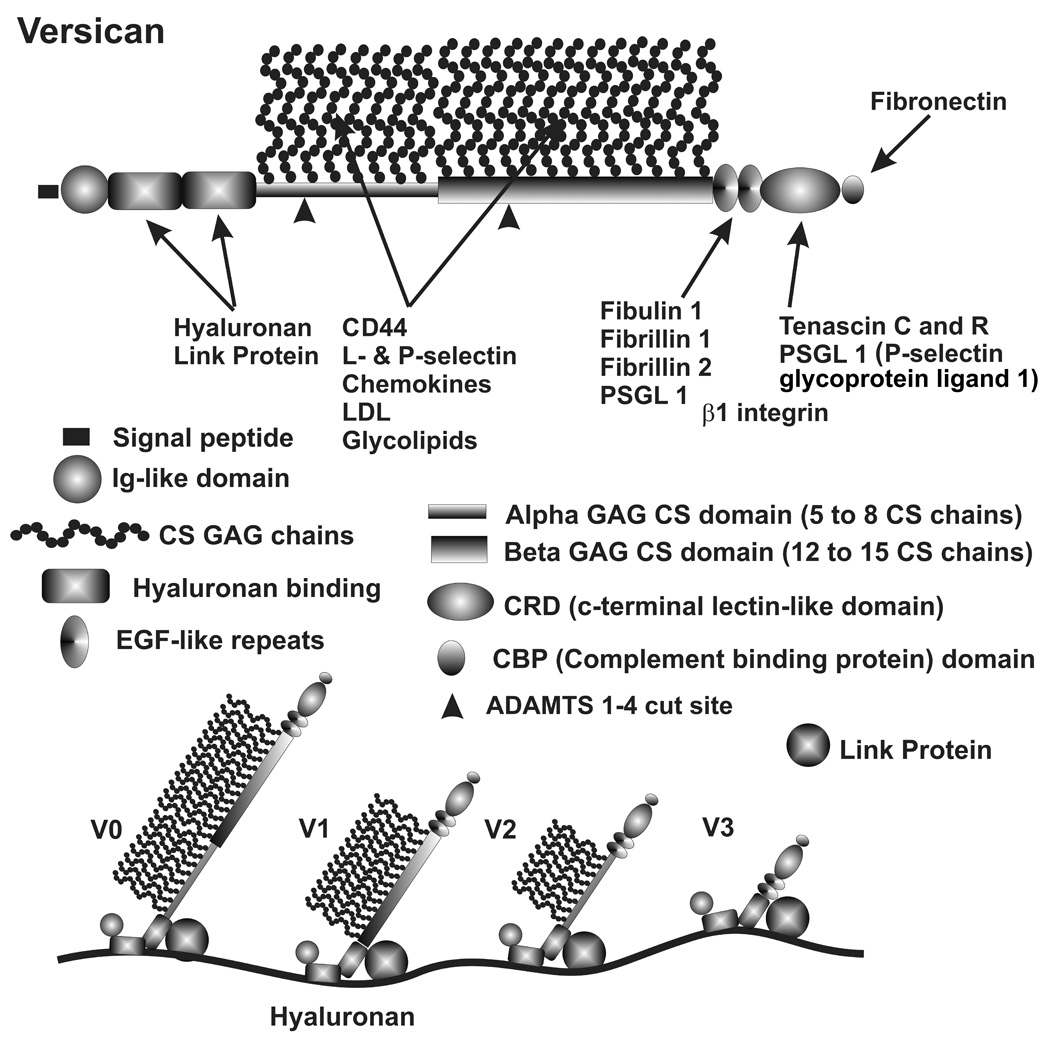
Versican structural domains, binding sites and alternative splicing. Key shows versican domains, positions of GAG chain attachments and position of ADAMTS 1–4 cut site. Various ECM molecules bind to these domains as indicated. The four alternative splice forms of versican are shown binding to hyaluronan with a stabilizing interaction with link protein. (Diagram modified from (Wight 2002; Wu et al. 2005)
Other ECM components identified in the TM
Using molecular, biochemical and immunohistochemical approaches, a variety of other ECM proteins have been identified in the TM, or are expressed by TM cells. A select group is listed in Table 2. These ECM proteins and glycoproteins serve a variety of divergent functions in other tissues, although most have been studied in little detail in the TM.
Table 2.
Other ECM components found in the TM/SC
| ECM Component | Isoforms | Family |
|---|---|---|
| Fibronectin | Several splice variants | ECM organization, cell interaction, fibrils |
| Laminin | α2–4, β1–3, γ1–3 | Basement membrane |
| Nidogen | 1 | Basement membrane, laminin |
| Fibrillar collagen | I, III & V | ECM structure, tensile strength |
| Fibril network collagen IV | α1, α2, α4, α5 & α6 | Basement membrane |
| Filamentous collagen | VI | Microfibril structure |
| Non-fibrillar collagens | VII, VIII, XI, XV & XXII | ECM organization |
| Fibril-associated collagen | XII & XIV | FACIT, ECM organization |
| Elastin | Elastic fibrils | |
| Fibulin | 1, 5 & 6 | Elastic microfibril, ECM organizer |
| Fibrillin | 1 & 2 | Elastic microfibril protein |
| Microfibril-associated | 1,2 & 4 | Elastic microfibril protein |
| glycoprotein (MAGP) | ||
| Vitronectin | ECM organization, cell interaction | |
| Tenascin | C | Matricellular, ECM reorganization |
| SPARC/osteonectin | Matricellular, ECM reorganization | |
| Thrombospondin | 1, 2 & 4 | Matricellular, ECM reorganization |
| NEL-like | 1 & 2 | ECM organization |
| Galectin | 1, 3, 6 & 8 | Lectin, galactose binding protein |
| Periostin | Heparan binding cell adhesion | |
| Osteopontin | 1 | Matricellular, Integrin binding |
| Oculoglycin, opticin | SLRP | |
| TNF-stimulated gene | TSG-6 | ECM & hyaluronan stabilization |
| Inter-α-trypsin inhibitor | H1, H2, H3, MEG3 | ECM & hyaluronan stabilization |
| Cartilage link protein | 1 | Versican & hyaluronan binding |
Basement Membrane
Several intrinsic basement membrane proteins, particularly laminin, type IV collagen and perlecan, are expressed in the outflow pathway and have been localized to basement membranes of TM beam cells, SC inner wall cells and the JCT cells. (Murphy et al. 1987; Marshall et al. 1990; Hernandez and Gong 1996; Yue 1996; Zhou et al. 1998a; Brilakis et al. 2001; Ueda et al. 2002; Ueda and Yue 2003; Fuchshofer et al. 2006) Interestingly, these components are not strictly limited to the basal lamina of the JCT but are also found dispersed throughout the JCT, perhaps indicating something novel about the origins of the JCT ECM. Laminins have a cruciform shape composed of a heterotrimer of different isoforms of α, β and γ subunits. (Scheele et al. 2007) The central shaft of the cross is a long coiled-coil formed by interaction of α-helix domains in the three subunits. The arms and head are formed by the individual subunits. The foot of the cross contains cell receptor binding sites and the various individual arms have binding sites for other cell-surface receptors, a number of ECM proteins, and several proteoglycans. Laminin can also form extended networks. The cellular laminin receptors include a number of integrins, the α-subunit of dystroglycan, specific HS GAG chains and sulfatides. (Scheele et al. 2007) Nidogen/entactin is a glycoprotein that binds to laminin and is thought to link it to type IV collagen as part of stabilizing basement membrane ECM networks. (Gersdorff et al. 2007) Nidogen 1 is expressed in TM. (Fuchshofer et al. 2006) Type IV collagen, a non-fibrillar collagen with one noncollagenous domain in the mature form, is also an integral component of basement membranes. It exists as a trimer of various α-subunits and can form larger three-dimensional networks. (Linsenmayer 1991) Several different α subunits have been identified in the TM. (Fuchshofer et al. 2006) Perlecan, the large basement membrane HSPG, is also an integral component of TM ECMs.
Fibronectin and Vitronectin
Fibronectin (Fig. 3) is present in basement membranes, but is also made by cells without basement membranes and found in other ECMs. It is found abundantly in TM beams and the JCT region, with slightly higher levels in the basement membranes of TM beam cells, JCT cells and SC inner wall cells (Keller et al, manuscript in review). (Floyd et al. 1985; Murphy et al. 1987; Zhou et al. 1998a; Bachmann et al. 2006; Keller et al. 2007a) In the JCT at the ultrastructural level, fibronectin is found in the amorphous material associated with the basement membranes and with the peripheral SD material of the elastic microfibrils. (Yue 1996; Brilakis et al. 2001; Ueda et al. 2002; Ueda and Yue 2003) In addition to forming a thin fibrous array that appears to encircle TM cells in culture, (Zhou et al. 1998b) fibronectin also forms thick fibrous strands. (Keller et al. 2007a) Fibronectin fibril formation has been studied in other tissues and involves interactions with cell surface integrins, proteoglycans and tenascin C. It produces a fibril with considerable elasticity. (Baneyx et al. 2002; Mao and Schwarzbauer 2005; Abu-Lail et al. 2006) This elasticity appears to come from unfolding domains of fibronectin and shifting to extended molecular conformations. (Baneyx et al. 2002; Abu-Lail et al. 2006; Vogel and Sheetz 2006) As discussed later, mechanical stretching of TM cells and their ECMs triggers homeostatic responses that reduce the outflow resistance. These fibronectin fibrils, which would have a number of cellular integrin attachments for each fibril, would provide an ideal sensing structure to communicate IOP-induced mechanical stretch to TM cells. Fibronectin has tandem repeats of three types of domains of around 50 amino acids each, which form protein-, GAG-, and integrin cell-binding domains. Three alternative splice regions are known, and TM cells express EIIIA, EIIIB and the variable regions V1, V2 and V3 of IIICS under different conditions. (Li et al. 2000; Vittal et al. 2005) Inclusion of the V1–V3 domains within the IIICS domain introduces a number of binding sites for heparin and several additional integrin α4β1 sites. (Vittal et al. 2005) Vitronectin has also been identified in the TM, associating with the cores and SD material of the elastic microfibrils. (Yue 1996; Ueda et al. 2002; Ueda and Yue 2003) Attachment of TM cells to vitronectin also has effects on cell properties. (Yue 1996)
Fig. 3.
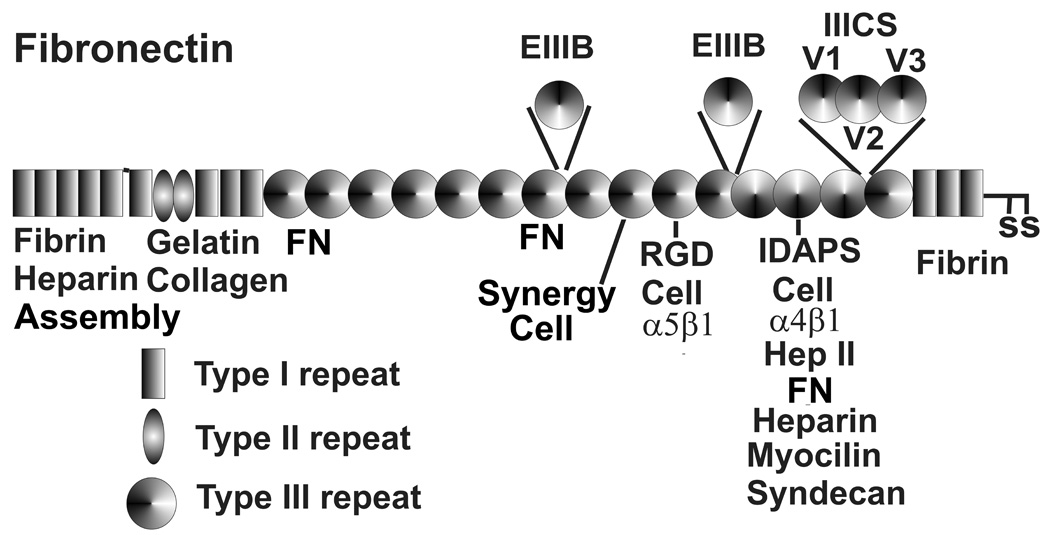
Fibronectin repeat type I, II and III domains, binding sites and alternative splicing. RGD, synergy and IDAPS sequences bind α5β1 and α4β1 integrins. Alternative splicing domains EIIIB, EIIIA and V1, V2 and V3 of IIICS are inserted as indicated. V1, V2 and V2 produce additional heparin, α4β1 integrin and a zinc binding site. FN and assembly represent fibril formation sites.
Collagens
In addition to the type IV basement membrane collagen mentioned earlier, the TM expresses a variety of other collagens. The classic interstitial fibrillar collagens, types I and III, form the standard collagen fibrils with a periodic 67-nm pattern, seen throughout the center of TM beams and JCT region. (Murphy et al. 1987; Marshall et al. 1991; Hernandez and Gong 1996; Yue 1996; Zhou et al. 1998a; Zhou et al. 1998b) They typically form evenly-spaced fibrils that serve primarily as a source of tensile strength for tissues. As mentioned above, type IV forms networks somewhat reminiscent of the hexagonal lattices in chicken wire, which coupled with other basement membrane proteins, provide a stable support for cell attachments.
Types V and VI collagens have been found in both the TM beams and JCT region. They have been observed associated with basement membranes in areas where collagen or elastic fibrils intersect or link to them; associated with the striated collagen fibrils forming a fine filamentous lattice around and between fibrils; associated with the SD microfibrils and to a lesser extent with the dense elastic cores; and associated with long-spaced collagen. (Lütjen-Drecoll et al. 1989; Marshall et al. 1991; Hernandez and Gong 1996; Lutjen-Drecoll 1999; Ueda et al. 2002; Ueda and Yue 2003) Some variations in their precise localizations were observed, probably reflecting differential epitope availability.
Type VI appears to be a key component of the connecting fibrils that have been observed between SC or JCT cells and the cribriform elastic-like plexus, which permeates the JCT region. (Lutjen-Drecoll et al. 1989 ; Tripathi, BJ et al. 1994; Lütjen-Drecoll and Rohen 1996; Lutjen-Drecoll 1999; Ueda et al. 2002; Ueda and Yue 2003; Fuchshofer et al. 2006) Type VI chains have a collagenous triple helix domain for assembly into a disulfide-bonded heterotrimeric type VI monomer, which then forms tetramers that assemble into beaded microfibrils (Fig. 4). Type VI collagen is a major component of 3–5 nm diameter microfibrils (Kielty et al. 1991) and fibrillin is a central component of the beaded 8–12 nm microfibrils discussed later. (Sakai et al. 1991) Several models for tetramer and higher level collagen VI microfibril have been proposed and one is shown in Fig. 4. (Bruns et al. 1986; Engvall et al. 1986; Ball et al. 2001; Reale et al. 2001; Baldock et al. 2003; Ball et al. 2003; Knupp et al. 2006) In the TM, type VI immunostaining of both heteromorphic aggregates and interband longitudinal sheets has been observed, supporting the model in Fig. 4. (Reale et al. 2001) This may relate to the 100 nm repeat structures in the JCT, although this has not been comprehensively studied in detail. (Hessle and Engvall 1984; Bruns et al. 1986; Engvall et al. 1986; Knupp et al. 2006) The type VI collagen N-terminal repeats bind hyaluronan and heparin, while the C-terminal domains are involved in self-assembly. (Kielty et al. 1992b; Specks et al. 1992; Ball et al. 2001; Ball et al. 2003) Decorin and biglycan associate with the N-terminal end of the central collagenous domain, affecting fibril formation. This domain also contains RGD sequences, which are responsible for cell-association. (Kielty et al. 1992a; Fitzgerald et al. 2001; Wiberg et al. 2001; Wiberg et al. 2002; Wiberg et al. 2003) It also interacts with a membrane-associated CSPG 4 and microfibril-associated glycoprotein (MAGP) 1. (Kreis and Vale 1999) Finally, type VI collagen exhibits a number of alternative mRNA splice variants, some of which are marked in Fig. 4. (Chu et al. 1989; Zanussi et al. 1992)
Fig. 4.
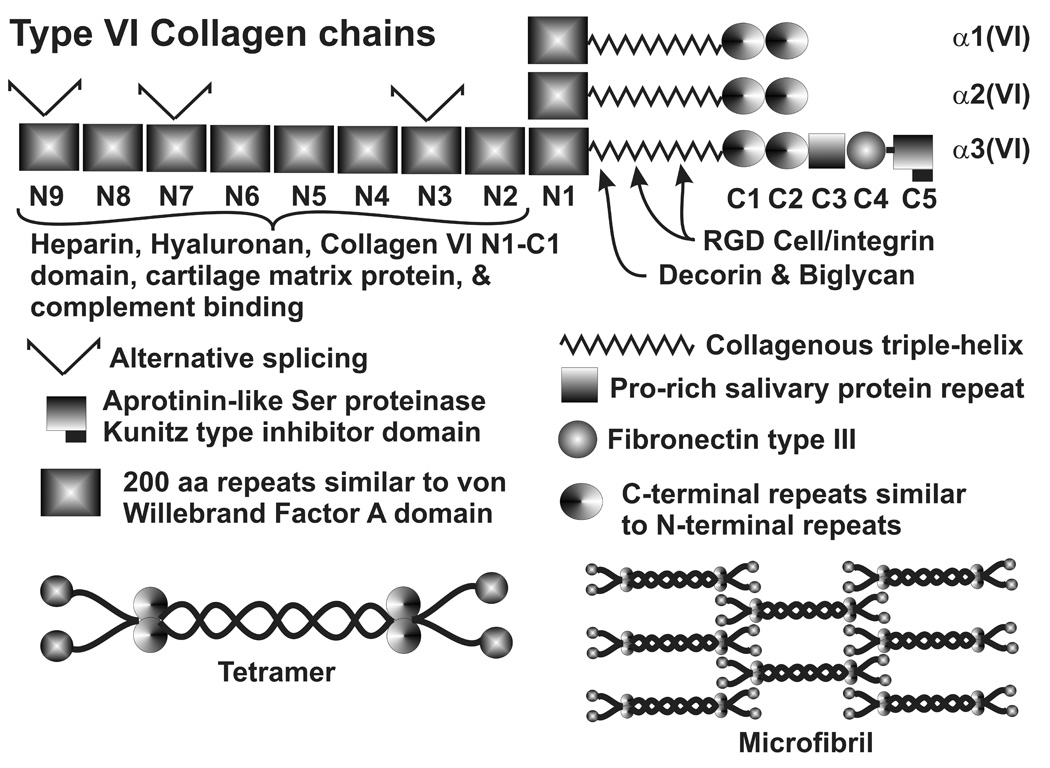
Type VI collagen chains, domain structure, binding domains and proposed microfibril organization.
Non-fibrillar collagens type VII, VIII, XI, XV and XXII are produced by TM cells, as are fibril-associated collagens type XII and XIV (Chen et. al., manuscript in preparation). (Yue 1996; Zhao et al. 2004; Vittal et al. 2005; Fuchshofer et al. 2006; Keller et al. 2007b) Collagen VIII is a short-chain collagen also found in Descemet’s membrane and in subendothelial ECMs that forms hexagonal lattice structures and may serve to provide an open, porous structure that can withstand compressive force. (Kreis and Vale 1999) Collagen type XV is among the multiplexins and is normally found in basement membranes forming short triple helices. (Kreis and Vale 1999) Additional specific details about the functions of these collagens in the outflow pathway have not been determined.
The FACIT (fibril associated collagens with interrupted triple helices), including type XII and XIV collagens, contain small collagenous domains. Their longer, non-collagenous (NC) domains are comprised of a number of typical ECM binding domains, including one for the DS chains of decorin (Fig. 5). (Kreis and Vale 1999) They are found in dense connective tissue associated with other collagen fibrils and appear to be important in fibril organization and interactions. They also often exhibit alternative mRNA splicing (Keller et al. 2007b) and type XII can have GAG chains. (Kreis and Vale 1999) Fig. 5 shows the domain structure, multimerization, and binding domains of type XII collagen, which is very similar to type XIV. (Kania et al. 1999) Alternative mRNA splice variants in the TM include short and long versions of NC3 and NC1. (Kania et al. 1999) Normally, the TM predominantly expresses the short form of both, but mechanical stretching increases expression of the long NC3 form and a novel alternative splice form missing exons 29 and 30 from the C-terminal end of NC3. (Keller et al. 2007b)
Fig. 5.
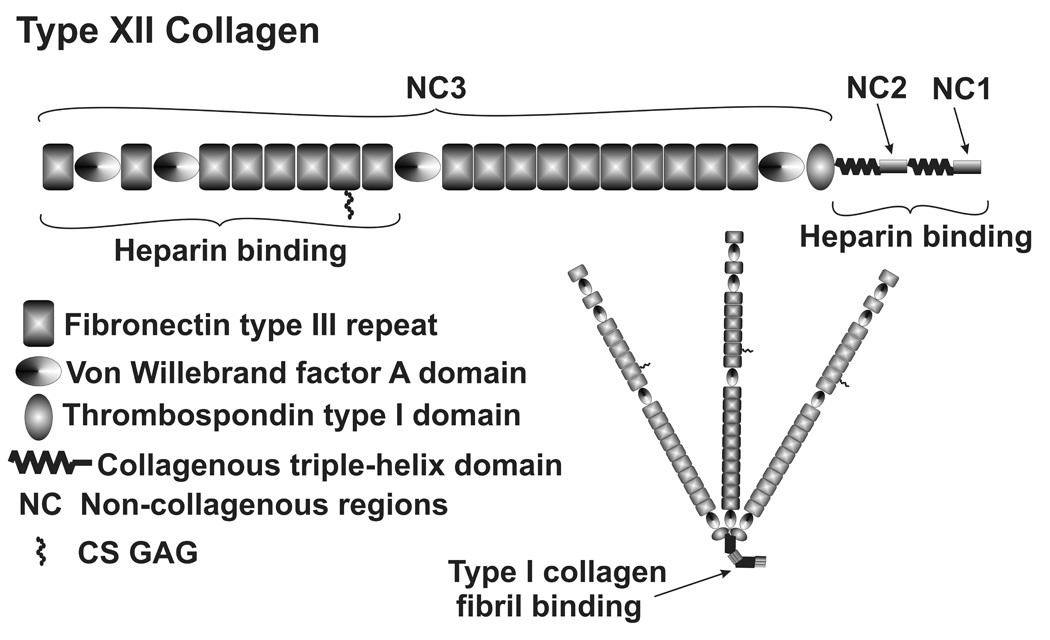
Type XII FACIT collagen domain structure, multimerization and binding domains.
Elastin and Microfibrils
The TM and JCT region has an extensive elastin-containing microfibril network that has been described as a cribriform elastic-like plexus. (Lütjen-Drecoll et al. 1981; Lütjen-Drecoll and Rohen 1996; Lutjen-Drecoll 1999) The elastin-containing microfibrils appear to attach to JCT cells and SC endothelial cells via the small connecting fibrils mentioned earlier that appear to contain type VI collagen. This elastic microfibril plexus also appears to attach to longitudinal processes of the scleral spur and eventually the ciliary muscle tendons. (Lütjen-Drecoll et al. 1981; Lütjen-Drecoll and Rohen 1996; Lutjen-Drecoll 1999; Fuchshofer et al. 2006) A contractile cell has also been identified in the scleral spur, which may have some function in modulating outflow via this elastic microfibril plexus. (Tamm et al. 1992, 1993; Lütjen-Drecoll and Rohen 1996; Lutjen-Drecoll 1999) TM beam and JCT cells appear to be highly similar or even identical, except that the latter expresses αB crystallin, both in situ and in primary cell culture. (Fuchshofer et al. 2006) Since TM cells do express αB crystallin in response to mechanical stretching (Gonzalez et al. 2000; Vittal et al. 2005), this expression difference in situ was interpreted as evidence that JCT cells experience chronic mechanical stress/stretching/tension via this elastic microfibril plexus. (Fuchshofer et al. 2006)
Elastin has been localized to the electron lucent central core of these microfibril structures in the TM beams and the JCT. (Gong et al. 1989; Umihira et al. 1994; Hernandez and Gong 1996; Ueda et al. 2002; Ueda and Yue 2003) The sheath-derived material shows a regular banding pattern and has been referred to as type III plaques. (Lütjen-Drecoll et al. 1981; Lütjen-Drecoll and Rohen 1996; Lutjen-Drecoll 1999) Fibulin 1, fibrillin 1 and 2, and microfibril-associated glycoproteins (MAGP) 1, 2 and 4 are apparent components of this microfibril sheath. (Ueda et al. 2002; Ueda and Yue 2003; Liton et al. 2006) A number of other ECM proteins have also been localized to this SD material, including fibronectin, versican and decorin. (Ueda et al. 2002; Ueda and Yue 2003) Binding interactions between various domains of all of these molecules, including several cell surface receptors, have been identified and a number of models developed to explain aspects of the structures. However, these models are complex and not completely in agreement and no details for the outflow pathway have been established. (Aspberg et al. 1999; Trask et al. 1999; Trask et al. 2000; Olin et al. 2001; Isogai et al. 2002; Kielty et al. 2002a; Kielty et al. 2002b; Segade et al. 2002; Timpl et al. 2003; Kielty et al. 2005) Several models could be proposed that would link this system to the ECM components that may be involved in the outflow resistance and to specific cellattachment components.
Matricellular Proteins
Matricellular proteins are a group of modular ECM proteins that are often involved in reorganization of ECM and changes in ECM-cell interactions or relationships. (Bornstein and Sage 2002) Typically, they interact with both other ECM components and cell-surface receptors, often intervening in their interactions. They are often highly expressed during differentiation, wound healing, tissue remodeling and similar processes. In these processes, they are often anti-adhesive or interfere with binding interactions to allow changes in ECM organization. By contrast, they can also exhibit adhesive properties. Their functions and effects on cells and ECM are very cell type and context-sensitive, producing very different actions in different tissues under various conditions. With increased scrutiny, they continue to exhibit new and different functions and properties. The TM expresses a number of matricellular proteins.
SPARC/osteonectin (secreted protein, acidic and rich in cysteines) is expressed throughout the TM and JCT region and by TM cells in culture. (Wirtz et al. 1997; Rhee et al. 2003) Testicans 1 and 3, proteoglycan members of the SPARC family, are expressed in TM, as discussed earlier. (Chen et al. 2005) The domain structure of members of this family is shown in Figure 6.
Fig. 6.
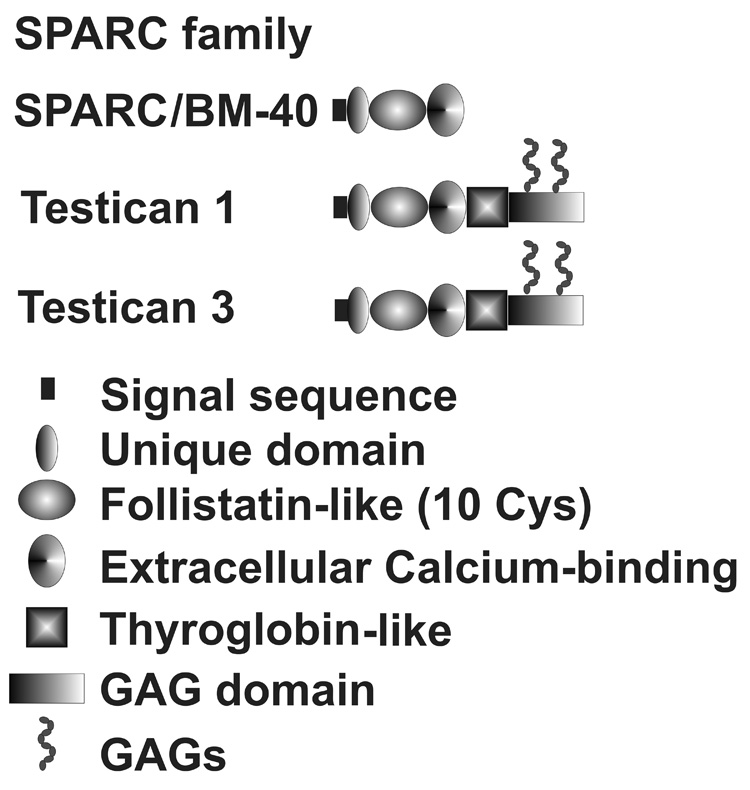
Structural domains of several SPARC family member expressed by outflow pathway cells.
Thrombospondins 1–4 are expressed in the TM. (Tripathi et al. 1991; Si et al. 2003; Flugel-Koch et al. 2004; Zhao et al. 2004; Chen et al. 2005; Fleenor et al. 2006; Liton et al. 2006; Fuchshofer et al. 2007) Figure 7 shows the domain structures and binding motifs of the thrombospondins. Thrombospondins 1 and 2 form homotrimeric structures and are considered to be matricellular proteins, while thrombospondins 3 and 4 are structurally somewhat different and are not considered to be matricellular proteins. (Bornstein and Sage 2002) Thrombospondin 1 is thought to be a major activator of TGFβ, most isoforms of which are present in latent forms that need activation for function. (Crawford et al. 1998) Since TGFβ is thought to be of significance in affecting the normal and glaucomatous ECMs of the outflow pathway and has been shown to modify outflow facility, this may be of considerable importance. (Tripathi, RC et al. 1994; Welge-Lussen et al. 1999; Li et al. 2000; Welge-Lussen et al. 2000; Lutjen-Drecoll and Rohen 2001; Gottanka et al. 2004) Immunostaining for thrombospondin 1 is observed throughout the TM, with the greatest density in the JCT region.(Flugel-Koch et al. 2004)
Fig. 7.
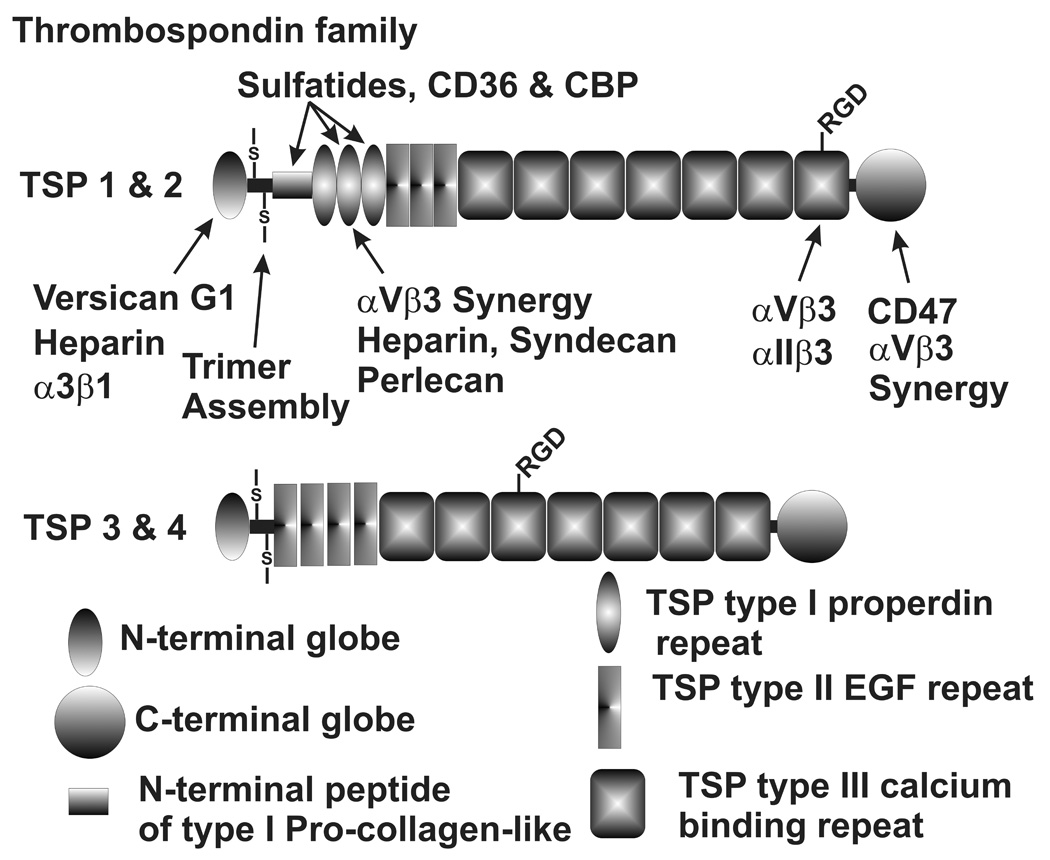
Thrombospondin family domain structures and binding sites. N- and C-terminal globe domains, a multimerization domain resembling the N-terminal region of type I procollagen and three types of thrombospondin (TSP) repeat domains are shown.
Tenascin C or hexabrachion is another matricellular protein expressed in the TM. (Chen et al. 2005; Vittal et al. 2005; Keller et al. 2007b) Tenascin C is composed of numerous repeat binding domains and forms a hexamer by disulfide bonding of the N-terminal tenascin assembly domain using heptad repeats (Fig. 8). (Ghert et al. 2001) It has both adhesive and anti-adhesive effects and is involved in cell migration, ECM remodeling, development, tumorogenesis and wound healing. (Chiquet-Ehrismann et al. 1988; Vainio et al. 1989; Ghert et al. 2001; Bornstein and Sage 2002; Chiquet-Ehrismann and Chiquet 2003; Chiquet-Ehrismann 2004) Some of these processes occur via tenascin C’s association with fibronectin fibrils and effects on fibronectin-cell interactions. (Chung et al. 1995; Ghert et al. 2001) In addition to exhibiting changes in levels with various treatments that modify outflow facility (see later), tenascin C also undergoes a number of changes in mRNA splicing (Keller et al, manuscript in preparation). (Keller et al. 2007b) The inclusion of various partial or complete fibronectin type III domains between exons 5 and 6 of tenascin C introduces a number of heparin and other types of binding domains. Although the nuances of these changes on the properties of the outflow pathway have not been established, the potential seems significant.
Fig. 8.
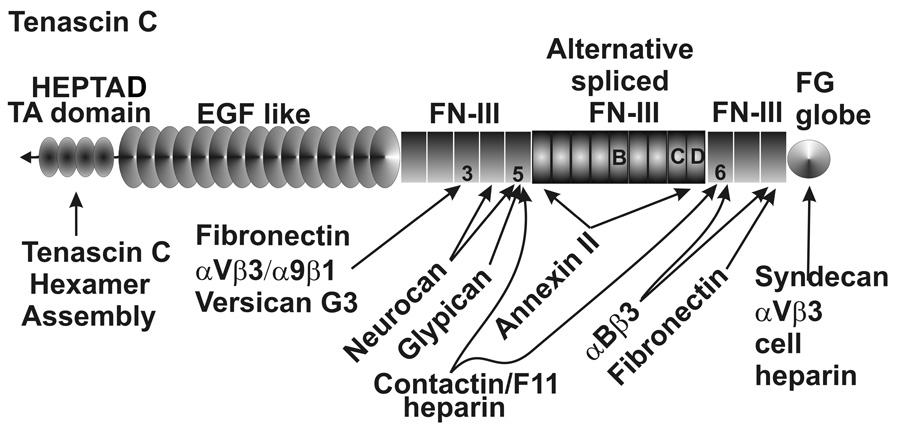
Tenascin C domain structure, binding sites and alternative splicing. Heptad repeat hexamer assembly domain, EGF-like domains, type III fibronectin and fibrin globular domains are as indicated.
Osteopontin is expressed by TM cells and has recently been classed as a matricellular protein. (Bornstein and Sage 2002; Chen et al. 2005) Unlike the other matricellular proteins, it also may serve some structural roles. (Denhardt and Guo 1993; O'Regan and Berman 2000; Bornstein and Sage 2002) It is a secreted, highly acidic and phosphorylated sialoprotein, which binds to cells via GRDGS interactions with αVβ3 integrin. (Denhardt and Guo 1993; Sodek et al. 2000) Thrombin cleavage releases two peptides with different functions. It interacts with fibronectin, type I collagen and osteocalcin (bone Gla protein). Additional details on its functional involvements are emerging. (Denhardt and Guo 1993; O'Regan and Berman 2000; Sodek et al. 2000; Bornstein and Sage 2002)
Other ECM Proteins
Periostin or osteoblast specific factor 2, although not classed as a matricellular protein, appears to exhibit some characteristics and functions common to this group. Periostin is expressed by TM cells. (Vittitow and Borras 2004; Zhao et al. 2004; Chen et al. 2005; Vittal et al. 2005; Liton et al. 2006) Periostin contains several fasciclin-like binding domains, binds several different integrins, including αVβ3, αVβ5, and affects cell adhesion and migration. (Gillan et al. 2002; Litvin et al. 2004) It is expressed during development and active ECM remodeling, particularly where collagen is being modified. (Litvin et al. 2004; Kii et al. 2006) Initially involved in osteoblast function in mineralized tissues, it appears to have other functions in other tissues, such as the TM.
As may pertain to this latter pair of proteins, osteopontin and periostin (both thought initially to be bone-specific and limited to osteoblast mineralization functions) matrix GLA protein is also expressed by TM cells. (Vittitow and Borras 2004; Chen et al. 2005; Vittal et al. 2005) In this protein, a number of glutamate residues are converted post-translationally to γ-carboxylglutamic acid (GLA). Mediated by calcium-binding, this creates a domain, which binds bone morphogenic protein (BMP) 2. The exact mechanism of matrix GLA protein involvement in the outflow pathway remains unclear, but some type of calcium deposition event has been hypothesized. (Xue et al. 2006; Xue et al. 2007) Since it can inhibit BMP2-induced calcification, its presence and modulation by a number of modulators of outflow is intriguing. Matrix GLA protein is secreted and found in bone and cartilage, serving a regulator role in calcification in these tissues and in the vascular system, where it is involved in arterial wall stiffness. In the TM, it has been shown to affect levels of calcification markers. (Xue et al. 2006; Xue et al. 2007)
A number of other ECM proteins are expressed in the outflow pathway, but important functional roles have not been established for most of them.
Cell-surface ECM receptors of the TM
Various typical ECM cell-surface receptors are expressed in the TM. Integrins α1, α2, α3, α4, α5, α6, αV, β1, β3, β4 and β5 subunits and α5β1, αVβ3 and α4β1 dimeric complexes have been specifically identified. (Yue 1996; Zhou et al. 1996; Zhou et al. 1998b; Zhou et al. 1999c; Vittal et al. 2005) Different combinations of α and β subunits create a 3-dimensional recognition site, providing a wide array of partially-overlapping specificities for different ECM macromolecules. Thus, it seems certain that a number of additional integrin pair combinations are also present on TM cells. The integrins are involved in fibronectin fibril formation, cell-ECM attachments and both inside-out and outside-in signaling processes. For many of these functions, they form large heterogeneous aggregates at the cell surface in which multiple integrin pairs, accessory and adaptor proteins, ECM macromolecules, signaling molecules and cytoskeletal attachments coalesce. Focal adhesions, focal contacts, podosomes and CLANs are among the more well-defined coalesced structures. (Geiger and Bershadsky 2001; Geiger, B et al. 2001; Geiger, B. et al. 2001) Attachment of TM cells to various integrins produces differential cellular responses. (Yue 1996; Zhou et al. 1996; Zhou et al. 1998b, 1999a,b; Zhou et al. 1999c; Zhou et al. 2000; Filla et al. 2006) Additionally, both β1 and β3 integrins are involved in CLAN formation under some sets of conditions. (Filla et al. 2006) Integrins are thought to serve a key function in sensing the mechanical stretching of the JCT, which reflects changes in IOP and serves as a signal for homeostatic outflow resistance adjustments. (Acott 1994b; Acott and Wirtz 1996; Bradley et al. 2001; Borras et al. 2002; Bradley et al. 2003)
Anterior segment perfusion with the second heparin binding domain (Hep II) of fibronectin, composed of numbers 12–14 of the type III fibronectin repeat domains (Fig. 3), reversibly increases outflow facility. (Santas et al. 2003) The Hep II domain of fibronectin interacts with α4β1 integrin and the heparan sulfate chains of syndecans. After the Hep II domain was washed out of the system and the outflow facility had returned to normal, micrographs showed that Schlemm’s inner wall lining cells were missing in some areas and their basement membranes in these areas were not intact. The remainder of the TM, including the JCT cells, seemed normal and relatively unaffected in both light and electron micrographs. A similar study using an RGD peptide (GRGDSP), which is from the 10th fibronectin type III domain and is the binding site for α5β1 integrin, showed similar disruptions of Schlemm’s inner wall endothelium, but did not significantly change outflow facility. (Bahler et al. 2004) The remainder of the TM was not visibly affected.
Syndecans 1–4 are another family of cell-surface ECM receptors expressed in the TM (Table I). (Filla et al. 2004; Chen et al. 2005; Vittal et al. 2005; Filla et al. 2006) The syndecans are transmembrane HS proteoglycans that cooperate with specific integrins, bridge to specific cytoskeletal structures, and are involved in inside-out and outside-in signaling. (Couchman and Woods 1999; Couchman et al. 2001; Couchman 2003; Yoneda and Couchman 2003) As HS proteoglycans, they can modulate growth factor activity and availability. They are involved in focal adhesions (particularly syndecan-4), stress fiber formation (particularly syndecan-2), cytoskeletal organization and other cellular processes. (Couchman and Woods 1999; Couchman et al. 2001; Santas et al. 2002; Couchman 2003; Yoneda and Couchman 2003; FFilla et al. 2004; Peterson et al. 2005; Filla et al. 2006) Syndecan-4 also appears to localize with the verticies of CLANs and syndecan-2 localizes with podosomes in the TM (Aga, et. al. manuscript in preparation). (Filla et al. 2006) They clearly serve several important functions in the outflow pathway, but their exact roles here are only beginning to emerge. (Santas et al. 2002; Filla et al. 2006) The glypicans are another family of cellsurface HS proteoglycans. They are similar to the syndecans, except that they attach to the cell surface by glycosylphosphatidylinositol (GPI) lipid anchors instead of by a transmembrane tail. (Filmus and Selleck 2001; Song and Filmus 2002) Glypican 3, 4 and 5 are expressed in TM. (Zhao et al. 2004; Chen et al. 2005) The 6 glypicans share a conserved pattern of 14 Cys and 2–4 HS attachment sites near the C-terminal hydrophobic domain where the GPI anchor is attached. (De Cat and David 2001; Song and Filmus 2002) They are critical in development, serve a variety of roles in adult tissues, and are involved in Wnt, TGFβ and BMP signaling. (Hartwig et al. 2005; Song et al. 2005) Their GAGs share a pattern of iduronate and sulfate distributions that resemble the syndecans and facilitate comparable fibronectin Hep II domain interactions. (Tumova et al. 2000) Details of glypican involvement in the TM are unknown.
Another cell-surface ECM receptor, CD44, which is a hyaluronan receptor, has been studied in the TM in some detail and clearly plays an important role, directly or indirectly, in outflow and TM function. (Knepper, P et al. 1998; Knepper, P et al. 2002; Choi et al. 2005; Knepper et al. 2005) An important impact of the truncated soluble form of CD44 on TM cells has been reported. Alternative mRNA splicing of CD44 in the TM is also observed. (Jumper et al. 1998; Keller et al. 2007b)} The N-terminal portion of CD44 is coded by exons 1–5 and the transmembrane C-terminal portion is coded by exons 6–10. Between these two is an alternative splicing site where variable domains V1–V10 can be inserted. Some of these variable domains contain CS or HS GAG chains and can affect growth factor functions by binding and modulating their availability. CD44 is also directly involved in cellular signaling, where it can trigger several processes. It can also serve as a co-receptor and docking site for MMP-7, MMP-9 and MMP-14.
VCAM-1 is expressed by TM cells and PECAM-1 is expressed by SC cells. (Heimark et al. 2002; Chen et al. 2005; Vittal et al. 2005) A variety of other cell-surface receptors and cell-cell interaction proteins are expressed by TM/SC cells. These include sarcoglycan β and ε, dystroglycan and sarcospan, which are members of the dystroglycan glycoprotein complex that can serve as a laminin receptor in some tissues. (Kreis and Vale 1999) Fibronectin leucine-rich transmembrane proteins (FLRTs) 2 and 3 are also expressed by TM cells, although their functional significance has not been determined. (Chen et al. 2005; Liton et al. 2006)
ECM turnover and outflow resistance
Several stimuli that affect outflow facility change the activity of a family of enzymes, the matrix metalloproteinases (MMPs), which are responsible for initiating most ECM turnover. (Parks and Mecham 1998) These stimuli include TNFα, IL-1α, TGFβ, dexamethasone, elevated pressure, and/or mechanical stretch. (Samples et al. 1993; Clark et al. 1995b; Alexander et al. 1998; Bradley et al. 1998; Bradley et al. 2000; Bradley et al. 2001; Gottanka et al. 2004; Chen et al. 2005; Clark et al. 2005; Vittal et al. 2005; Fleenor et al. 2006; Kelley et al. 2007) The process of ECM turnover is a concerted and highly coordinated program of degradation and biosynthetic replacement of specific ECM components. IOP homeostasis appears to be maintained, at lease in part by ECM turnover triggered in response to pressure changes or mechanical stretch or distortion, sensed by cells within the JCT. This triggers corrective steps by which these cells adjust the outflow resistance and restore the IOP to normal ranges.
Regulators of aqueous outflow and ECM component expression
A number of agents or modulators have been shown to affect aqueous humor outflow facility. Although the direct mode of action of each of these agents has been hypothesized, essentially all of them affect multiple aspects of TM cell behavior. Evaluating the TM cellular responses to several of these agents might provide insights into the mechanism(s) and actual components involved in the outflow resistance. Microarrays, proteomics or similar types of studies may thus provide some guidance for this approach.
Elevated IOP, mechanical stretching and IOP homeostasis
We and others have developed evidence that TM cells can mount a homeostatic response to elevated IOP, adjusting the outflow resistance and restoring the IOP level. (Bradley et al. 1998; Bradley et al. 2001; Borras et al. 2002) When the flow rate in perfused anterior segment organ culture is doubled, the measured pressure immediately doubles. However, TM cells respond by increasing MMP enzyme activity and changing the expression of a number of genes. Over the next few days, the TM adjusts the outflow resistance and restores the initial IOP, although the flow rate is maintained at twice the initial value. (Bradley et al. 2001) TM cells sense mechanical stretching or distortion, probably of their ECMs via their cell-surface integrins and other cell-surface receptors, as an indication that the outflow resistance is too high. In addition to adjusting MMP activity, TM cells change expression levels of a number of ECM components, a few of which are shown below (Table 3). (Vittal et al. 2005) A number of changes in mRNA splicing patterns also occur. (Vittal et al. 2005; Keller et al. 2007b) Other cellular changes have also been observed. (Tumminia et al. 1998)
Table 3.
Changes in expression of a few select ECM genes in response to some treatments that modify aqueous outflow facility. Approximate fold changes are shown from microarray, quantitative RT-PCR, or proteomics analyses. NS indicates that changes were not statistically or biologically significant, N indicates information not available, and S indicates alternative mRNA splicing has been reported. Values are at the times of maximum change and range from 12 to 72 hrs for stretch, cytokines or TGFβ and up to 10 days for dex.
| ECM Component | Stretch | TNFα | IL-1α | TGFβ | Dex |
|---|---|---|---|---|---|
| Versican | 0.45, S | 0.47, S | 0.46, S | 4.8–9, S | 0.034 |
| Tenascin C | 2.6, S | 4.5, S | 3.7, S | N | N |
| Fibronectin | 1.5, S | NS, S | NS, S | 3–4, S | 2, S |
| CD44 | 1.8, S | 1.7, S | 1.36, NS, S | N | N |
| Syndecan 1 | 0.77, NS | 0.33 | 0.23 | N | N |
| Syndecan 2 | 1.5 | 0.32 | 0.46 | N | N |
| Synedcan 4 | NS | 2.1 | 1.87 | N | N |
| Glypican 3 | N | 1.98 | 2.78 | N | N |
| SPARC | 2.4 | 0.21 | 0.48 | N | N |
| Testican 1 | N | 0.37 | 0.48 | N | N |
| Testican 3 | N | 1.79 | NS | N | N |
| Thrombospondin 1 | NS | 0.32 | 0.49 | 2.4–5 | N |
| Thrombospondin 2 | 0.68, NS | 0.12 | 0.29 | N | N |
| TSG-6 | N | 10.24 | 24.5 | 3.3 | N |
| VCAM-1 | 1.69 | 47 | 28 | 0.31 | N |
| Collagen XIV | 3.25 | NS | NS | 0.38 | N |
| Periostin | 0.49–2.2 | 4.63 | 4.93 | 5.3 | N |
| Matrix Gla protein | 2.5–7 | 0.29 | 0.31 | N | N |
| Link protein | 1.3, NS | NS | 1.34, NS | 2.1 | N |
| MMP-2 | 1.4 | NS | NS | 4.9 | N |
| MMP-3 | NS | 49.5 | 54.6 | 8 | N |
| ADAMTS 4 | N | 25.7 | 36.6 | N | N |
| ADAMTS 5 | NS | 8.3 | 18.3 | 4.0 | N |
IL-1 and TNF
Laser trabeculoplasty, a common glaucoma treatment, triggers IL-1 and TNF release within a few hours. (Bradley et al. 2000) These cytokines then stimulate sustained MMP-3 increases specifically within the juxtacanalicular region. (Parshley et al. 1995; Parshley et al. 1996) Adding either cytokine to perfused anterior segment organ cultures increases MMP-3 and outflow facility. (Bradley et al. 1998; Kelley et al. 2007) Thus, MMP-3 stimulation by these cytokines is probably a critical component of the effect of this treatment on outflow facility. Microarray analysis of TM cells treated with TNFα or IL-1α allows the identification of a number of ECM components that are changed and thus may be involved in the ECM remodeling and adjustment of the outflow resistance (Table 3) (Chen, et. al., Invest Ophthalmol Vis Sci 46: e-abstract #1349, 2005).
TGFβ
Glaucomatous aqueous humor has been reported to contain elevated levels of TGFβ2. (Tripathi, RC et al. 1994) TGFβ2 perfusion reduces outflow facility and is generally thought to increase ECM production. (Gottanka et al. 2004; Fleenor et al. 2006) In TM cells, a number of ECM genes are modulated by TGFβ1 and/or TGFβ2 (Table 3). (Zhao et al. 2004; Fleenor et al. 2006) Earlier, it was shown that TGFβ1 β2, β3 and β5 all increased MMP-3 expression by the TM. (Alexander et al. 1998) In addition to these and many other changes observed, versican splicing was affected, as discussed later. (Zhao and Russell 2005)
Glucocorticoids. Dexamethasone has been shown to reduce outflow facility. (Clark et al. 1995b; Clark et al. 2005) Much of this effect may be due to dramatic cytoskeleton changes, i.e., the formation of cross-linked actin networks (CLANs) (Clark et al. 1994; Clark et al. 1995a), or to a number of other TM cell responses, e.g., increased expression of myocilin. (Polansky et al. 1997; Stone et al. 1997; Polansky and Nguyen 1998; Polansky et al. 2000; Lo et al. 2003) Myocilin has been shown to affect outflow facility, although the exact mechanism appears to be complex. (Borras et al. 2002; Goldwich et al. 2003; Zillig et al. 2005; Fautsch et al. 2006) Myocilin also binds several ECM components, including the hep II domain of fibronectin, (Filla et al. 2002) and is found in the JCT and SC region. (Ueda et al. 2002; Ueda and Yue 2003) An association with elastic microfibril SD plaques was also observed. Several ECM genes’ expression was changed in response to dexamethasone treatment, including a reduction in versican and an increase in fibronectin levels. (Steely et al. 1992; Liu et al. 2003; Lo et al. 2003) Glucocorticoids also increase ECM deposition in the TM, producing a fingerprint-like arrangement somewhat resembling basement membrane material. (Johnson et al. 1997) This pattern differs from the SD plaques seen with glaucoma (see later). These studies may provide clues to the molecular mechanisms involved in steroid-induced glaucoma.
Alternative splicing of ECM components
In addition to the changes in expression levels of a number of potentially relevant ECM components with these modulations of outflow facility, several components exhibit unusual mRNA splicing in the TM or change the relative amounts of different splice variants in response to outflow modulation. Some of these are indicated by the # symbol in Table 3. The presumed consequence is that the modified isoform will have an increase or decrease in some specific binding domain or possibly GAG attachment sites. In response to mechanical stretching, fibronectin mRNA splicing shifts to an increased proportion of transcripts containing the V1 and V3 regions. (Vittal et al. 2005) This introduces two additional α4β1 integrin binding sites and has been shown to affect fibronectin’s Hep II domain interactions with cells. (Santas et al. 2002; Peterson et al. 2005) TNF and IL-1 have modest effects and TGFβ and dexamethasone have stronger effects of the EIIIA and EIIIB splice isoforms, changing availability of integrin α9 sites (Keller et. al. Manuscript in preparation). (Li et al. 2000)
Tenascin C, which is upregulated by mechanical stretch, TNFα and IL-1α treatments, also exhibits a wide variety of splice variants in the TM (Gregory, et. al., Invest Ophthalmol Vis Sci 46: e-abstract #1341, 2005). (Keller et al. 2007b) Some of these variants were not previously reported and several are changed by one or more of these treatments. Changes in TM cell splicing variations have also been identified in CD44, introducing a CS and a HS GAG attachment site, and in collagen type XII, introducing a heparin binding domain and a GAG attachment site. (Keller et al. 2007b)
Versican expression and alternative mRNA splicing is also modified with mechanical stretch, TNFα, IL-1α and TGFβ treatments (Chen, et. al., Invest Ophthalmol Vis Sci 46: e-abstract #1349, 2005). (Lo et al. 2003; Zhao et al. 2004; Vittal et al. 2005; Keller et al. 2007b) In addition, TGFβ has been shown to produce a shift in the proportion of TM versican mRNA splice variants increasing domains containing additional CS GAG attachment sites (Fig. 2). (Zhao and Russell 2005) Shifts in the relative amounts of these splice forms have also been detected with mechanical stretching (Keller et al. 2007b), TNFα and IL-1α treatment of TM cells (Keller, et. al. unpublished studies).
Glaucoma and ECM
In glaucoma, an increase in plaque-like material with the ECM has been identified and studied in some detail. (Segawa 1979; Lütjen-Drecoll et al. 1981; Rohen et al. 1981; Rohen 1983; Lütjen-Drecoll et al. 1986; Lütjen-Drecoll and Tamm 1987; Lutjen-Drecoll and Rohen 2001) The exact molecular nature of this material is not clear and a direct causal link to the increased outflow resistance has been difficult to establish. An additional caveat to these glaucoma studies is that, with few exceptions, (Rohen et al. 1993) the tissue is from eyes that have been on various glaucoma medications for extended periods. None the less, valuable information can be gleaned from such studies. At least a portion of the plaque material appears to be from the elastic microfibril SD material. Three types of plaques have been identified by their appearance and properties. Increases with age are apparent, although less dramatic than with glaucoma. Morphometric and hydrodynamic analyses suggests that these plaques are not the direct source of the increased outflow resistance (Johnson 2006), but our inability to identify the normal or glaucomatous outflow resistance adds a caveat to this conclusion. Whether direct or indirect, this material seems likely to play some role in the elevated outflow resistance.
Flow segmentation appears to be higher in older eyes and particularly in glaucoma (Buller and Johnson 1994), although individual variability is high and a molecular cause has not yet been established. Long-lived, primarily ECM-related herniations of the JCT and SC inner wall at collector channels in glaucoma have also been identified, although their molecular nature is not established (Gong, et. al., Invest Ophthalmol Vis Sci 48:e-abstract #2079, 2007).
Gene expression analysis comparing glaucomatous and normal eyes and cells also shows a number of potentially important differences. (Wang et al. 2001; Diskin et al. 2006; Liton et al. 2006) The cell surface receptor ELAM-1 was found to be elevated in eyes with glaucoma, probably reflecting a chronic stress response that produces a sustained cellular phenotype change. (Wang et al. 2001) In one microarray study, the expression levels of mimecan, lumican, syndecan 4, ICAM-1 and several ECM glycolconjugate biosynthetic and degradative enzymes were changed. (Diskin et al. 2006) In another study, changes were observed in the expression of ECM proteins, including matrix Gla protein, chitinase 3-like 1 (glycoprotein 39), TIMP2, fibronectin, cartilage link protein, MAGP 2, collagens type I, III, V, VI, XI, XII and XIV, thrombospondin 3, periostin, MMP-1, -3 and -10, integrin α9, cochlin and myocilin. (Liton et al. 2006) The fibronectin change and perhaps other aspects of this study could be related to TGFβ2, BMP-4 and Gremlin studies recently reported. (Wordinger et al. 2007)
In other studies of glaucomatous outflow tissue, calcification markers relating to matrix Gla protein and BMP-2 action appear to be modified, although mechanisms remain to be established. (Xue et al. 2006; Xue et al. 2007) Increases in soluble CD44, particularly the phosphorylated form, have also been recently linked to glaucoma, although causal relationships remain to be established. (Knepper, PA et al. 1998; Knepper, P et al. 2002; Knepper, PA et al. 2002; Knepper et al. 2005) In an extension of the GAG stratification within the JCT region, discussed earlier, large changes in GAG distributions have also been observed in glaucoma. HA was decreased and CS was increased in all layers of the JCT of primary open-angle glaucoma eyes. (Knepper et al. 1996b)
Acknowledgements
Support was provided by NIH EY003279, EY008247, EY010572, and by an unrestricted grant to Casey Eye Institute from Research to Prevent Blindness, New York, NY. We thank Genevieve Long, PhD, for editorial assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abu-Lail NI, Ohashi T, et al. Understanding the elasticity of fibronectin fibrils: unfolding strengths of FN-III and GFP domains measured by single molecule force spectroscopy. Matrix Biol. 2006;25:175–184. doi: 10.1016/j.matbio.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Acott TS. Trabecular extracellular matrix regulation. In: Drance SM, Van Buskirk EM, Neufeld AH, editors. Pharmacology of Glaucoma. Baltimore: Williams & Wilkins; 1992. pp. 125–157. [Google Scholar]
- Acott TS. Biochemistry of aqueous humor outflow. In: Kaufman PL, Mittag TW, editors. Textbook of Ophthalmology. Vol. 7. London: Mosby; 1994a. pp. 1.47–1.78. [Google Scholar]
- Acott TS. Receptor biology and glaucoma - Integrins in the eye. In: Anderson DR, Drance SM, editors. Encounters in glaucoma research 1: Receptor biology and glaucoma. Milano: Foglizza Editore; 1994b. pp. 97–135. [Google Scholar]
- Acott TS, Kingsley PD, et al. Human trabecular meshwork organ culture: Morphology and glycosaminoglycan synthesis. Invest Ophthalmol Vis Sci. 1988;29:90–100. [PubMed] [Google Scholar]
- Acott TS, Westcott M, et al. Trabecular meshwork glycosaminon human and cynomolgus monkey eye. Invest Ophthalmol Vis Sci. 1985;26:1320–1329. [PubMed] [Google Scholar]
- Acott TS, Wirtz MK. Biochemistry of aqueous outflow. In: Ritch R, Shields MB, Krupin T, editors. The Glaucomas. I. St Louis: Mosby; 1996. pp. 281–305. [Google Scholar]
- Alexander JP, Samples JR, et al. Growth factor and cytokine modulation of trabecular meshwork matrix metalloproteinase and TIMP expression. Curr Eye Res. 1998;17:276–285. doi: 10.1076/ceyr.17.3.276.5219. [DOI] [PubMed] [Google Scholar]
- Aspberg A, Adam S, et al. Fibulin-1 is a ligand for the C-type lectin domains of aggrecan and versican. J Biol Chem. 1999;274:20444–20449. doi: 10.1074/jbc.274.29.20444. [DOI] [PubMed] [Google Scholar]
- Bachmann B, Birke M, et al. Ultrastructural and biochemical evaluation of the porcine anterior chamber perfusion model. Invest Ophthalmol Vis Sci. 2006;47:2011–2020. doi: 10.1167/iovs.05-1393. [DOI] [PubMed] [Google Scholar]
- Bahler CK, Hann CR, et al. Pharmacologic disruption of Schlemm's canal cells and outflow facility in anterior segments of human eyes. Invest Ophthalmol Vis Sci. 2004;45:2246–2254. doi: 10.1167/iovs.03-0746. [DOI] [PubMed] [Google Scholar]
- Baldock C, Sherratt MJ, et al. The supramolecular organization of collagen VI microfibrils. J Mol Biol. 2003;330:297–307. doi: 10.1016/s0022-2836(03)00585-0. [DOI] [PubMed] [Google Scholar]
- Ball S, Bella J, et al. Structural basis of type VI collagen dimer formation. J Biol Chem. 2003;278:15326–15332. doi: 10.1074/jbc.M209977200. [DOI] [PubMed] [Google Scholar]
- Ball SG, Baldock C, et al. The role of the C1 and C2 a-domains in type VI collagen assembly. J Biol Chem. 2001;276:7422–7430. doi: 10.1074/jbc.M002816200. [DOI] [PubMed] [Google Scholar]
- Baneyx G, Baugh L, et al. Fibronectin extension and unfolding within cell matrix fibrils controlled by cytoskeletal tension. Proc Natl Acad Sci U S A. 2002;99:5139–5143. doi: 10.1073/pnas.072650799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bárány E. In vitro studies of the resistance to flow through the angle of the anterior chamber. Acta Soc Med Upsal. 1953;59:260–276. [PubMed] [Google Scholar]
- Bárány EH, Scotchbrook S. Influence of testicular hyaluronidase on the resistance to flow through the angle of the anterior chamber. Acta Physiol Scand. 1954;30:240–248. doi: 10.1111/j.1748-1716.1954.tb01092.x. [DOI] [PubMed] [Google Scholar]
- Bárány EH, Scotchbrook S. Influence of testicular hyaluronidase on the resistance to flow through the angle of the anterior chamber. Acta Physiol Scand. 1954;30:240–248. doi: 10.1111/j.1748-1716.1954.tb01092.x. [DOI] [PubMed] [Google Scholar]
- Bárány EH, Woodin AM. Hyaluronic acid and hyaluronidase in the aqueous humour and the angle of the anterior chamber. Acta Physiol Scand. 1954;33:257–290. doi: 10.1111/j.1748-1716.1955.tb01208.x. [DOI] [PubMed] [Google Scholar]
- Bhatt K, Gong H, et al. Freeze-fracture studies of interendothelial junctions in the angle of the human eye. Invest Ophthalmol Vis Sci. 1995;36:1379–1389. [PubMed] [Google Scholar]
- Bill A, Maepea O. Mechanisms and routes of aqueous humor drainage. In: Albert DM, Jakotiec FA, editors. Principles and Practices of Ophthalmology. Philadelphia: W B Saunders; 1994. pp. 206–226. [Google Scholar]
- Bornstein P, Sage EH. Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol. 2002;14:608–616. doi: 10.1016/s0955-0674(02)00361-7. [DOI] [PubMed] [Google Scholar]
- Borras T. Gene expression in the trabecular meshwork and the influence of intraocular pressure. Prog Retin Eye Res. 2003;22:435–463. doi: 10.1016/s1350-9462(03)00018-1. [DOI] [PubMed] [Google Scholar]
- Borras T, Rowlette LL, et al. Effects of elevated intraocular pressure on outflow facility and TIGR/MYOC expression in perfused human anterior segments. Invest Ophthalmol Vis Sci. 2002;43:33–40. [PubMed] [Google Scholar]
- Bradley JM, Kelley MJ, et al. Signaling pathways used in trabecular matrix metalloproteinase response to mechanical stretch. Invest Ophthalmol Vis Sci. 2003;44:5174–5181. doi: 10.1167/iovs.03-0213. [DOI] [PubMed] [Google Scholar]
- Bradley JMB, Anderssohn AM, et al. Mediation of laser trabeculoplasty-induced matrix metalloproteinase expression by IL-1βand TNFα. Invest Ophthalmol Vis Sci. 2000;41:422–430. [PubMed] [Google Scholar]
- Bradley JMB, Kelley MJ, et al. Effects of mechanical stretching on trabecular matrix metalloproteinases. Invest Ophthalmol Vis Sci. 2001;42:1505–1513. [PubMed] [Google Scholar]
- Bradley JMB, Vranka JA, et al. Effects of matrix metalloproteinase activity on outflow in perfused human organ culture. Invest Ophthalmol Vis Sci. 1998;39:2649–2658. [PubMed] [Google Scholar]
- Brilakis HS, Hann CR, et al. A comparison of different embedding media on the ultrastructure of the trabecular meshwork. Curr Eye Res. 2001;22:235–244. doi: 10.1076/ceyr.22.3.235.5520. [DOI] [PubMed] [Google Scholar]
- Brubaker RF. The measurement of pseudofacility and true facility by constant pressure perfusion in the normal rhesus monkey eye. Invest Ophthalmol. 1970;9:42–52. [PubMed] [Google Scholar]
- Brubaker RF. Flow of aqueous humor in humans. Invest Ophthalmol Vis Sci. 1991;32:3145–3166. [PubMed] [Google Scholar]
- Bruns RR, Press W, et al. Type VI collagen in extracellular, 100-nm periodic filaments and fibrils: identification by immunoelectron microscopy. J Cell Biol. 1986;103:393–404. doi: 10.1083/jcb.103.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller C, Johnson D. Segmental variability of the trabecular meshwork in normal and glaucomatous eyes. Invest Ophthalmol Vis Sci. 1994;35:3841–3851. [PubMed] [Google Scholar]
- Chen Y, Kelley MJ, et al. DNA microarray analysis of gene expression in trabecular meshwork cells in response to TNFα and IL-1α. Invest Ophthalmol Vis Sci. 2005;46 doi: 10.1167/iovs.05-0075. (Abstract), #1349. [DOI] [PubMed] [Google Scholar]
- Chiquet-Ehrismann R. Tenascins. Int J Biochem Cell Biol. 2004;36:986–990. doi: 10.1016/j.biocel.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Chiquet-Ehrismann R, Chiquet M. Tenascins: regulation and putative functions during pathological stress. J Pathol. 2003;200:488–499. doi: 10.1002/path.1415. [DOI] [PubMed] [Google Scholar]
- Chiquet-Ehrismann R, Kalla P, et al. Tenascin interferes with fibronectin action. Cell. 1988;53:383–390. doi: 10.1016/0092-8674(88)90158-4. [DOI] [PubMed] [Google Scholar]
- Choi J, Miller AM, et al. Soluble CD44 is cytotoxic to trabecular meshwork and retinal ganglion cells in vitro. Invest Ophthalmol Vis Sci. 2005;46:214–222. doi: 10.1167/iovs.04-0765. [DOI] [PubMed] [Google Scholar]
- Chu ML, Pan TC, et al. Sequence analysis of alpha 1(VI) and alpha 2(VI) chains of human type VI collagen reveals internal triplication of globular domains similar to the A domains of von Willebrand factor and two alpha 2(VI) chain variants that differ in the carboxy terminus. EMBO J. 1989;8:1939–1946. doi: 10.1002/j.1460-2075.1989.tb03598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Zardi L, et al. Binding of tenascin-C to soluble fibronectin and matrix fibrils. J Biol Chem. 1995;270:29012–29017. doi: 10.1074/jbc.270.48.29012. [DOI] [PubMed] [Google Scholar]
- Clark AF, Brotchie D, et al. Dexamethasone alters F-actin architecture and promotes cross-linked actin network formation in human trabecular meshwork tissue. Cell Motil Cytoskeleton. 2005;60:83–95. doi: 10.1002/cm.20049. [DOI] [PubMed] [Google Scholar]
- Clark AF, Miggans ST, et al. Cytoskeletal changes in cultured human glaucoma trabecular meshwork cells. J Glaucoma. 1995a;4:183–188. [PubMed] [Google Scholar]
- Clark AF, Wilson K, et al. Dexamethasone-induced ocular hypertension in perfusion-cultured human eyes. Invest Ophthalmol Vis Sci. 1995b;36:478–489. [PubMed] [Google Scholar]
- Clark AF, Wilson K, et al. Glucocorticoid-induced formation of cross-linked actin networks in cultured human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 1994;35:281–294. [PubMed] [Google Scholar]
- Couchman JR. Syndecans: proteoglycan regulators of cell-surface microdomains? Nat Rev Mol Cell Biol. 2003;4:926–937. doi: 10.1038/nrm1257. [DOI] [PubMed] [Google Scholar]
- Couchman JR, Chen L, et al. Syndecans and cell adhesion. Int Rev Cytol. 2001;207:113–150. doi: 10.1016/s0074-7696(01)07004-8. [DOI] [PubMed] [Google Scholar]
- Couchman JR, Woods A. Syndecan-4 and integrins: combinatorial signaling in cell adhesion. J Cell Sci. 1999;112(Pt 20):3415–3420. doi: 10.1242/jcs.112.20.3415. [DOI] [PubMed] [Google Scholar]
- Crawford SE, Stellmach V, et al. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998;93:1159–1170. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- De Cat B, David G. Developmental roles of the glypicans. Semin Cell Dev Biol. 2001;12:117–125. doi: 10.1006/scdb.2000.0240. [DOI] [PubMed] [Google Scholar]
- Denhardt DT, Guo X. Osteopontin: a protein with diverse functions. FASEB J. 1993;7:1475–1482. [PubMed] [Google Scholar]
- Diskin S, Kumar J, et al. Detection of differentially expressed glycogenes in trabecular meshwork of eyes with primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2006;47:1491–1499. doi: 10.1167/iovs.05-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellingsen BA, Grant WM. Trabeculotomy and sinusotomy in enucleated human eyes. Invest Ophthalmol. 1972;11:21–28. [PubMed] [Google Scholar]
- Engvall E, Hessle H, et al. Molecular assembly, secretion, and matrix deposition of type VI collagen. J Cell Biol. 1986;102:703–710. doi: 10.1083/jcb.102.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein D, Anderson P. The Biochemistry of Outflow Mechanisms. In: Drance S, editor. Glaucoma: Applied Pharmacology in Medical Treatment. New York: Grune & Stratton; 1984. pp. 135–150. [Google Scholar]
- Epstein D, Rowlette L-L, et al. Acto-myosin drug effects and aqueous outflow function. Invest Ophthalmol Vis Sci. 1999;40:74–81. [PubMed] [Google Scholar]
- Epstein DL, Hashimoto JM, et al. Effect of iodoacetamide perfusion on outflow facility and metabolism of the trabecular meshwork. Invest Ophthalmol Vis Sci. 1981;20:625–631. [PubMed] [Google Scholar]
- Epstein DL, Rohen JW. Morphology of the trabecular meshwork and inner-wal endothelium after cationized ferritin perfusioin in the monkey eye. Invest Ophthalmol Vis Sci. 1991;32:160–171. [PubMed] [Google Scholar]
- Ethier CR. The inner wall of Schlemm's canal. Exp Eye Res. 2002;74:161–172. doi: 10.1006/exer.2002.1144. [DOI] [PubMed] [Google Scholar]
- Ethier CR, Chan D-H. Cationic ferritin changes outflow facility in human eyes whereas anionic ferritin does not. Invest Ophthalmol Vis Sci. 2001;42:1795–1802. [PubMed] [Google Scholar]
- Ethier CR, Kamm RD, et al. Calculations of flow resistance in the juxtacanalicular meshwork. Invest Ophthalmol Vis Sci. 1986;27:1741–1750. [PubMed] [Google Scholar]
- Ethier CR, Read AT, et al. Biomechanics of Schlemm's canal endothelial cells: influence on F-actin architecture. Biophys J. 2004;87:2828–2837. doi: 10.1529/biophysj.103.038133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fautsch MP, Bahler CK, et al. Perfusion of his-tagged eukaryotic myocilin increases outflow resistance in human anterior segments in the presence of aqueous humor. Invest Ophthalmol Vis Sci. 2006;47:213–221. doi: 10.1167/iovs.05-0334. [DOI] [PubMed] [Google Scholar]
- Fautsch MP, Johnson DH. Aqueous humor outflow: what do we know? Where will it lead us? Invest Ophthalmol Vis Sci. 2006;47:4181–4187. doi: 10.1167/iovs.06-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filla MS, David G, et al. Distribution of syndecans 1–4 within the anterior segment of the human eye: expression of a variant syndecan-3 and matrix-associated syndecan-2. Exp Eye Res. 2004;79:61–74. doi: 10.1016/j.exer.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Filla MS, Liu X, et al. In vitro localization of TIGR/MYOC in trabecular meshwork extracellular matrix and binding to fibronectin. Invest Ophthalmol Vis Sci. 2002;43:151–161. [PubMed] [Google Scholar]
- Filla MS, Woods A, et al. Beta1 and beta3 integrins cooperate to induce syndecan-4-containing cross-linked actin networks in human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2006;47:1956–1967. doi: 10.1167/iovs.05-0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filmus J, Selleck SB. Glypicans: proteoglycans with a surprise. J Clin Invest. 2001;108:497–501. doi: 10.1172/JCI13712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald J, Morgelin M, et al. The N-terminal N5 subdomain of the alpha 3(VI) chain is important for collagen VI microfibril formation. J Biol Chem. 2001;276:187–193. doi: 10.1074/jbc.M008173200. [DOI] [PubMed] [Google Scholar]
- Fleenor DL, Shepard AR, et al. TGFbeta2-induced changes in human trabecular meshwork: implications for intraocular pressure. Invest Ophthalmol Vis Sci. 2006;47:226–234. doi: 10.1167/iovs.05-1060. [DOI] [PubMed] [Google Scholar]
- Floyd BB, Cleveland PH, et al. Fibronectin in human trabecular drainage channels. Invest Ophthalmol Vis Sci. 1985;26:797–804. [PubMed] [Google Scholar]
- Flugel-Koch C, Ohlmann A, et al. Thrombospondin-1 in the trabecular meshwork: localization in normal and glaucomatous eyes, and induction by TGF-beta 1 and dexamethasone in vitro. Exp Eye Res. 2004;79:649–663. doi: 10.1016/j.exer.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Francois J. The importance of the mucopolysaccharides in intraocular pressure regulation. Invest Ophthalmol. 1975;14:173–176. [PubMed] [Google Scholar]
- Friedman JS, Ducharme R, et al. Isolation of a novel iris-specific and leucine-rich repeat protein (oculoglycan) using differential selection. Invest Ophthalmol Vis Sci. 2000;41:2059–2066. [PubMed] [Google Scholar]
- Friedman JS, Faucher M, et al. Protein localization in the human eye and genetic screen of opticin. Hum Mol Genet. 2002;11:1333–1342. doi: 10.1093/hmg/11.11.1333. [DOI] [PubMed] [Google Scholar]
- Fuchshofer R, Welge-Lussen U, et al. Biochemical and morphological analysis of basement membrane component expression in corneoscleral and cribriform human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2006;47:794–801. doi: 10.1167/iovs.05-0292. [DOI] [PubMed] [Google Scholar]
- Fuchshofer R, Yu AH, et al. Bone morphogenetic protein-7 is an antagonist of transforming growth factor-beta2 in human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2007;48:715–726. doi: 10.1167/iovs.06-0226. [DOI] [PubMed] [Google Scholar]
- Funderburgh JL. Keratan sulfate biosynthesis. IUBMB Life. 2002;54:187–194. doi: 10.1080/15216540214932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A. Assembly and mechanosensory function of focal contacts. Curr Opin Cell Biol. 2001;13:584–592. doi: 10.1016/s0955-0674(00)00255-6. [DOI] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A, et al. Transmembrane extracellular matrix-cytoskeleton crosstalk. Nature Mol Cell Biol. 2001;2:793–806. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A, et al. Transmembrane crosstalk between the extracellular matrix--cytoskeleton crosstalk. Nat Rev Mol Cell Biol. 2001;2:793–805. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- Gersdorff N, Otto S, et al. The absence of one or both nidogens does not alter basement membrane composition in adult murine kidney. Histol Histopathol. 2007;22:1077–1084. doi: 10.14670/HH-22.1077. [DOI] [PubMed] [Google Scholar]
- Ghert MA, Qi WN, et al. Tenascin-C splice variant adhesive/anti-adhesive effects on chondrosarcoma cell attachment to fibronectin. Cell Struct Funct. 2001;26:179–187. doi: 10.1247/csf.26.179. [DOI] [PubMed] [Google Scholar]
- Gillan L, Matei D, et al. Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha(V)beta(3) and alpha(V)beta(5) integrins and promotes cell motility. Cancer Res. 2002;62:5358–5364. [PubMed] [Google Scholar]
- Goldwich A, Ethier CR, et al. Perfusion with the olfactomedin domain of myocilin does not affect outflow facility. Invest Ophthalmol Vis Sci. 2003;44:1953–1961. doi: 10.1167/iovs.02-0863. [DOI] [PubMed] [Google Scholar]
- Gong H, Freddo TF, et al. Age-related changes of sulfated proteoglycans in the normal human trabecular meshwork. Exp Eye Res. 1992;55:691–709. doi: 10.1016/0014-4835(92)90174-q. [DOI] [PubMed] [Google Scholar]
- Gong H, Overby D, et al. Human outflow pathway viewed by quick freeze deep etch. Invest Ophthalmol Vis Sci. 2001;42:S749. [Google Scholar]
- Gong H, Ruberti J, et al. A new view of the human trabecular meshwork using quick-freeze, deep-etch electron microscopy. Exp Eye Res. 2002;75:347–358. [PubMed] [Google Scholar]
- Gong H, Trinkaus-Randall V, et al. Ultrastructural immunoctochemical localization of elastin in normal human trabecular meshwork. Cur Eye Res. 1989;8:1071–1082. doi: 10.3109/02713688908997400. [DOI] [PubMed] [Google Scholar]
- Gonzalez P, Epstein DL, et al. Genes upregulated in the human trabecular meshwork in response to elevated intraocular pressure. Invest Ophthalmol Vis Sci. 2000;41:352–361. [PubMed] [Google Scholar]
- Gottanka J, Chan D, et al. Effects of TGF-beta2 in perfused human eyes. Invest Ophthalmol Vis Sci. 2004;45:153–158. doi: 10.1167/iovs.03-0796. [DOI] [PubMed] [Google Scholar]
- Gottanka J, Johnson DH, et al. Beta-adrenertic blocker therapy and the trabecular meshwork. Graefes Arch Clin Exp Ophthalmol. 2001;239:138–144. doi: 10.1007/s004170000231. [DOI] [PubMed] [Google Scholar]
- Grant SM. Further studies on facility of flow through the trabecular meshwork. Arch Ophthalmol. 1958;60:523–533. doi: 10.1001/archopht.1958.00940080541001. [DOI] [PubMed] [Google Scholar]
- Grant WM. Facility of flow through the trabecular meshwork. Arch Ophthalmol. 1956;54:245–248. doi: 10.1001/archopht.1955.00930020251012. [DOI] [PubMed] [Google Scholar]
- Grant WM. Experimental aqueous perfusion in enucleated human eyes. Arch Ophthalmol. 1963;69:783–801. doi: 10.1001/archopht.1963.00960040789022. [DOI] [PubMed] [Google Scholar]
- Grierson I, Calthorpe CM. Proceedings of the Fourth Iinternational Symposium of the Northern Eye Institute. K. B. Mills, Oxford: Pergamon Press; Characteristics of meshwork cells and age changes in the outflow system of the eye: Their relevance to primary open-angle glaucoma. Glaucoma; pp. 12–31. [Google Scholar]
- Grierson I, Lee WR. Junctions between the cells of the trabecular meshwork. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1974;192:89–104. doi: 10.1007/BF00410696. [DOI] [PubMed] [Google Scholar]
- Grierson I, Lee WR. Acid mucopolysaccharides in the outflow apparatus. Experimental Eye Research. 1975;21:417–431. doi: 10.1016/0014-4835(75)90124-4. [DOI] [PubMed] [Google Scholar]
- Grierson I, Lee WR, et al. Associations between the cells of the walls of Schlemm's canal. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1978;208:33–47. doi: 10.1007/BF00406980. [DOI] [PubMed] [Google Scholar]
- Hann CR, Bahler CK, et al. Cationic ferritin and segmental flow through the trabecular meshwork. Invest Ophthalmol Vis Sci. 2005;46:1–7. doi: 10.1167/iovs.04-0800. [DOI] [PubMed] [Google Scholar]
- Hann CR, Johnson DH. Trabecular meshwork pigmentation and collector channels - areas of higher flow? Invest Ophthalmol Vis Sci. 2007 ARVO e-Abstracts, #2053. [Google Scholar]
- Hartwig S, Hu MC, et al. Glypican-3 modulates inhibitory Bmp2-Smad signaling to control renal development in vivo. Mech Dev. 2005;122:928–938. doi: 10.1016/j.mod.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Heimark RL, Kaochar S, et al. Human Schlemm's canal cells express the endothelial adherens proteins, VE-cadherin and PECAM-1. Curr Eye Res. 2002;25:299–308. doi: 10.1076/ceyr.25.5.299.13495. [DOI] [PubMed] [Google Scholar]
- Hernandez MR, Gong H. Extracellular matrix of the trabecular meshwork and optic nerve head. St. Louis: Mosby-Year Book, Inc.; 1996. [Google Scholar]
- Hessle H, Engvall E. Type VI collagen. Studies on its localization, structure, and biosynthetic form with monoclonal antibodies. J Biol Chem. 1984;259:3955–3961. [PubMed] [Google Scholar]
- Hogan MJ, Alvarado JA, et al. An atlas and textbook. Philadelphia: W. B. Saunders Company; 1971. Histology of the human eye. [Google Scholar]
- Hubbard W, Johnson M, et al. Intraocular pressure and outflow facility are unchanged following acute and chronic intracameral chondroitinase ABC and hyaluronidase in monkeys. Exp Eye Res. 1997;65:177–190. doi: 10.1006/exer.1997.0319. [DOI] [PubMed] [Google Scholar]
- Inomata H, Bill A, et al. Aqueous humor pathways through the trabecular meshwork and into Schlemm's canal in the cynomolgus monkey (Macaca irus): An electron microscopic study. Am J Ophthalmol. 1972;73:760. doi: 10.1016/0002-9394(72)90394-7. [DOI] [PubMed] [Google Scholar]
- Ishibashi T, Takagi Y, et al. cDNA microarray analysis of gene expression changes induced by dexamethasone in cultured human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2002;43:3691–3697. [PubMed] [Google Scholar]
- Isogai Z, Aspberg A, et al. Versican interacts with fibrillin-1 and links extracellular microfibrils to other connective tissue networks. J Biol Chem. 2002;277:4565–4572. doi: 10.1074/jbc.M110583200. [DOI] [PubMed] [Google Scholar]
- Itano N, Kimata K. Mammalian hyaluronan synthases. IUBMB Life. 2002;54:195–199. doi: 10.1080/15216540214929. [DOI] [PubMed] [Google Scholar]
- Johnson D. The effect of cytochalasin D on outflow facility and the trabecular meshwork of the human eye in perfusion organ culture. Invest Ophthalmol Vis Sci. 1997;38:2790–2799. [PubMed] [Google Scholar]
- Johnson D, Bahler C. Heparitinase increases outflow facility in the human eye. Invest Ophthalmol Vis Sci. 1999;40:S504. Abstract. [Google Scholar]
- Johnson D, Gottanka J, et al. Ultrastructural changes in the trabecular meshwork of human eyes treated with corticosteroids. Arch Ophthalmol. 1997;115:375–383. doi: 10.1001/archopht.1997.01100150377011. [DOI] [PubMed] [Google Scholar]
- Johnson M. 'What controls aqueous humour outflow resistance?'. Exp Eye Res. 2006;82:545–557. doi: 10.1016/j.exer.2005.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M, Erickson K. Mechanisms and routes of aqueous humor drainage. In: Albert D, Jakobiec F, editors. Principles and Practice of Ophthalmology. Philadelphia: W.B. Saunders; 2000. pp. 2577–2595. [Google Scholar]
- Johnson M, Shapiro A, et al. Modulation of outflow resistance by the pores of the inner wall endothelium. Invest Ophthalmol Vis Sci. 1992;33:1670–1675. [PubMed] [Google Scholar]
- Johnstone MA. The aqueous outflow system as a mechanical pump: evidence from examination of tissue and aqueous movement in human and non-human primates. J Glaucoma. 2004;13:421–438. doi: 10.1097/01.ijg.0000131757.63542.24. [DOI] [PubMed] [Google Scholar]
- Jumper MD, Stern R, et al. Expression of CD44 isoforms in cultured human trabecular meshwork cells. Ophthalmic Res. 1998;30:314–320. doi: 10.1159/000055490. [DOI] [PubMed] [Google Scholar]
- Kania AM, Reichenberger E, et al. Structural variation of type XII collagen at its carboxyl-terminal NC1 domain generated by tissue-specific alternative splicing. J Biol Chem. 1999;274:22053–22059. doi: 10.1074/jbc.274.31.22053. [DOI] [PubMed] [Google Scholar]
- Kaufman PL, Erickson KA. Cytochalasin B and D dose-outflow facility response relationships in the cynomolgus monkey. Invest Ophthalmol Vis Sci. 1982;23:646–650. [PubMed] [Google Scholar]
- Keller KE, Bradley JM, et al. Effect of modifiers of glycosaminoglycan biosynthesis on outflow facility in perfusion culture. Invest Ophthalmol Vis Sci. 2007a;48 doi: 10.1167/iovs.07-0903. e-Abstract #2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller KE, Kelley MJ, et al. Extracellular matrix gene alternative splicing by trabecular meshwork cells in response to mechanical stretching. Invest Ophthalmol Vis Sci. 2007b;48:1164–1172. doi: 10.1167/iovs.06-0875. [DOI] [PubMed] [Google Scholar]
- Kelley MJ, Rose AY, et al. Synergism of TNF and IL-1 in the induction of matrix metalloproteinase-3 in trabecular meshwork. Invest Ophthalmol Vis Sci. 2007;48:2634–2643. doi: 10.1167/iovs.06-1445. [DOI] [PubMed] [Google Scholar]
- Kenagy RD, Plaas AH, et al. Versican degradation and vascular disease. Trends Cardiovasc Med. 2006;16:209–215. doi: 10.1016/j.tcm.2006.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielty CM, Baldock C, et al. Fibrillin: from microfibril assembly to biomechanical function. Philos Trans R Soc Lond B Biol Sci. 2002a;357:207–217. doi: 10.1098/rstb.2001.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielty CM, Cummings C, et al. Isolation and ultrastructural analysis of microfibrillar structures from foetal bovine elastic tissues. Relative abundance and supramolecular architecture of type VI collagen assemblies and fibrillin. J Cell Sci. 1991;99(Pt 4):797–807. doi: 10.1242/jcs.99.4.797. [DOI] [PubMed] [Google Scholar]
- Kielty CM, Sherratt MJ, et al. Fibrillin microfibrils. Adv Protein Chem. 2005;70:405–436. doi: 10.1016/S0065-3233(05)70012-7. [DOI] [PubMed] [Google Scholar]
- Kielty CM, Wess TJ, et al. Fibrillin-rich microfibrils: elastic biopolymers of the extracellular matrix. J Muscle Res Cell Motil. 2002b;23:581–596. [PubMed] [Google Scholar]
- Kielty CM, Whittaker SP, et al. Attachment of human vascular smooth muscles cells to intact microfibrillar assemblies of collagen VI and fibrillin. J Cell Sci. 1992a;103(Pt 2):445–451. doi: 10.1242/jcs.103.2.445. [DOI] [PubMed] [Google Scholar]
- Kielty CM, Whittaker SP, et al. Type VI collagen microfibrils: evidence for a structural association with hyaluronan. J Cell Biol. 1992b;118:979–990. doi: 10.1083/jcb.118.4.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kii I, Amizuka N, et al. Periostin is an extracellular matrix protein required for eruption of incisors in mice. Biochem Biophys Res Commun. 2006;342:766–772. doi: 10.1016/j.bbrc.2006.02.016. [DOI] [PubMed] [Google Scholar]
- Knepper P, Goossens W, et al. CD44H localization in primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 1998;39:673–680. [PubMed] [Google Scholar]
- Knepper P, Mayanil C, et al. Aqueous humor in primary open-angle glaucoma contains increased levels of CD44S. Invest Ophthalmol Vis Sci. 2002;43:133–139. [PubMed] [Google Scholar]
- Knepper PA, Farbman AI, et al. Aqueous outflow pathway glycosaminoglycans. Exp Eye Res. 1981;32:265–277. doi: 10.1016/0014-4835(81)90032-4. [DOI] [PubMed] [Google Scholar]
- Knepper PA, Farbman AI, et al. Exogenous hyaluronidases and degradation of hyaluronic acid in the rabbit eye. Invest Ophthalmol Vis Sci. 1984;25:286–293. [PubMed] [Google Scholar]
- Knepper PA, Goossens W, et al. Glycosaminoglycans of the human trabecular meshwork in primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 1996a;37:1360–1367. [PubMed] [Google Scholar]
- Knepper PA, Goossens W, et al. CD44H localization in primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 1998;39:673–680. [PubMed] [Google Scholar]
- Knepper PA, Goossens W, et al. Glycosaminoglycan stratification of the juxtacanalicular tissue in normal and primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 1996b;37:2414–2425. [PubMed] [Google Scholar]
- Knepper PA, Mayanil CS, et al. Aqueous humor in primary open-angle glaucoma contains an increased level of CD44S. Invest Ophthalmol Vis Sci. 2002;43:133–139. [PubMed] [Google Scholar]
- Knepper PA, Miller AM, et al. Hypophosphorylation of aqueous humor sCD44 and primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2005;46:2829–2837. doi: 10.1167/iovs.04-1472. [DOI] [PubMed] [Google Scholar]
- Knupp C, Pinali C, et al. Structural correlation between collagen VI microfibrils and collagen VI banded aggregates. J Struct Biol. 2006;154:312–326. doi: 10.1016/j.jsb.2006.03.023. [DOI] [PubMed] [Google Scholar]
- Kreis T, Vale R, editors. Guidebook to the Extracellular Matrix, Anchor, and Adhesion Proteins. Oxford: Oxford University Press; 1999. [Google Scholar]
- Lerner LE, Polansky JR, et al. Hyaluronan in the human trabecular meshwork. Invest Ophthalmol Vis Sci. 1997;38:1222–1228. [PubMed] [Google Scholar]
- Li J, Tripathi BJ, et al. Modulation of pre-mRNA splicing and protein production of fibronectin by TGF-beta2 in porcine trabecular cells. Invest Ophthalmol Vis Sci. 2000;41:3437–3443. [PubMed] [Google Scholar]
- Linsenmayer TF. Collagen. In: Hay ED, editor. Cell Biology of Extracellular Matrix. New York: Plenum Press; 1991. pp. 7–44. [Google Scholar]
- Liton PB, Luna C, et al. Genome-wide expression profile of human trabecular meshwork cultured cells, nonglaucomatous and primary open angle glaucoma tissue. Mol Vis. 2006;12:774–790. [PMC free article] [PubMed] [Google Scholar]
- Litvin J, Selim AH, et al. Expression and function of periostin-isoforms in bone. J Cell Biochem. 2004;92:1044–1061. doi: 10.1002/jcb.20115. [DOI] [PubMed] [Google Scholar]
- Liu X, Wu Z, et al. Low dose latrunculin-A inhibits dexamethasone-induced changes in the actin cytoskeleton and alters extracellular matrix protein expression in cultured human trabecular meshwork cells. Exp Eye Res. 2003;77:181–188. doi: 10.1016/s0014-4835(03)00118-0. [DOI] [PubMed] [Google Scholar]
- Lo WR, Rowlette LL, et al. Tissue differential microarray analysis of dexamethasone induction reveals potential mechanisms of steroid glaucoma. Invest Ophthalmol Vis Sci. 2003;44:473–485. doi: 10.1167/iovs.02-0444. [DOI] [PubMed] [Google Scholar]
- Lütjen-Drecoll E. Functional morphology of the trabecular meshwork in primate eyes. Prog Retin Eye Res. 1999;18:91–119. doi: 10.1016/s1350-9462(98)00011-1. [DOI] [PubMed] [Google Scholar]
- Lütjen-Drecoll E, Futa R, et al. Ultrahistochemical studies on tangential sections of the trabecular meshwork in normal and glaucomatous eyes. Invest Ophthalmol Vis Sci. 1981;21:563–573. [PubMed] [Google Scholar]
- Lütjen-Drecoll E, Rittig M, et al. Immunomicroscopical study of type VI collagen in the trabecular meshwork of normal and glaucomatous eyes. Exp Eye Res. 1989;48:139–147. doi: 10.1016/0014-4835(89)90027-4. [DOI] [PubMed] [Google Scholar]
- Lütjen-Drecoll E, Rohen JW. Anatomy of aqueous humor formation and drainage. In: Podos SM, Yanoff M, editors. Textbook of Ophthalmology. Vol. 7. London: Mosby; 1994. pp. 1.1–1.16. [Google Scholar]
- Lütjen-Drecoll E, Rohen JW. Functional Morphology of the Trabecular Meshwork. In: Tasman W, Jaeger EA, editors. Duane's Foundations of Clinical Ophthalmology. Philadelphia: J.B. Lippincott Company; 2001. pp. 1–30. [Google Scholar]
- Lütjen-Drecoll E, Rohen JW. Morphology of aqueous outflow pathways in normal and glaucomatous eyes. In: Ritch R, Shields MB, Krupin T, editors. The Glaucomas. I. St. Louis: Mosby-Year Book, Inc.; 1996. pp. 89–124. [Google Scholar]
- Lütjen-Drecoll E, Schenholm M, et al. Visualization of hyaluronic acid in the anterior segment of rabbit and monkey eyes. Exp Eye Res. 1990;51:55–63. doi: 10.1016/0014-4835(90)90170-y. [DOI] [PubMed] [Google Scholar]
- Lütjen-Drecoll E, Shimizu T, et al. Quantitative analysis of "plaque material" in the inner- and outer wall of Schlemm's canal in normal and glaucomatous eyes. Experimental Eye Research. 1986;42:443–455. doi: 10.1016/0014-4835(86)90004-7. [DOI] [PubMed] [Google Scholar]
- Lütjen-Drecoll E, Tamm E, editors. Glaucoma Update III. Berlin Heidelberg: Springer-Verlag; 1987. Differences in the amount of 'plaque-material' in the outflow system of eyes with chronic simple and exfoliation glaucoma. [Google Scholar]
- Maepea O, Bill A. Pressures in the juxtacanalicular tissue and Schlemm's canal in monkeys. Exp Eye Res. 1992;54:879–883. doi: 10.1016/0014-4835(92)90151-h. [DOI] [PubMed] [Google Scholar]
- Mao Y, Schwarzbauer JE. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol. 2005;24:389–399. doi: 10.1016/j.matbio.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Marshall GE, Konstas AG, et al. Immunogold localization of type IV collagen and laminin in the aging human outflow system. Exp Eye Res. 1990;51:691–699. doi: 10.1016/0014-4835(90)90054-x. [DOI] [PubMed] [Google Scholar]
- Marshall GE, Konstas AGP, et al. Immunogold ultrastructural localization of collagens in the aged human outflow system. Ophthalmology. 1991;98:692–700. doi: 10.1016/s0161-6420(91)32232-2. [DOI] [PubMed] [Google Scholar]
- McEwen WK. Application of Poiseuille's law to aqueous outflow. Arch Ophthalmol. 1958;60:290–294. doi: 10.1001/archopht.1958.00940080306017. [DOI] [PubMed] [Google Scholar]
- Morrison JC, Acott TS. Anatomy and Physiology of Aqueous Humor Outflow. In: Morrison JC, Pollack IP, editors. Glaucoma: Science and Practice. New York: Thieme; 2003. pp. 34–41. [Google Scholar]
- Murphy CG, Yun AJ, et al. Localization of extracellular proteins of the human trabecular meshwork by indirect immunofluorescence. Am J Ophthalmol. 1987;104:33–43. doi: 10.1016/0002-9394(87)90290-x. [DOI] [PubMed] [Google Scholar]
- O'Regan A, Berman JS. Osteopontin: a key cytokine in cell-mediated and granulomatous inflammation. Int J Exp Pathol. 2000;81:373–390. doi: 10.1046/j.1365-2613.2000.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olin AI, Morgelin M, et al. The proteoglycans aggrecan and Versican form networks with fibulin-2 through their lectin domain binding. J Biol Chem. 2001;276:1253–1261. doi: 10.1074/jbc.M006783200. [DOI] [PubMed] [Google Scholar]
- Overby D, Gong H, et al. The mechanism of increasing outflow facility during washout in the bovine eye. Invest Ophthalmol Vis Sci. 2002;43:3455–3464. [PubMed] [Google Scholar]
- Parc C, Johnson D, et al. Giant vacuoles are found preferentially near collector channels. Invest Ophthalmol Vis Sci. 2000;41:2984–2990. [PubMed] [Google Scholar]
- Parks W, Mecham R, editors. Matrix Metalloproteinases. San Diego: Academic Press; 1998. [Google Scholar]
- Parshley DE, Bradley JMB, et al. Laser trabeculoplasty induces stromelysin expression by trabecular juxtacanalicular cells. Invest Ophthalmol Vis Sci. 1996;37:795–804. [PubMed] [Google Scholar]
- Parshley DE, Bradley JMB, et al. Early changes in matrix metalloproteinases and inhibitors after in vivo laser treatment to the trabecular meshwork. Cur Eye Res. 1995;14:537–544. doi: 10.3109/02713689508998400. [DOI] [PubMed] [Google Scholar]
- Pedler C. The relationship of hyaluronidase to aqueous outflow resistance. Trans Opthal Soc U K. 1956;76:51–63. [PubMed] [Google Scholar]
- Peterson JA, Sheibani N, et al. Heparin II domain of fibronectin uses alpha4beta1 integrin to control focal adhesion and stress fiber formation, independent of syndecan-4. J Biol Chem. 2005;280:6915–6922. doi: 10.1074/jbc.M406625200. [DOI] [PubMed] [Google Scholar]
- Peterson JA, Tian B, et al. In cynomolgus monkeys Latrunculin (LAT)-A, LAT-B increase outflow facility, LQT-A decreased intraocular pressure and initially increases aqueous humor formation. Invest Ophthalmol Vis Sci. 1997;38 Suppl:S243. [Google Scholar]
- Peterson WS, Jocson VL. Hyaluronidase effects on aqueous outflow resistance. Quantitative and localizaing studies in the rhesus monkey eye. Am J Ophthalmol. 1974;77:573–577. doi: 10.1016/0002-9394(74)90473-5. [DOI] [PubMed] [Google Scholar]
- Polansky JR, Fauss DJ, et al. Cellular pharmacology and molecular biology of the trabecular meshwork inducible glucocorticoid response gene product. Ophthalmologica. 211;1997:126–139. doi: 10.1159/000310780. [DOI] [PubMed] [Google Scholar]
- Polansky JR, Fauss DJ, et al. Regulation of TIGR/MYOC gene expression in human trabecular meshwork cells. Eye. 2000;14(Pt 3B):503–514. doi: 10.1038/eye.2000.137. [DOI] [PubMed] [Google Scholar]
- Polansky JR, Nguyen TD. The TIGR gene, pathogenic mechanisms, and other recent advances in glaucoma genetics. Curr Opin Ophthalmol. 1998;9:15–23. doi: 10.1097/00055735-199804000-00004. [DOI] [PubMed] [Google Scholar]
- Quigley HA. Open-angle glaucoma. New England J Med. 1993;328:1097–1106. doi: 10.1056/NEJM199304153281507. [DOI] [PubMed] [Google Scholar]
- Quigley HA. Number of people with glaucoma worldwide. Brit J Ophthalmol. 1996;80:389–393. doi: 10.1136/bjo.80.5.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA, Vitale S. Models of open-angle glaucoma prevalence and incidence in the United States. Invest Ophthalmol Vis Sci. 1997;38:83–91. [PubMed] [Google Scholar]
- Rao P, Deng P-F, et al. Modulation of aqueous humor outflow facility by the Rho kinase-specific inhibitor Y-27632. Invest Ophthalmol Vis Sci. 2001;42:1029–1037. [PubMed] [Google Scholar]
- Rao PV, Deng P, et al. Expression of dominant negative Rho-binding domain of Rho-kinase in organ cultured human eye anterior segments increases aqueous humor outflow. Mol Vis. 2005;11:288–297. [PubMed] [Google Scholar]
- Raviola G, Raviola E. Paracellular route of aqueous outflow in the trabecular meshwork and canal of Schlemm. A freeze-fracture study of the endothelial junctions in the sclerocorneal angel of the macaque monkey eye. Invest Ophthalmol Vis Sci. 1981;21:52–72. [PubMed] [Google Scholar]
- Reale E, Groos S, et al. In the mammalian eye type VI collagen tetramers form three morphologically different aggregates. Matrix Biol. 2001;20:37–51. doi: 10.1016/s0945-053x(00)00132-3. [DOI] [PubMed] [Google Scholar]
- Rhee DJ, Fariss RN, et al. The matricellular protein SPARC is expressed in human trabecular meshwork. Exp Eye Res. 2003;77:601–607. doi: 10.1016/s0014-4835(03)00190-8. [DOI] [PubMed] [Google Scholar]
- Richardson TM. Distribution of glycosaminoglycans in the aqueous outflow system of the cat. Invest Ophthalmol Vis Sci. 1982;22:319–329. [PubMed] [Google Scholar]
- Rohen JW. Why is intraocular pressure elevated in chronic simple glaucoma? Ophthalmology. 1983;90:758–765. doi: 10.1016/s0161-6420(83)34492-4. [DOI] [PubMed] [Google Scholar]
- Rohen JW, Futa R, et al. The fine structure of the cribriform meshwork in normal and glaucomatous eyes as seen in tangential sections. Invest Ophthalmol Vis Sci. 1981;21:574–585. [PubMed] [Google Scholar]
- Rohen JW, Lutjen-Drecoll E. Biology of the trabecular meshwork. Stuttgard: F. K. Schattauer, Verlap; 1982. [Google Scholar]
- Rohen JW, Lütjen-Drecoll E. Ageing- and non-ageing processes within the connective tissues of the anterior segment of the eye. Weisbaden: Steiner Verlag; 1981. [Google Scholar]
- Rohen JW, Lutjen-Drecoll E, et al. Ultrastructure of the trabecular meshwork in untreated cases of primary open-angle glaucoma (POAG) Exp Eye Res. 1993;56:683–692. doi: 10.1006/exer.1993.1085. [DOI] [PubMed] [Google Scholar]
- Rohen JW, Schachtschabel DO, et al. Histoautoradiographic and biochemical studies on human and monkey trabecular meshwork and ciliary body in short-term explant culture. Graefes Arch Clin Exp Ophthalmol. 1984;221:199–206. doi: 10.1007/BF02134140. [DOI] [PubMed] [Google Scholar]
- Rosenquist R, Epstein D, et al. Outflow resistance of enucleated human eyes at two different perfusion pressures and different extents of trabeculotomy. Curr Eye Res. 1989;8:1233–1240. doi: 10.3109/02713688909013902. [DOI] [PubMed] [Google Scholar]
- Sabanay I, Gabelt B, et al. H-7 effects on the structure and fluid conductance of monkey trabecular meshwork. Arch Ophthalmol. 2000;118:955–962. [PubMed] [Google Scholar]
- Sakai LY, Keene DR, et al. Purification and partial characterization of fibrillin, a cysteine-rich structural component of connective tissue microfibrils. J Biol Chem. 1991;266:14763–14770. [PubMed] [Google Scholar]
- Samples JR, Alexander JP, et al. Regulation of the levels of human trabecular matrix metalloproteinases and inhibitor by interleukin-1 and dexamethasone. Invest Ophthalmol Vis Sci. 1993;34:3386–3395. [PubMed] [Google Scholar]
- Sanka K, Maddala R, et al. Influence of actin cytoskeletal integrity on matrix metalloproteinase-2 activation in cultured human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2007;48:2105–2114. doi: 10.1167/iovs.06-1089. [DOI] [PubMed] [Google Scholar]
- Santas AJ, Bahler C, et al. Effect of heparin II domain of fibronectin on aqueous outflow in cultured anterior segments of human eyes. Invest Ophthalmol Vis Sci. 2003;44:4796–4804. doi: 10.1167/iovs.02-1083. [DOI] [PubMed] [Google Scholar]
- Santas AJ, Peterson JA, et al. Alternative splicing of the IIICS domain in fibronectin governs the role of the heparin II domain in fibrillogenesis and cell spreading. J Biol Chem. 2002;277:13650–13658. doi: 10.1074/jbc.M111361200. [DOI] [PubMed] [Google Scholar]
- Sawaguchi S, Lam TT, et al. Effects of glycosaminoglycan-degrading enzymes on bovine trabecular meshwork in organ culture. J Glaucoma. 1993;2:80–86. [PubMed] [Google Scholar]
- Sawaguchi S, Yue B, et al. Effects of intracameral injection of chondroitinase ABC in vivo. Arch Ophthalmol. 1992;110:110–117. doi: 10.1001/archopht.1992.01080130112037. [DOI] [PubMed] [Google Scholar]
- Schachtschabel DO, Bigalke B, et al. Production of glycosaminoglycans by cell cultures of the trabecular meshwork of the primate eye. Exp Eye Res. 1977;24:71–80. doi: 10.1016/0014-4835(77)90286-x. [DOI] [PubMed] [Google Scholar]
- Schachtschabel DO, Rohen JW, et al. Synthesis and composition of glycosaminoglycans by cultured human trabecular meshwork cells. Graefe's Arch Clin Exp Ophthalmol. 1982;218:113–117. doi: 10.1007/BF02215647. [DOI] [PubMed] [Google Scholar]
- Schachtschabel U, Lindsey JD, et al. The mechanism of action of prostaglandins on uveoscleral outflow. Curr Opin Ophthalmol. 2000;11:112–115. doi: 10.1097/00055735-200004000-00008. [DOI] [PubMed] [Google Scholar]
- Scheele S, Nystrom A, et al. Laminin isoforms in development and disease. J Mol Med. 2007;85:825–836. doi: 10.1007/s00109-007-0182-5. [DOI] [PubMed] [Google Scholar]
- Segade F, Trask BC, et al. Identification of a matrix-binding domain in MAGP1 and MAGP2 and intracellular localization of alternative splice forms. J Biol Chem. 2002;277:11050–11057. doi: 10.1074/jbc.M110347200. [DOI] [PubMed] [Google Scholar]
- Segawa K. Electron microscopic changes of the trabecular tissue in primary open-angle glaucoma. Ann Ophthalmol. 1979;11:49–54. [PubMed] [Google Scholar]
- Si Z, Palkama A, et al. Distribution of thrombospondin-4 in the bovine eye. Curr Eye Res. 2003;27:165–173. doi: 10.1076/ceyr.27.3.165.16050. [DOI] [PubMed] [Google Scholar]
- Silbert JE, Sugumaran G. Biosynthesis of chondroitin/dermatan sulfate. IUBMB Life. 2002;54:177–186. doi: 10.1080/15216540214923. [DOI] [PubMed] [Google Scholar]
- Sodek J, Ganss B, et al. Osteopontin. Crit Rev Oral Biol Med. 2000;11:279–303. doi: 10.1177/10454411000110030101. [DOI] [PubMed] [Google Scholar]
- Song HH, Filmus J. The role of glypicans in mammalian development. Biochim Biophys Acta. 2002;1573:241–246. doi: 10.1016/s0304-4165(02)00390-2. [DOI] [PubMed] [Google Scholar]
- Song HH, Shi W, et al. The loss of glypican-3 induces alterations in Wnt signaling. J Biol Chem. 2005;280:2116–2125. doi: 10.1074/jbc.M410090200. [DOI] [PubMed] [Google Scholar]
- Specks U, Mayer U, et al. Structure of recombinant N-terminal globule of type VI collagen alpha 3 chain and its binding to heparin and hyaluronan. EMBO J. 1992;11:4281–4290. doi: 10.1002/j.1460-2075.1992.tb05527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steely HT, Browder SL, et al. The effects of dexamethasone on fibronectin expression in cultured human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 1992;33:2242–2250. [PubMed] [Google Scholar]
- Stone EM, Fingert JH, et al. Identification of a gene that causes primary open angle glaucoma. Scinece. 1997;275:668–670. doi: 10.1126/science.275.5300.668. [DOI] [PubMed] [Google Scholar]
- Sugahara K, Kitagawa H. Heparin and heparan sulfate biosynthesis. IUBMB Life. 2002;54:163–175. doi: 10.1080/15216540214928. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Anderson P. A temperature dependent action of fluoride on aqueous outflow facility of the calf eye. Curr Eye Res. 1993;12:1–7. doi: 10.3109/02713689308999489. [DOI] [PubMed] [Google Scholar]
- Tamm E, Flugel C, et al. Contractile cells in the human scleral spur. Exp Eye Res. 1992;54:531–543. doi: 10.1016/0014-4835(92)90132-c. [DOI] [PubMed] [Google Scholar]
- Tamm E, Flugel C, et al. Immunocytochemical and ultrastructural studies of human scleral spur. Characterization of a contractile cell population. Ophthalmologe. 1993;90:66–72. [PubMed] [Google Scholar]
- Tanihara H, Ohira A, et al. Localization and possible gene expression of proteoglycan decorin in the trabecular meshwork. Curr Eye Res. 1995;14:727–730. doi: 10.3109/02713689508998501. [DOI] [PubMed] [Google Scholar]
- Tawara A, Inomata H. Distribution and characterization of sulfated proteoglycans in the trabecular tissue of goniodysgenetic glaucoma. Am J Ophthalmol. 1994;117:741–755. doi: 10.1016/s0002-9394(14)70317-4. [DOI] [PubMed] [Google Scholar]
- Tawara A, Varner HH, et al. Distribution and characterization of sulfated proteoglycans in the human trabecular tissue. Invest Ophthalmol Vis Sci. 1989;30:2215–2231. [PubMed] [Google Scholar]
- Tian B, Kaufman PL, et al. H-7 disrupts the actin cytoskeleton and increases outflow facility. Arch Ophthalmol. 1998;116:633–643. doi: 10.1001/archopht.116.5.633. [DOI] [PubMed] [Google Scholar]
- Timpl R, Sasaki T, et al. Fibulins: a versatile family of extracellular matrix proteins. Nat Rev Mol Cell Biol. 2003;4:479–489. doi: 10.1038/nrm1130. [DOI] [PubMed] [Google Scholar]
- Trask BC, Trask TM, et al. The microfibrillar proteins MAGP-1 and fibrillin-1 form a ternary complex with the chondroitin sulfate proteoglycan decorin. Mol Biol Cell. 2000;11:1499–1507. doi: 10.1091/mbc.11.5.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trask TM, Ritty TM, et al. N-terminal domains of fibrillin 1 and fibrillin 2 direct the formation of homodimers: a possible first step in microfibril assembly. Biochem J. 1999;340(Pt 3):693–701. [PMC free article] [PubMed] [Google Scholar]
- Tripathi BJ, Hansen M, et al. Identification of type VI collagen in the trabecular meshwork and expression of its mRNA by trabecular cells. Exp Eye Res. 1994;58:181–187. doi: 10.1006/exer.1994.1006. [DOI] [PubMed] [Google Scholar]
- Tripathi BJ, Tripathi RC, et al. Synthesis of a thrombospondin-like cytoadhesion molecule by cells of the trabecular meshwork. Invest Ophthalmol Vis Sci. 1991;32:181–188. [PubMed] [Google Scholar]
- Tripathi RC. Mechanism of the aqueous outflow across the trabecular wall of Schlemm's canal. Exp Eye Res. 1971;11:116–121. doi: 10.1016/s0014-4835(71)80073-8. [DOI] [PubMed] [Google Scholar]
- Tripathi RC. The functional morphology of the outflow systems of ocular and cerebrospinal fluids. Exp Eye Res. 1977;25 Suppl:65–116. doi: 10.1016/s0014-4835(77)80010-9. [DOI] [PubMed] [Google Scholar]
- Tripathi RC, Li J, et al. Aqueous humor in glaucomatous eyes contains an increased level of TGF-beta 2. Exp Eye Res. 1994;59:723–727. doi: 10.1006/exer.1994.1158. [DOI] [PubMed] [Google Scholar]
- Tschumper RC, Johnson DH, et al. Glycosaminoglycans of human trabecular meshwork in perfusion organ culture. Curr Eye Res. 1990;9:363–369. doi: 10.3109/02713689008999624. [DOI] [PubMed] [Google Scholar]
- Tumminia SJ, Mitton KP, et al. Mechanical stretch alters the actin cytoskeletal network and signal transduction in human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 1998;39:1361–1371. [PubMed] [Google Scholar]
- Tumova S, Woods A, et al. Heparan sulfate chains from glypican and syndecans bind the Hep II domain of fibronectin similarly despite minor structural differences. J Biol Chem. 2000;275:9410–9417. doi: 10.1074/jbc.275.13.9410. [DOI] [PubMed] [Google Scholar]
- Ueda J, Wentz-Hunter K, et al. Distribution of myocilin and extracellular matrix components in the juxtacanalicular tissue of human eyes. Invest Ophthalmol Vis Sci. 2002;43:1068–1076. [PubMed] [Google Scholar]
- Ueda J, Yue BY. Distribution of myocilin and extracellular matrix components in the corneoscleral meshwork of human eyes. Invest Ophthalmol Vis Sci. 2003;44:4772–4779. doi: 10.1167/iovs.02-1002. [DOI] [PubMed] [Google Scholar]
- Umihira J, Nagata S, et al. Localization of elastin in the normal and glaucomatous human trabecular meshwork. Invest Ophthalmol Vis Sci. 1994;35:486–494. [PubMed] [Google Scholar]
- Vainio S, Jalkanen M, et al. Syndecan and tenascin expression is induced by epithelial-mesenchymal interactions in embryonic tooth mesenchyme. J Cell Biol. 1989;108:1945–1953. doi: 10.1083/jcb.108.5.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Buskirk EM, Brant WM. Influence of temparature and the question of involvement of cellular metabolism in aqueous outflow. Am J Ophthalmol. 1974;77:565–572. doi: 10.1016/0002-9394(74)90472-3. [DOI] [PubMed] [Google Scholar]
- Van Buskirk EM, Brett J. The canine eye: in vitro dissolution of the barriers to aqueous outflow. Invest Ophthalmol Vis Sci. 1978;17:258–271. [PubMed] [Google Scholar]
- Vittal V, Rose A, et al. Changes in gene expression by trabecular meshwork cells in response to mechanical stretching. Invest Ophthalmol Vis Sci. 2005;46:2857–2868. doi: 10.1167/iovs.05-0075. [DOI] [PubMed] [Google Scholar]
- Vittitow J, Borras T. Genes expressed in the human trabecular meshwork during pressure-induced homeostatic response. J Cell Physiol. 2004;201:126–137. doi: 10.1002/jcp.20030. [DOI] [PubMed] [Google Scholar]
- Vittitow JL, Garg R, et al. Gene transfer of dominant-negative RhoA increases outflow facility in perfused human anterior segment cultures. Mol Vis. 2002;8:32–44. [PubMed] [Google Scholar]
- Vogel V, Sheetz M. Local force and geometry sensing regulate cell functions. Nat Rev Mol Cell Biol. 2006;7:265–275. doi: 10.1038/nrm1890. [DOI] [PubMed] [Google Scholar]
- Wang N, Chintala SK, et al. Activation of a tissue-specific stress response in the aqueous outflow pathway of the eye defines the glaucoma disease phenotype. Nat Med. 2001;7:304–309. doi: 10.1038/85446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb RN. Uveoscleral outflow: the other outflow pathway. J Glaucoma. 2000;9:343–345. doi: 10.1097/00061198-200010000-00001. [DOI] [PubMed] [Google Scholar]
- Weinreb RN, Toris CB, et al. Effects of prostaglandins on the aqueous humor outflow pathways. Surv Ophthalmol. 2002;47 Suppl 1:S53–S64. doi: 10.1016/s0039-6257(02)00306-5. [DOI] [PubMed] [Google Scholar]
- Welge-Lussen U, May CA, et al. AlphaB-crystallin in the trabecular meshwork is inducible by transforming growth factor-beta. Invest Ophthalmol Vis Sci. 1999;40:2235–2241. [PubMed] [Google Scholar]
- Welge-Lussen U, May CA, et al. Induction of tissue transglutaminase in the trabecular meshwork by TGF-beta1 and TGF-beta2. Invest Ophthalmol Vis Sci. 2000;41:2229–2238. [PubMed] [Google Scholar]
- Wiberg C, Hedbom E, et al. Biglycan and decorin bind close to the n-terminal region of the collagen VI triple helix. J Biol Chem. 2001;276:18947–18952. doi: 10.1074/jbc.M100625200. [DOI] [PubMed] [Google Scholar]
- Wiberg C, Heinegard D, et al. Biglycan organizes collagen VI into hexagonal-like networks resembling tissue structures. J Biol Chem. 2002;277:49120–49126. doi: 10.1074/jbc.M206891200. [DOI] [PubMed] [Google Scholar]
- Wiberg C, Klatt AR, et al. Complexes of matrilin-1 and biglycan or decorin connect collagen VI microfibrils to both collagen II and aggrecan. J Biol Chem. 2003;278:37698–37704. doi: 10.1074/jbc.M304638200. [DOI] [PubMed] [Google Scholar]
- Wight TN. Versican: a versatile extracellular matrix proteoglycan in cell biology. Curr Opin Cell Biol. 2002;14:617–623. doi: 10.1016/s0955-0674(02)00375-7. [DOI] [PubMed] [Google Scholar]
- Wirtz MK, Bradley JMB, et al. Proteoglycan expression by human trabecular meshworks. Curr Eye Res. 1997;16:412–421. doi: 10.1076/ceyr.16.5.412.7040. [DOI] [PubMed] [Google Scholar]
- Wordinger RJ, Fleenor DL, et al. Effects of TGF-beta2, BMP-4, and gremlin in the trabecular meshwork: implications for glaucoma. Invest Ophthalmol Vis Sci. 2007;48:1191–1200. doi: 10.1167/iovs.06-0296. [DOI] [PubMed] [Google Scholar]
- Wu YJ, La Pierre DP, et al. The interaction of versican with its binding partners. Cell Res. 2005;15:483–494. doi: 10.1038/sj.cr.7290318. [DOI] [PubMed] [Google Scholar]
- Xue W, Comes N, et al. Presence of an established calcification marker in trabecular meshwork tissue of glaucoma donors. Invest Ophthalmol Vis Sci. 2007;48:3184–3194. doi: 10.1167/iovs.06-1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Wallin R, et al. Matrix GLA protein function in human trabecular meshwork cells: inhibition of BMP2-induced calcification process. Invest Ophthalmol Vis Sci. 2006;47:997–1007. doi: 10.1167/iovs.05-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda A, Couchman JR. Regulation of cytoskeletal organization by syndecan transmembrane proteoglycans. Matrix Biol. 2003;22:25–33. doi: 10.1016/s0945-053x(03)00010-6. [DOI] [PubMed] [Google Scholar]
- Yue BYJT. The extracellular matrix and its modulation in the trabecular meshwork. Survey Ophthalmol. 1996;40:379–390. doi: 10.1016/s0039-6257(96)80066-x. [DOI] [PubMed] [Google Scholar]
- Yue BYJT, Elvart JL. Biosynthesis of glycosaminoglycans by trabecular meshwork cells in vitro. Curr Eye Res. 1987;6:959–967. doi: 10.3109/02713688709034867. [DOI] [PubMed] [Google Scholar]
- Zanussi S, Doliana R, et al. The human type VI collagen gene. mRNA and protein variants of the alpha 3 chain generated by alternative splicing of an additional 5-end exon. J Biol Chem. 1992;267:24082–24089. [PubMed] [Google Scholar]
- Zhao X, Ramsey KE, et al. Gene and protein expression changes in human trabecular meshwork cells treated with transforming growth factor-beta. Invest Ophthalmol Vis Sci. 2004;45:4023–4034. doi: 10.1167/iovs.04-0535. [DOI] [PubMed] [Google Scholar]
- Zhao X, Russell P. Versican splice variants in human trabecular meshwork and ciliary muscle. Mol Vis. 2005;11:603–608. [PubMed] [Google Scholar]
- Zhou L, Cheng EL, et al. Signal transduction mediated by adhesion of human trabecular meshwork cells to extracellular matrix. Exp Eye Res. 2000;70:457–465. doi: 10.1006/exer.1999.0806. [DOI] [PubMed] [Google Scholar]
- Zhou L, Higginbotham EJ, et al. Effects of ascorbic acid on levels of fibronectin, laminin and collagen type 1 in bovine trabecular meshwork in organ culture. Curr Eye Res. 1998a;17:211–217. doi: 10.1076/ceyr.17.2.211.5608. [DOI] [PubMed] [Google Scholar]
- Zhou L, Li Y, et al. Glucocorticoid effects on extracellular matrix proteins and integrins in bovine trabecular meshwork cells in relation to glaucoma. Int J Mol Med. 1998b;1:339–346. [PubMed] [Google Scholar]
- Zhou L, Li Y, et al. Alteration of cytoskeletal structure, integrin distribution, and migratory activity by phagocytic challenge in cells from an ocular tissue--the trabecular meshwork. In Vitro Cell Dev Biol Anim. 1999a;35:144–149. doi: 10.1007/s11626-999-0016-6. [DOI] [PubMed] [Google Scholar]
- Zhou L, Li Y, et al. Oxidative stress affects cytoskeletal structure and cell-matrix interactions in cells from an ocular tissue: the trabecular meshwork. J Cell Physiol. 1999b;180:182–189. doi: 10.1002/(SICI)1097-4652(199908)180:2<182::AID-JCP6>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Zhou L, Maruyama I, et al. Expression of integrin receptors in the human trabecular meshwork. Curr Eye Res. 1999c;19:395–402. doi: 10.1076/ceyr.19.5.395.5297. [DOI] [PubMed] [Google Scholar]
- Zhou LL, Zhang S-R, et al. Adhesion of human trabecular meshwork cells to extracellular matrix proteins: Roles and distribution of integrin receptors. Invest Ophthalmol Vis Sci. 1996;37:104–113. [PubMed] [Google Scholar]
- Zillig M, Wurm A, et al. Overexpression and properties of wild-type and Tyr437His mutated myocilin in the eyes of transgenic mice. Invest Ophthalmol Vis Sci. 2005;46:223–234. doi: 10.1167/iovs.04-0988. [DOI] [PubMed] [Google Scholar]