

Abstract
The tissue kallikrein–kinin system is important in regulating cardiovascular and renal function, and dysregulation of the system has been implicated in heart and kidney pathologies. These findings suggest that if balance can be restored to the kallikrein-kinin axis, then associated disease progression may be attenuated. To test this hypothesis, recombinant adeno-associated virus (rAAV)-mediated human tissue kallikrein (HK) expression was induced in a rodent model of chronic renal failure involving 5/6 nephrectomy. rAAV-HK treatment attenuated the rise in blood pressure, glomerular sclerosis, and tubulointerstitial injury observed in this model. rAAV-HK treatment also attenuated renal function decline as measured by urinary microalbumin, osmolarity and cGMP levels. RT-PCR analysis showed that rAAV-HK treated rats had higher levels of bradykinin-B2 receptor (B2R) and dopamine D1-like receptor mRNAs. In contrast, angiotensin II receptor 1, endothelin ETA receptor, and vasopressin V2 receptor mRNAs were markedly downregulated in kidneys from HK-treated rats. Bradykinin induced similar changes in receptor levels and prevented TGF-β1-induced tubulointerstitial fibrosis. The effects of bradykinin could be reversed by the B2R antagonist HOE-140. Together, these findings suggest that restoration of the kallikrein-kinin system reduces kidney injury and protects renal function in 5/6 nephrectomized rats via changes in the expression and activation of G-protein coupled receptors including B2R.
Keywords: recombinant adeno-associated virus vector, human tissue kallikrein, gene therapy, chronic renal failure, 5/6 nephrectomized rat
Introduction
Chronic renal failure is the final stage of various renal diseases and is the most common and serious complication of hypertension and diabetes. In the last two decades, more effective medicines have been developed to control blood pressure and blood glucose, and various treatment strategies have been used to protect the kidneys from damage due to these conditions. However, chronic renal failure still tends to progress steadily to end-stage renal disease, and the morbidity and mortality of renal failure is rising [1, 2]. Extensive studies have documented a reduction in urinary kallikrein levels in patients and animals with hypertension and renal injury [3, 4]. Indeed, an inverse relationship exists between urinary kallikrein excretion and blood pressure [5-8]. Dahl-salt sensitive rats, which have salt-induced hypertension, exhibit a dysfunctional kallikrein-kinin system (KKS) and are more susceptible to kidney injury than spontaneously hypertensive rats which have similar blood pressure [9]. Decreased kallikrein expression in the kidney of 5/6 nephrectomized rats was recently reported [10]. Dr. Yu and colleagues demonstrated that polymorphisms in the human tissue kallikrein gene promoter are etiologic effectors of hypertension and hypertension-associated end-stage renal failure [11].
In a previous study, we delivered human tissue kallikrein (HK) to spontaneously hypertensive rats (SHR) via a recombinant adeno-associated virus vector (rAAV). Delivery of the HK gene induced a mild reduction in systolic blood pressure (∼15 mmHg) and resulted in renal protection [12]. Previous experiments have also indicated that adenovirus mediated HK gene delivery significantly attenuated gentamycin-induced nephrotoxicity and enhanced renal function [13]. The renal protective effects of the KKS have been demonstrated in several hypertensive rat models with progressive renal injury [10, 14-17]. Together, these findings suggest that a repressed renal KKS may play an important role in the pathogenesis of chronic renal failure and that delivery of the HK gene may represent a novel approach to treat chronic renal failure.
The rAAV vector offers several unique advantages over other vectors, including stable transduction and long-term expression of the transgene, the ability to infect both dividing and non-dividing cells, and the lack of an inflammatory response [18, 19]. Improvement in packaging capacity and the recent establishment of package and replication systems independent of adenovirus helper make rAAV an appealing vector for long-term gene therapy for the treatment of chronic disease in humans [12, 18, 20].
Our previous studies documented that rAAV-HK delivery rendered a long-term and stable reduction in hypertension and provided protection against renal injury in the spontaneously hypertensive and the streptozotocin-induced type 2 diabetic rat models [12, 19, 21]. However, the therapeutic effect of HK on chronic renal failure has not been studied. In the present investigation, we evaluated the therapeutic effects of rAAV-HK treatment of chronic renal failure in 5/6 nephrectomized rats. We found that the long-term and stable expression of HK not only offered a sustained reduction in blood pressure and improvement of renal function, but also attenuated glomerular sclerosis and tubulointerstitial injury. These therapeutic effects were associated with activation of bradykinin receptor 2 (B2R), enhanced cGMP production and altered expression of various G-protein coupled receptors in the kidney, including, dopamine receptor 1 (D1R), angiotensin II receptor type 1 (AT1R), endothelin receptor A (ETAR) and vasopressin receptor 2 (V2R).
Materials and Methods
Production of rAAV serotype-2 vectors
The rAAV vector plasmid (pXXUF1) and the rAAV plasmid containing the green fluorescent protein (GFP) reporter gene (pdxII-GFP) were made as previously described [12]. An 860-bp fragment (Not I to Not I) containing the HK open reading frame was subcloned into pXXUF1 downstream of CMV to produce the plasmid pUF1.HK. The packaging plasmid pXX2 (for serotype-2) and the adenovirus helper plasmid pXX6 were made as previously described [18]. The packaging and production of the rAAV–HK and rAAV-GFP were performed in cultured human embryonic kidney 293 (HEK293) cells as described previously [12]. The packed rAAV–HK and rAAV-GFP were purified using a single-step gravity-flow column purification method [22] .
Animal treatment and gene delivery
Twenty-four male Wistar rats, two-month old, weighing 200−250g, were randomly divided into two groups. Six control rats underwent sham operation consisting of laparotomy and manipulation of the renal pedicles. The remaining eighteen rats underwent laparotomy with 5/6 nephrectomy using methods published previously by Hinojosa-Labord [23]. Resection of approximately two thirds of left kidney was followed by one week of recovery before a second operation was performed to remove the entire right kidney. Anesthesia for surgical procedures was accomplished by intraperitoneal injection of pentobarbital (50 mg/kg). All experimental animal protocols were approved by the Institutional Animal Research Committees of the Medical College of Tongji Medical College, and carried out according to the guidelines of the National Institutes of Health. One month after the second surgery, all eighteen 5/6-nephrectomized rats were randomly divided into three treatment groups. The control group (n=6) received 1 ml of saline solution intravenously. The GFP-treated group (GFP, n=6) was injected with a single dose of rAAV-GFP and the HK-treated group (HK, n=6) was injected with a single dose of rAAV-HK (1011 virions per rat in 1 ml saline solution) via the tail vein.
Blood pressure measurement and collection of serum and urine
Before and after the operations and rAAV injections, systolic blood pressures were measured weekly with a manometer–tachometer (Rat Tail NIBP System, ADI Instruments, Sydney, Australia) using the tail-cuff method as described previously [12, 24]. Every two weeks, 24-hour urine samples were collected from rats in metabolic cages and placed into 500 μl toluene to prevent the degradation. Approximately 1 ml blood was drawn from the tail vein and sera were collected after coagulation and centrifugation. All the samples were stored at −80°C until assayed.
Measurements of serum creatinine, 24-hour urinary microalbumin, urinary osmolarity and cGMP levels
Serum creatinine (SCr) levels were measured via autobiochemistry assay with an Abbott Aeroset (Abbott, Abbott Park, IL). The 24-hour urinary microalbumin content was measured usingan enzyme -linked immunosorbent assay (ELISA) as previously described [12]. Urine osmolarity was measured usinga model 3D3 osmometer (Advanced Instruments Norwood, MA). Urinary guanosine 3',5'-cyclic monophosphate (cGMP) levels were measured using an ELISA kit (Cayman Chemical, Ann Arbor, MI) as previously described [25].
RNA and protein extraction
50−100mg of kidney, heart, liver and aorta tissues were homogenized in TRIZOL (Life Technologies, Burlington, Ontario, Canada) and tissue RNA and protein was extracted according to the manufacturer`s instructions. Protein concentration was measured by the Bradford method with bovine serum albumin as standard.
Expression of human tissue kallikrein in rats
Reverse transcription-polymerase chain reactions (RT-PCR) were carried out for human tissue kallikrein (5’ primer, 5’-CCACCATGGGGTTCCTGGTT-3’; 3’ primer, 5’-CGCGGATCCACATTTGATTT-3’) and β-actin mRNAs (5’ primer, 5’-GGAGAAGATGACCCAGATC-3’, 3’ primer, 5’-GATCTTCATGAGGTAGTCAG-3’) using an RT-PCR kit (TAKARA Biotechnology Co. Ltd, Otsu, Japan) as described previously [12]. Urine HK levels were detected by ELISA as described previously [12]t. The ELISA reagents specific for human tissue kallikrein were a generous gift from Dr. Lee Chao (Department of Biochemistry and Molecular Biology, Medical University of South Carolina). Western blotting was performed to detect HK expression levels in liver, aorta, kidney and heart. Briefly, samples containing 20 μg of total protein were loaded in 10% SDS-polyacrylamide gels, and transferred onto nitrocellulose membranes after electrophoresis. Following incubation in blocking buffer (5% nonfat dry milk in Tris-buffered saline, pH 7.6, and 0.1% Tween 20) (TBST), the membrane s were incubated with anti-HK antibody (1:500 dilution) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or anti-β-actin antibody (1:3000) (Sigma Chemical Co., St. Louis, MO) overnight at 4°C. The membranes were then washed three times by TBST and incubated at room temperature for 1 hour with horseradish peroxidase-conjugated anti-rabbit IgG solution (1:8000 dilution) (New England Biolab, Beverly, MA, USA) in the blocking solution. Immunoreactive bands were visualized using an enhanced chemiluminescence (ECL) detection system (Amersham, Piscataway, NJ).
Real-time quantitative PCR to detect mRNAs for B2R, D1R, AT1R, ETAR and V2R
The expression of B2R, D1R, AT1R, ETAR and V2R mRNAs in kidney were detected by real-time quantitative PCR. 1μg of total RNA was reverse-transcribed in a 20 μL reaction mixture containing 1 μL AMV reverse transcriptase, 20 pmol oligo-dT, 2 μL 10 mmol/L dNTP mix, 2 μL 10× RT-PCR buffer (0.1 M Tris–HCl, pH 8.3, 500 mM KCl), 4 μl 25 mM MgCl2, 0.5 μL RNase inhibitor, 8.5 μL RNAase free H2O. The mixture was incubated at 30°C for 10 min, at 42°C for 30 min, and at 99°C for 5 min to allow synthesis of the first cDNA strand. Then, 5 μl cDNA was amplified in a 25 μL reaction mixture containing 4 pmol of the forward and reverse primers, 2 μL of 10× PCR buffer, 1μL of 25 mM MgCl2, and 0.5 μL Taq DNA polymerse. Amplification was for 35 cycles of a denaturing phase of 1 min at 94°C, annealing phase of 30 s at 55−58°C and extension phase of 1 min at 72°C. The forward and reverse primers are as follows: 5’-AGCAGGTCCAGACGGAGAAGA-3’ and 5’-CGATGGGCTTGTGTTCACTGC-3’ for B2R; 5’-CGATTCCATCACCTTCGATGTG-3’ and 5’-GGAATGCTGTCCACTGTGTGTG-3’ for D1R; 5’-CGATTCCATCACCTTCGATGTG-3’ and 5’-GGAATGCTGTCCACTGTGTGTG-3’ for AT1R; 5’-ACTTTGCCACTATGGGCTGTC-3’ and 5’-GCGGGACTTCATTGGGTG-3’ for ETAR; 5’-GATTGCCCTGATGGTGTTCGT-3’ and 5’-GCCCAGCACAGCACATAGAC-3 for V2R; and 5’-GGAGAAGATGACCCAGATC-3’ and 5’-GATCTTCATGAGGTAGTCAG-3’ for β-actin. The resulting PCR products were retrieved after electrophoresis as described in the DNA retrieve reagent kit (Yuanpinghao Company, Beijing, China). The retrieved DNAs were diluted to copy numbers of 108/μL; 107/μL; 106/μL; 105/μL for standard solution preparation. SYBR Green I real-time PCR was performed with 1 μl/well of RT products (10 ng total RNA). Fifteen μL reaction system per well was used including 0.6 μl SYBR Green I (10×) (Open-tech), 0.35 mM of primers, 0.25mM of dNTP-mix, 2 units of TaKaRa Ex Taq HS (Takara). Amplifications were as follows: 95 °C for 2 min followed by 40 cycles of 95°C for 2 s, 57°C for 5 s and 72°C for 10 s on the Light-Cycler System (Roche, Basel, Switzerland). The reverse-transcribed cDNA samples were amplified and cycle threshold (Ct) values were determined. All Ct values were normalized to the housekeeping gene β-actin. The half-life of eNOS mRNA was calculated by linear regression with the least squares method [26]
Analysis of protein expression in the kidney
To investigate the effects of rAAV-HK treatment on renal protein expression, proteins were extracted, quantified and separated by SDS-PAGE as described above. Western blots were probed with antibodies against B2R, and Akt and PI3K p308-Akt (Cell Signaling Technology, Boston, MA), AT1R (Santa Cruz Biotechnology, Santa Cruz, CA), and β-actin (Sigma).
Morphological analysis of the kidney
Kidney tissues not used for biochemical analyses were preserved in zinc formalin solution (10% zinc dichromate, 4% formaldehyde, 0.9% NaCl), embedded in paraffin, sectioned into 4 μm thick slices, and stained with either Periodic Acid-Schiff (PAS) reagent, Hematoxylin and Eosin (HE) or Masson's Trichrome (MT). The histologic sections were then observed microscopically and analyzed by HMIAS-2000 medicine chart analysis system. The severity of glomerular sclerosis was estimated according to the mesangial injury scoring (MIS) system established by Raij et al. [27]. Briefly, the severity was graded according to the percentage of glomeruli involved: 0 = no lesions; 1+ = 1%−25%; 2+=25%−50%; 3+ = 50%−75%; 4+ = 75%−100%. MIS was obtained by multiplying the severity score (0 to 4+) by the percentage of glomeruli which display the same degree of injury.
Influence of bradykinin on receptor mRNA levels in cultured HEK293 cells
HEK293 cells were maintained in DMEM containing 10% fetal calf serum in a 37°C incubator under 5% CO2. To determine optimal experimental conditions, different concentrations of bradykinin (10−4, 10−6, 10−8, 10−10 M) (Sigma-Aldrich, St. Louis, MO) were tested. Subsequent studies were done with bradykinin 10−8 M. The HEK293 cells were incubated with acetic acid (vehicle), bradykinin 10−8 M, or bradykinin (10−8 M) plus the B2R antagonist HOE-140 (10−8 M) for 24 hrs. Total cellular RNA was extracted from the cultures with TRIZOL. Real-time RT-PCR was performed as described above.
Effects of bradykinin on TGF-β1-induced tubulointerstitial fibrosis
Human tubular cells (HK-2 cell line) were cultured with DMEM/F12 with 10% fetal calf serum under 5% CO2 at 37°C. Tubular epithelial-mesenchymal transdifferentiation (TEMT) was induced in tubular cells by treating with TGF-β1 (10 ng/ml) for 72 hours. In some studies, cells were simultaneously treated with bradykinin (10−8 M) or bradykinin (10−8 M) plus HOE-140 (10−8 M). Tubulointerstitial fibrosis was assessed through morphological observation and analysis of E-cadherin and α-actin expression.
Statistical analysis
Statistical significance between the various groups was determined using SAS software. All the data are expressed as mean ± SEM, and differences were considered significant at p<0.05.
Results
Expression of human tissue kallikrein in 5/6 nephrectomized rats after kallikrein gene delivery
RT-PCR performed 3 months after intravenous injection of rAAV-HK showed that human tissue kallikrein mRNA was detected in liver, heart, kidney and aorta in 5/6 nephrectomized rats administered rAAV-HK, but not in 5/6 nephrectomized rats administered rAAV-GFP (Figure 1A). Western blots further indicated that human tissue kallikrein protein was expressed in liver, heart, kidney, and aorta in rAAV-HK treated rats, but not in rAAV-GFP treated rats (Figure 1B). Immunoreactive human kallikrein was also detected in urine (2.02±0.19 ng/ml) from the first month after injection and stable levels were maintained for up to 3 months (Figure 1C). Immunoreactive human tissue kallikrein levels were similar to pre-injection levels in the urine of rAAV-GFP treated rats (Figure 1C). These results suggested that the rAAV vector can efficiently mediate long-term and stable expression of exogenous genes in vivo in 5/6 nephrectomized rats.
Figure 1.
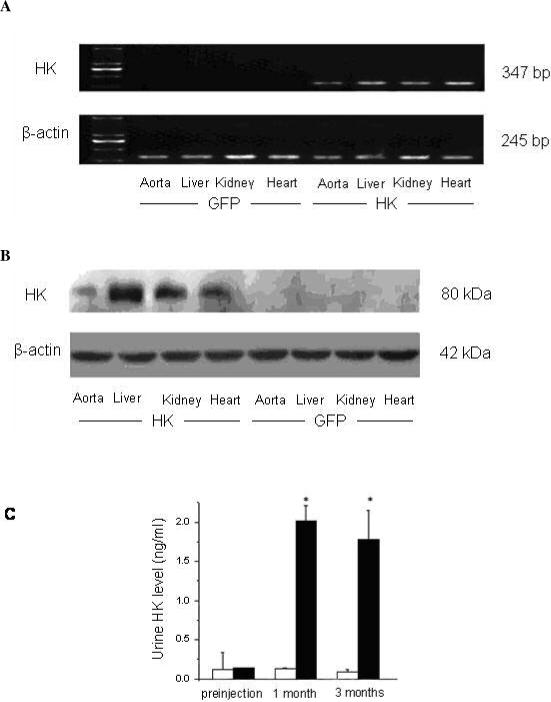
rAAV-HK mediates human tissue kallikrein expression in 5/6 nephrectomized rats. (A) HK mRNAs were detected by RT-PCR in 5/6 nephrectomized rats three months after gene delivery. RT-PCR was performed on 1 μg of total RNA from heart, aorta, kidney and liver. HK mRNA (374 bp) and β-actin control (245 bp) are shown. HK mRNA was detected in tissues from rAAV-HK-treated rats, but not rAAV-GFP treated rats. Results are representative of three independent experiments. (B) HK protein expression was determined by western blotting of tissues from 5/6 nephrectomized rats three months after gene delivery. HK protein was detected in tissues of rAAV-HK treated rats, but not in rAAV-GFP treated rats. The blot is representative of 4 independent experiments. (C) HK levels determined by ELISA on urine collected from rats prior to gene therapy and at one and three months after injection (white bars, rAAV-GFP; black bars, rAAV-HK). Urinary HK levels were detected only in rAAV-HK-treated rats. Results are expressed as mean ± SEM (n=6 per group); *p<0.01 vs. preinjection and rAAV-GFP.
Effect of rAAV-HK delivery on blood pressure in 5/6 nephrectomized rats
Systolic blood pressure of 5/6 nephrectomized rats was significantly higher than that of sham-operation control rats 2 weeks after surgery (182 ± 12 mmHg, n=18, vs. 103 ± 9 mmHg, n=6, p<0.001) (Figure 2A). HK gene delivery reduc ed blood pressure by ∼16 mmHg one month after gene delivery compared to pre-injection levels (Figure 2A). This hypotensive effect was maintained throughout the experiment (3 months). In contrast, systolic blood pressure remained elevatedin rAAV -GFP and saline solution treated rats (Figure 2A). Three months after gene delivery, systolic blood pressure of rAAV-HK treated rats was ∼50−60 mmHg less than saline or rAAV-GFP treated rats (163 ± 13 mmHg in rAAV-HK vs. 217 ± 16 mmHg in saline solution group or 220 ± 13 mmHg in rAAV-GFP, n=6 for each group, p<0.001).
Figure 2.

rAAV-HK gene delivery lowers systolic blood pressure and improves renal function in 5/6 nephrectomized rats. (A) Systolic blood pressure was measured in all rats at the indicated time points using the tail-cuff method. 5/6 nephrectomy results in an approximately 80 mmHg increase in systolic blood pressure. rAAV-HK gene delivery lowers systolic blood pressure, while blood pressure in saline and rAAV-GFP treated rats continues to rise. (B) Serum creatinine levels were measured in all rats at the indicated time points. 5/6 nephrectomy results in elevation in SCr levels and chronic and progressive renal failure. rAAV-HK treatment attenuates the renal failure in 5/6 nephrectomized rats. Values are expressed as mean ± SEM (n=6 for each group); * p<0.001 vs. sham, # p<0.001 vs. saline and rAAV-GFP.
Effects of rAAV-HK delivery on serum creatinine and urinary osmolarity, microalbumin, and cGMP levels in 5/6 nephrectomized rats
SCr level in 5/6 nephrectomized rats was significantly higher than that in sham-operation rats two weeks after the initial surgery (45 ± 7 μM, n=18 vs. 19 ± 6 μM, n=6, p<0.005) (Figure 2B). One month after gene delivery, there were no significant differences in SCr between rAAV-HK and rAAV-GFP treated rats. Two months after gene delivery, SCr levels in rAAV-GFP and saline treated rats tended to be higher than those in rAAV-HK treated rats (77 ± 9 and 84 ± 10 vs. 59 ± 10 μM, p=NS). At the three month time point, this trend reached statistical significance, with the SCr level in rAAV-HK treated rats significantly lower than that in rAAV-GFP and saline treated groups (59 ± 12 μM vs. 128 ± 20 and 133 ± 13 μM, n=6 for each, p<0.001) (Figure 2B). Similarly, three months after gene delivery, rAAV-HK treatment markedly decreased 24-hour urinary excretion of microalbumin (6.9 ± 1.8 mg/24h vs. 12.9 ± 2.5 mg/24h in saline group and 13.4 ± 3.1 mg/24h in rAAV-GFP group, n=6 for each, p<0.001) (Figure 3A). Urinary osmolarity was also increased in rAAV-HK treated rats compared to saline and rAAV-GFP treated rats (590 ± 74 Osmd/Kg vs. 414 ± 68 Osmd/Kg vs. 433 ± 81 Osmd/Kg, n=6, p<0.05) (Figure 3B). 5/6-nephrectomy significantly reduced urinary cGMP level in saline and rAAV-GFP treated rats, whereas rAAV-HK treatment normalized urinary cGMP levels (Figure 3C). Together, these data suggest that rAAV-HK treatment significantly attenuates the progression of chronic renal failure induced by 5/6-nephrectomy.
Figure 3.
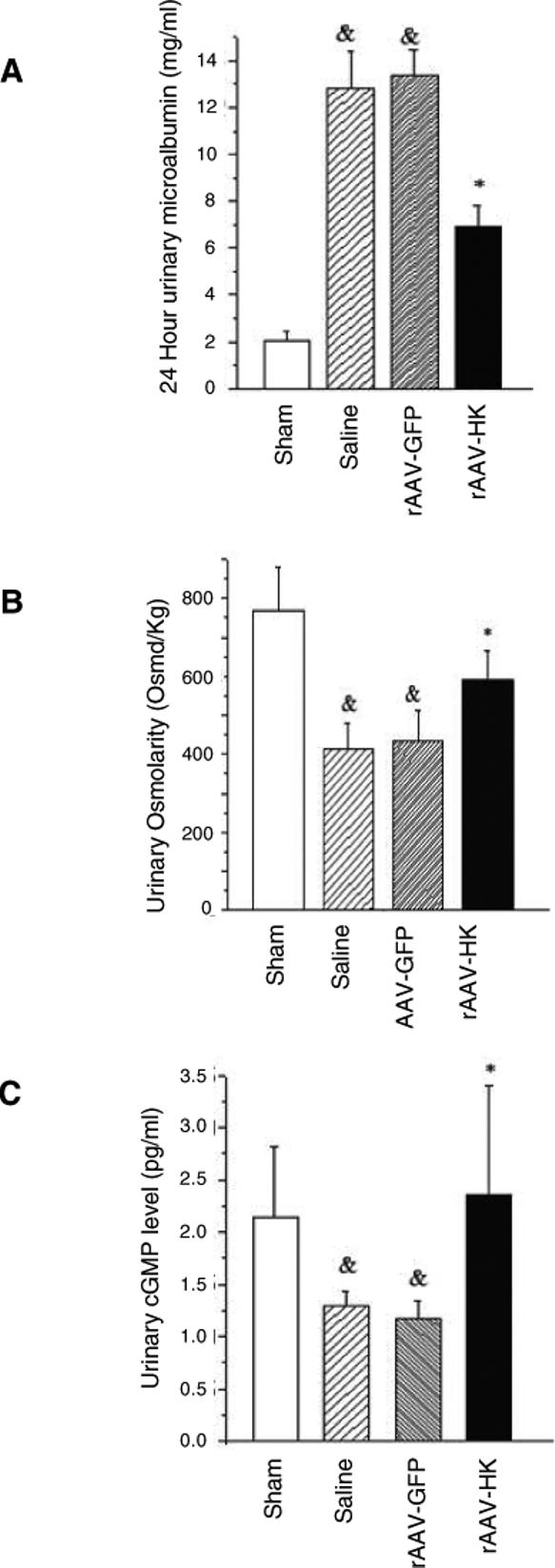
rAAV-HK gene therapy attenuates changes in urinary microalbumin excretion, osmolarity, and cGMP levels in 5/6 nephrectomized rats. Twenty-four hour urinary microalbumin excretion (A), urinary osmolarity (B) and urinary cGMP levels (C) were measured in 5/6 nephrectomized rats three months after gene delivery. rAAV-HK treatment attenuated the rise in microalbumin excretion and the reduction in urinary osmolarity and cGMP levels observed after 5/6 nephrectomy. Data are expressed as mean ± SEM (n=6 per group); # p<0.05 vs. sham; * p<0.05 vs. saline and rAAV-GFP.
Effects of rAAV-HK delivery on kidney morphology in 5/6 nephrectomized rats
Morphological assessment of kidneys was performed using three different staining methods. PAS and HE staining of saline and rAAV-GFP treated 5/6 nephrectomy rats revealed severe glomerular injury characterized by glomerular compensatory hypertrophy, mesangial expansion, loss of brush border, tubular dilation, formation of protein casts within tubules, and increased mesangial matrix accompanied by sclerotic changes. Interestingly, rAAV-HK treatment significantly attenuated these pathological changes (Figure 4A). Furthermore, Masson's Trichrome (MT) staining showed significant tubulointerstitial injury, including collagen deposition, in saline and rAAV-GFP treated rats. However, rAAV-HK treated rats showed significant protection from this injury (Figure 4A). Quantification of the degree of glomerular injury revealed that rAAV-HK treatment significantly decreased positive PAS-staining area (data not shown), glomerular cell number (data not shown), and MIS (Figure 4B) compared with saline and rAAV-GFP treated rats. Together, these data indicate that rAAV-HK treatment significantly attenuates glomerlular and tubulointerstitial injury induced by 5/6-nephrectomy.
Figure 4.
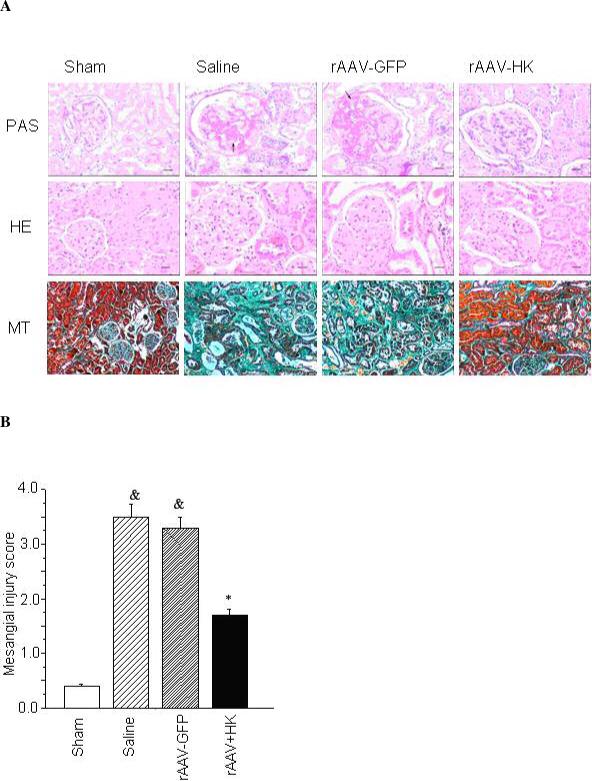
rAAV-HK gene therapy attenuates changes on kidney morphology associated with 5/6 nephrectomy. (A) Kidney tissue sections were stained with periodic acid-schiff (PAS) reagent, hematoxylin and eosin (HE) or Masson's Trichrome (MT) as indicated. PAS and HE stained sections are shown at 200× magnification (scale bar, 10 μm). Sections stained with MT are shown at 40× magnification (scale bar, 50 μm). (B) PAS stained sections were evaluated for the severity of glomerular sclerosis according to the MIS system established by Raij et al [27]. rAAV-HK treatment attenuated the morphological changes in the kidneys of 5/6 nephrectomized rats. Data are expressed as mean ± SEM (n=6 per group); # p<0.05 vs. sham; * p<0.05 vs. saline and rAAV-GFP.
The Effects of rAAV-HK delivery on PI3K/Akt signaling pathway
The effects of rAAV-HK delivery on the PI3K/Akt signaling pathway were examined. PI3 kinase expression and Akt phosphorylation levels were diminished in kidneys from saline and rAAV-GFP treated rats (Figure 5). Importantly, rAAV-HK gene delivery significantly reversed these effects (Figure 5), suggesting that overexpression of HK activated PI3K/Akt signaling and enhanced survival of renal cells.
Figure 5.
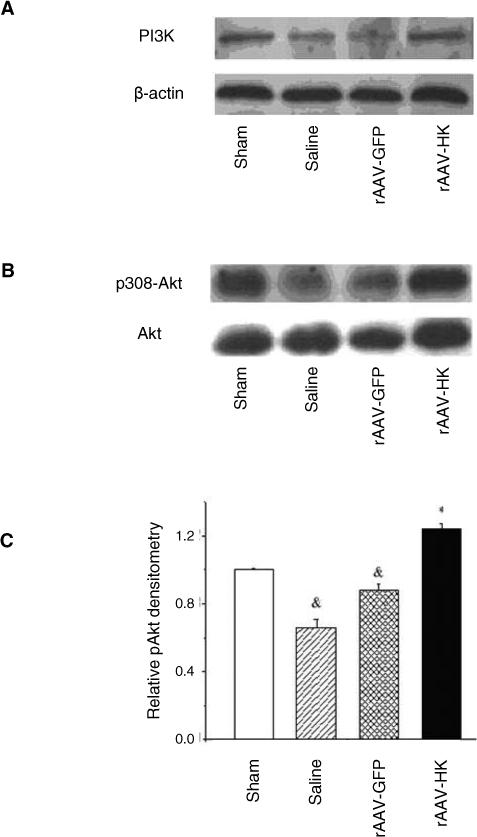
5/6 nephrectomy and rAAV-HK gene delivery alter renal PI3K/Akt signaling pathways. Representative western blots show that PI3K expression (A) and p308-Akt phosphorylation (B) are reduced in control 5/6 nephrectomy rats but restored by rAAV-HK treatment. (C) Densitometric analysis of p308-Akt phosphorylation detected by western blotting. Results are expressed as mean ± SEM (n=6 per group); * p<0.05 vs. saline and rAAV-GFP controls; # p<0.05 vs. sham control.
Effects of rAAV-HK delivery on expression of B2R, D1R, AT1R, ETAR, and V2R
Real-time quantitative PCR was carried out to investigate the effects of rAAV-HK treatment on renal expression of B2R, D1R, AT1R, ETAR and V2R using β-actin as a control. The mRNA levels of B2R and D1R genes were downregulated by approximately 50% in 5/6 nephrectomized rats treated with saline or rAAV-GFP compared with sham-operated rats (Figure 6A). rAAV-HK treatment reversed the downregulation observed in untreated 5/6 nephrectomized rats. In contrast, mRNA levels of AT1R, ETAR and V2R were significantly upregulated in 5/6 nephrectomized animals treated with saline or rAAV-GFP compared with sham-operated rats, while rAAV-HK treatment markedly attenuated this upregulation (Figure 6A). Thus, rAAV-HK reversed the loss of expression of genes that are favorable to renal function and prevented the upregulation of genes that are detrimental to renal function.
Figure 6.
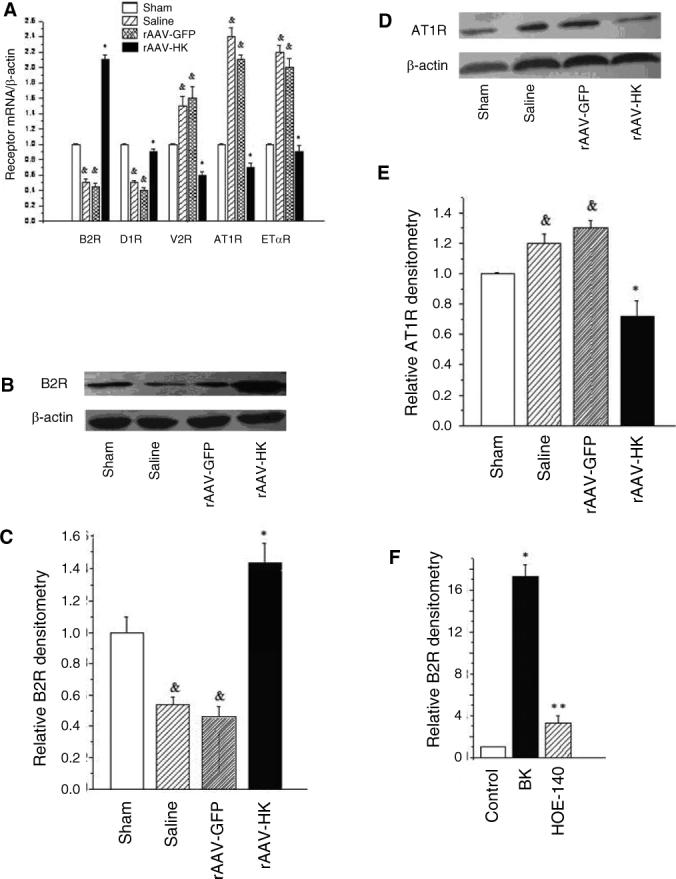
5/6 nephrectomy and rAAV-HK gene therapy alter rat kidney G-protein coupled receptor mRNA expression. (A) Real-time PCR showing mRNA levels for B2R, D1R, AT1R, ETAR, and V2R in kidneys of 5/6 nephrectomized rats three months after gene delivery. The ratio of each mRNA/β-actin is shown. Representative western blots of B2R (B) and AT1R (D) expression in kidney in 5/6 nephrectomized rats after three months of gene delivery. Densitometric quantification shows levels of B2R (C) and AT1R (E) expression (normalized to β-actin) in rat kidneys after three months of gene delivery. rAAV-HK treatment reverses the decrease of B2R and increase in AT1R observed after 5/6 nephrectomy. Data are expressed as mean ± SEM (n=4=5 per group); & p<0.05 vs. sham; * p<0.05 vs. saline and GFP controls. (F) HEK293 cells were treated with vehicle, bradykinin (BK) (10 nM), or BK and HOE-140 (10 nM each) for 72 hours and B2R expression was determined by real-time PCR. Results shown are mean ± SEM (n=4−5 per group); * p<0.01 vs. control; ** p<0.01 vs. bradykinin.
Western blotting confirmed that the HK-induced changes in receptor expression observed at the mRNA level also occurred at the protein level. Thus, rAAV-HK treatment significantly upregulated renal expression of B2R protein but downregulated renal expression of AT1R compared with saline and rAAV-GFP treated rats (Figure 6B-E). Furthermore, we incubated HEK293 cells in vitro with bradykinin (BK) and BK plus the B2R antagonist HOE-140 and assessed the expression of B2R mRNA by real-time RT-PCR. BK addition upregulated B2R mRNA level by 17-fold in HEK293 cells as compared with the control cells and the B2R antagonist HOE-140 significantly blocked this upregulation of B2R by BK (Figure 6F). These data suggest that BK affects B2R expression via activation of B2R.
Bradykinin attenuates TGF-β1-induced TEMT
Morphological observation showed that incubation of HK-2 tubular cells with TGF-β1 for 72 hrs induced significant phenotypic changes; cells changed from round or ellipse-like shape to long, spindle shaped, fibroblast-like cells, and intercellular connections were reduced (Figure 7A). Interestingly, BK addition prevented these morphological changes induced by TGF-β1, an effect that was attenuated by the B2R antagonist HOE-140 (Figure 7A). Staining with α-smooth muscle actin (α-SMA) showed that almost all cells were positive after treatment with TGF-β1, suggesting induction of TEMT (Figure 7B). In contrast, BK addition reduced the number of α-SMA positive cells induced by TGF-β1 treatment. This effect of BK could be blocked by the addition of B2R antagonist HOE-140 (Figure 7B).
Figure 7.
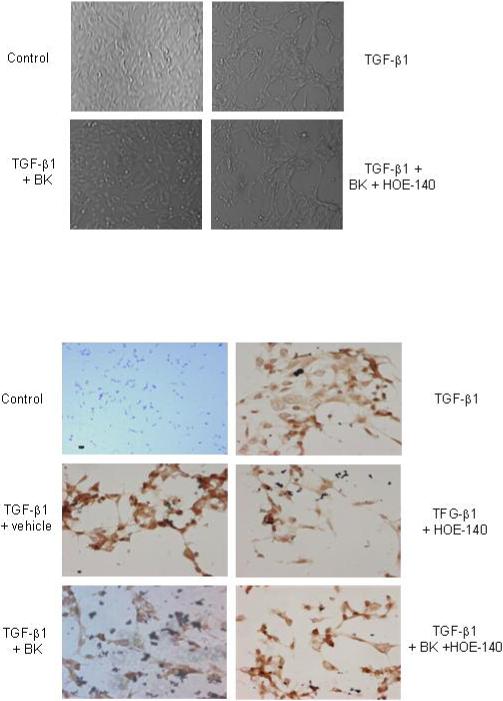
Bradykinin attenuates markers of tubular epithelial-mesenchymal transdifferentiation (TEMT) induced by TGF-β1 in HK-2 cells. (A) HK-2 cells were incubated for 48 hrs with TGF-β1 (10 ng/ml) with/without BK (10nM) and B2R antagonist HOE-140 (10nM) as indicated. (B) Anti-α-SMA staining of HK-2 cells treated with TGF-β1, BK, and/or HOE-140 as indicated. Cells were stained with antibodies. Control HK-2 cells are α-SMA negative, while TGF-β1 incubation results in TEMT, an effect that is attenuated by BK. BK effects are blocked by HOE-140. Results shown are representative of three independent experiments.
Western blots showed that incubation of HK-2 cells with TGF-β1 resulted in downregulation of E-cadherin expression, and upregulation of α-SMA and smad4 expression (Figure 8A-C). Treatment with BK attenuated each of these effects. Furthermore, the attenuation of changes in protein expression by BK was due to activation of B2R, as HOE-140 treatment prevented these effects (Figure 8A-C).
Figure 8.
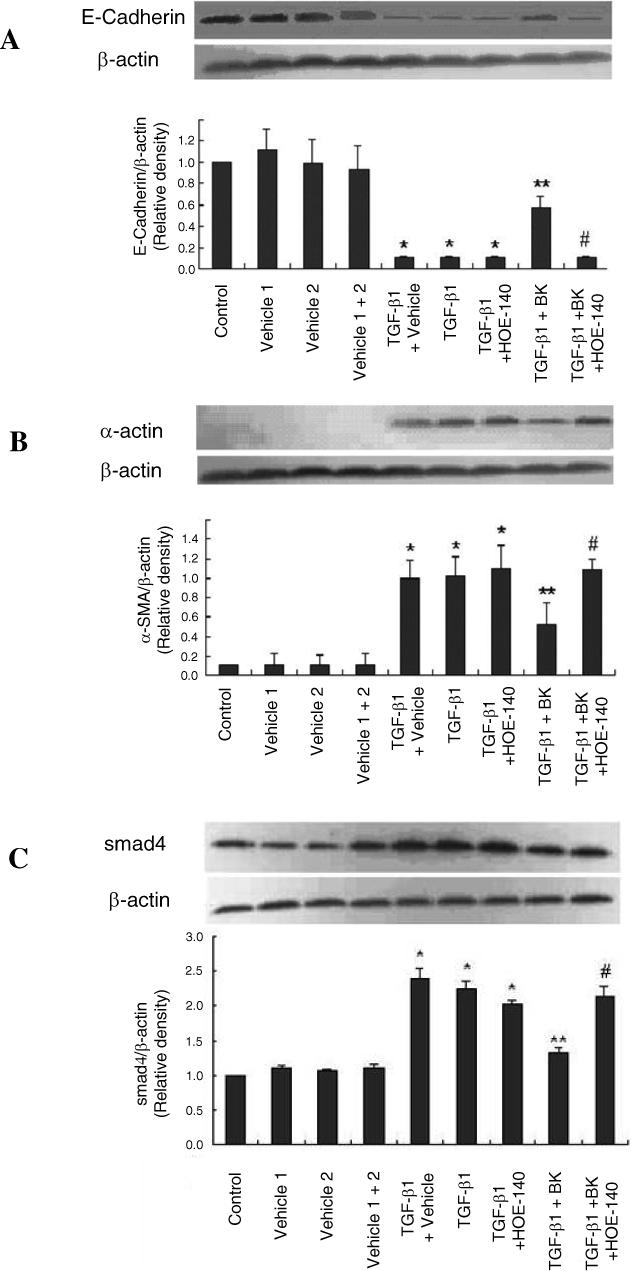
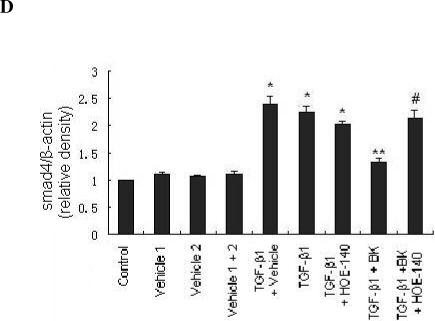
Bradykinin attenuates expression of TEMT markers induced by TGF-β1 in HK-2 cells through the B2R receptor. HK-2 cells were incubated for 48 hrs with TGF-β1 (10 ng/ml) with or without vehicle, BK (10nM) and B2R antagonist HOE-140 (10nM) as indicated. Representative western blots for E-cadherin (A), α-SMA (B), and smad4 (C) are shown. Densitometric quantification of western blots stained with E-cadherin, α-SMA, and smad4 antibodies is shown normalized to β-actin. Data are expressed as mean ± SEM (n=6 per group); * p<0.01 vs. all controls; ** p<0.01 vs. TGF-β1; # p<0.05 vs. TGF-β1 + BK.
We also investigated the effects of BK on cAMP and cGMP in incubation of HK-2 tubular cells and results showed that BK addition did not affect cAMP level in cultures but significantly increases cGMP level compared with control (17.35±0.72 vs. 15.34±0.64 nmol/L, p<0.05) and HOE-140 attenuated the effect (15.40±0.55 nmol/L).
Discussion
This study demonstrates effective delivery of the human tissue kallikrein cDNA using an rAAV-based vector to 5/6 nephrectomized rats and examines the effects on blood pressure, renal function and renal morphology. We found that a single intravenous injection of rAAV-HK provided prolonged attenuation of the detrimental effects associated with 5/6 nephrectomy. rAAV-HK reduced blood pressure, improved renal function, and attenuated glomerular sclerosis and tubulointerstitial injury. These data suggest that rAAV-mediated delivery of the HK cDNA protects the kidney against development and progression of chronic renal failure in this model.
Tissue kallikrein is a serine proteinase present in many tissues, including kidney, vasculature, lung, heart and adrenal gland – tissues that are critical for blood pressure regulation and cardiovascular function. Tissue kallikrein cleaves low-molecular-weight kininogen, leading to the release of potent vasodilator kinins. Intact BK exerts its bioactive functions via binding to G protein-coupled receptors [28-30]. To date, two types of kinin G protein-coupled receptors, B1 and B2, have been characterized. The B2R mediates the majority of the cardiovascular and renal actions of bradykinin [31, 32]. Acute blockade of the B2R substantially decreases renal blood flow and natriuretic response, and chronic blockade of the B2R increases susceptibility to Angiotensin II-induced hypertension [33, 34].
Transgenic mice which overexpress human B2R exhibit hypotension and augmented renal hemodynamics as manifested by increased renal blood flow, glomerular filtration rate and urine flow. Transgenic mice which overexpress human tissue kallikrein, in contrast, are permanently hypotensive throughout their lifetime compared with their control littermates. Administration of aprotinin, a tissue kallikrein inhibitor, or icatibant, a specific B2R antagonist, to the transgenic mice restores their blood pressure to normal levels [35, 36]. These findings suggest that enhanced expression of tissue kallikrein and B2R could significantly reduce blood pressure and improve renal function. In the present study, rAAV-HK delivery resulted in prolonged elevation of renal HK expression which lowered blood pressure and attenuated morphological changes in kidneys of 5/6 nephrectomized rats. Interestingly, the expression levels of B2R mRNA and protein were significantly higher in kidneys of rAAV-HK treated compared to rAAV-GFP and saline treated 5/6 nephrectomized rats. The high plasma level of HK and the HK-enhanced B2R expression also contributed to the reduction in blood pressure and renal protection. These findings suggest that the activation of kallikrein increased the production of BK, which in turn upregulated the B2R in kidney following HK gene transfer. These findings were supported by the in vitro observations that BK induced a significant upregulation of B2R mRNA expression in HEK293 cells.
The decrease in blood pressure by kallikrein gene transfer may also potentially contribute to renal protection and attenuation of renal dysfunction following 5/6 nephrectomy. On the other hand, it is a likely and important consideration that bradykinin directly protects the kidney from injury. We found that HK gene therapy significantly stimulates the PI3 kinase/Akt pathway in 5/6 nephrectomized rats which could enhance cell survival and improve renal function. In addition, B2R activation in kidney activates guanylyl cyclase to enhance cGMP production [37], which contributes to renal cell protection and improvement of renal function [38, 39].
rAAV-HK administration or addition of bradykinin significantly upregulated B2R and D1R and downregulated AT1R, ETAR and V2R both in vivo and in vitro. Although the precise mechanisms remain unclear, these changes should favor protection of the kidney from injury and chronic renal failure. It has been well documented that persistent activation of the renin-angiotensin-aldosterone system (RAS) and the endothelin (ET) receptor in the kidney and vasculature play key roles in clinical and experimental chronic renal failure [40, 41]. ET-1 is a potent vasoconstrictor peptide with pro-inflammatory, mitogenic and pro-fibrotic properties that is involved in both normal renal physiology and pathology. ET-1 exerts a wide variety of biological effects, including constriction of cortical and medullary vessels, mesangial cell contraction, stimulation of extracellular matrix production, and renal resistance vessel contraction [42]. Angiotensin II is also a potent vasoconstrictor and pro-inflammatory mediator. Increased angiotensin II in the kidney induces cell injury via AT1R, including oxidative stress damage, apoptosis, inflammation of vascular endothelial and renal tubular epithelial cells, and increase in arterial resistance [14, 15]. Treatment with angiotensin II-receptor antagonists is thought to prevent renal injury and improve renal function in clinical trials and animal models [43]. In addition to modulating renal resistance and retention of water and sodium via V2R [44, 45], vasopressin (AVP) causes contraction of mesangial and interstitial cells, proliferation and hypertrophy via activation of MAP kinase, PI3 kinase/Akt and protein kinase C [46, 47]. In contrast, low doses of dopamine and treatment with D1R agonists provide renal protection and have diuretic effects [48]. In addition to hypotensive effects, B2R stimulation by bradykinin activates MAP kinases, PI3 kinase/Akt, and JNK signaling pathways to enhance survival and reduce apoptosis [21]. rAAV-mediated delivery of the HK gene markedly upregulated B2R and D1R and downregulated AT1R, ETAR and V2R in both in vitro and in vivo experiments in this study. These changes in receptor expression may contribute to the observed effect of HK gene therapy on kidney morphology and renal failure in the 5/6 nephrectomy rat model.
Tubulointerstitial fibrosis is the ultimate common pathway to end stage renal disease. TEMT induced by TGF-β1 plays a crucial role in renal fibrosis [49]. E-cadherin is a characteristic marker of tubular epithelial cells and is regulated by β-catenin. Loss of E-cadherin is associated with loss of renal epithelial cell function and is an early and important step in TEMT [49, 50]. On the other hand, α-SMA is the characteristic marker of myofibroblasts and is not abundant in renal tubular epithelial cells. Its appearance in tubular cells suggests a transformation of cell phenotype to one that is fibroblast-like [49, 50]. The present study found that TGF-β1 induced transformation of the cellular phenotype from tubular epithelial cells to fibroblast-like cells. Smad4 is an important molecule of TGF-β/smad signaling pathway and TGF-β1 induced the upregulation of smad4 which is a marker of this signaling pathway. BK significantly attenuated this transformation as seen by changes in the cellular expression of E-cadherin, α-SMA and smad4. These further suggest a role for the kallikrein-kinin-bradykinin system in protection against renal disease and TEMT in this model of chronic renal failure. Additionally, tissue kallikrein may also activate bradykinin B2 receptor, independent of bradykinin release [51].
In summary, our data demonstrates that rAAV-mediated HK delivery significantly attenuates hypertension and chronic renal dysfunction, and protects the kidney from morphologic changes (glomerular sclerosis, tubulointerstitial injury and collagen deposition) following 5/6 nephrectomy in rats. Real time RT-PCR assays and western blots indicate that HK gene therapy or treatment with BK upregulate B2R and D1R and downregulate AT1R, ETAR and V2R in 5/6 nephrectomized rat kidneys and cultured kidney cells. No detectable adverse effects were found in rAAV-injected animals. Furthermore, B2R activation prevented tubular epithelial-mesenchymal transdifferentiation and tubulointerstitial fibrosis induced by TGF-β1. These results lend strong support to the potential application of rAAV-HK as a safe and effective therapeutic approach in the prophylaxis and treatment of renal diseases and chronic renal failure.
Acknowledgements
This work was supported by grants from National “973” projects (No. 2006CB503801) and International Project (2005DFA30880) and the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences. Drs. Ling Tu and Xizhen Xu contributed equally to this work.
References
- 1.Moeller S, Gioberge S, Brown G. ESRD patients in 2001: global overview of patients, treatment modalities and development trends. Nephrol. Dial. Transplant. 2002;17(12):2071–2076. doi: 10.1093/ndt/17.12.2071. [DOI] [PubMed] [Google Scholar]
- 2.Xue JL, et al. Forecast of the Number of Patients with End-Stage Renal Disease in the United States to the Year 2010. J Am Soc Nephrol. 2001;12(12):2753–2758. doi: 10.1681/ASN.V12122753. [DOI] [PubMed] [Google Scholar]
- 3.Scicli AG, C.O. Renal kallikrein-kinin system. Kidney Int. 1986;29(1):120–130. doi: 10.1038/ki.1986.14. [DOI] [PubMed] [Google Scholar]
- 4.Margolius HS. Tissue kallikreins and kinins: regulation and roles in hypertensive and diabetic diseases. Annu Rev Pharmacol Toxicol. 1989;29:343–64. doi: 10.1146/annurev.pa.29.040189.002015. [DOI] [PubMed] [Google Scholar]
- 5.Margolius HS, et al. Urinary kallikrein excretion in hypertensive man. Relationships to sodium intake and sodium-retaining steroids. Circ Res. 1974;35(6):820–5. doi: 10.1161/01.res.35.6.820. [DOI] [PubMed] [Google Scholar]
- 6.Zinner SH, et al. Stability of blood pressure rank and urinary kallikrein concentration in childhood: an eight-year follow-up. Circulation. 1978;58(5):908–15. doi: 10.1161/01.cir.58.5.908. [DOI] [PubMed] [Google Scholar]
- 7.Margolius HS, et al. Altered urinary kallikrein excretion in rats with hypertension. Circ Res. 1972;30(3):358–62. [PubMed] [Google Scholar]
- 8.Shimamoto K, et al. The role of renal natriuretic depressor systems on hypertensive mechanisms in reduced renal mass hypertensive rats. Hypertens Res. 1995;18(Suppl 1):S53–7. doi: 10.1291/hypres.18.supplementi_s53. [DOI] [PubMed] [Google Scholar]
- 9.Uehara Y, N.A., Hirawa N, Kawabata Y, Iwai J, Ono H, Matsuoka H, Takabatake Y, Yagi S, Sugimoto T. Antihypertensive effects of cicletanine and renal protection in Dahl salt-sensitive rats. J Hypertens. 1991;9(8):10. doi: 10.1097/00004872-199108000-00005. [DOI] [PubMed] [Google Scholar]
- 10.Ardiles LG, et al. Effect of mycophenolate mofetil on kallikrein expression in the kidney of 5/6 nephrectomized rats. Kidney Blood Press Res. 2002;25(5):289–95. doi: 10.1159/000066796. [DOI] [PubMed] [Google Scholar]
- 11.Yu H, S.Q., Freedman BI, Chao J, Chao L, Rich SS, Bowden DW. Association of the tissue kallikrein gene promoter with ESRD and hypertension. Kidney International. 2002;61:1030–1039. doi: 10.1046/j.1523-1755.2002.00198.x. [DOI] [PubMed] [Google Scholar]
- 12.Wang T, et al. Recombinant adeno-associated virus-mediated kallikrein gene therapy reduces hypertension and attenuates its cardiovascular injuries. Gene Ther. 2004;11(17):1342–50. doi: 10.1038/sj.gt.3302294. [DOI] [PubMed] [Google Scholar]
- 13.Murakami H, et al. Human kallikrein gene delivery protects against gentamycin-induced nephrotoxicity in rats. Kidney Int. 1998;53(5):1305–1313. doi: 10.1046/j.1523-1755.1998.00867.x. [DOI] [PubMed] [Google Scholar]
- 14.Mori T, Cowley AW, Jr., Ito S. Molecular mechanisms and therapeutic strategies of chronic renal injury: physiological role of angiotensin II-induced oxidative stress in renal medulla. J Pharmacol Sci. 2006;100(1):2–8. doi: 10.1254/jphs.fmj05003x2. [DOI] [PubMed] [Google Scholar]
- 15.Tamada S, et al. Molecular mechanisms and therapeutic strategies of chronic renal injury: the role of nuclear factor kappaB activation in the development of renal fibrosis. J Pharmacol Sci. 2006;100(1):17–21. doi: 10.1254/jphs.fmj05003x4. [DOI] [PubMed] [Google Scholar]
- 16.Dumont Y, et al. Blood pressure-independent effect of angiotensin AT1 receptor blockade on renal endothelin-1 production in hypertensive uremic rats. J Hypertens. 2001;19(8):1479–87. doi: 10.1097/00004872-200108000-00017. [DOI] [PubMed] [Google Scholar]
- 17.Wolf WC, et al. Human tissue kallikrein gene delivery attenuates hypertension, renal injury, and cardiac remodeling in chronic renal failure. Kidney Int. 2000;58(2):730–9. doi: 10.1046/j.1523-1755.2000.00219.x. [DOI] [PubMed] [Google Scholar]
- 18.Monahan PE, et al. Direct intramuscular injection with recombinant AAV vectors results in sustained expression in a dog model of hemophilia. Gene Ther. 1998;5(1):40–9. doi: 10.1038/sj.gt.3300548. [DOI] [PubMed] [Google Scholar]
- 19.Sun L, Li J, Xiao X. Overcoming adeno-associated virus vector size limitation through viral DNA heterodimerization. Nat Med. 2000;6(5):599–602. doi: 10.1038/75087. [DOI] [PubMed] [Google Scholar]
- 20.Xiao X, Li J, Samulski RJ. Production of High-Titer Recombinant Adeno-Associated Virus Vectors in the Absence of Helper Adenovirus. J. Virol. 1998;72(3):2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan G, et al. Tissue Kallikrein Reverses Insulin Resistance and Attenuates Nephropathy in Diabetic Rats by Activation of Phosphatidylinositol 3-Kinase/Protein Kinase B and Adenosine 5'-Monophosphate-Activated Protein Kinase Signaling Pathways. Endocrinology. 2007;148(5):2016–2026. doi: 10.1210/en.2006-0602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Auricchio A, et al. Isolation of Highly Infectious and Pure Adeno-Associated Virus Type 2 Vectors with a Single-Step Gravity-Flow Column. Human Gene Therapy. 2001;12(1):71–76. doi: 10.1089/104303401450988. [DOI] [PubMed] [Google Scholar]
- 23.Hinojosa-Laborde C, Frohlich BH, Cowley AW., Jr. Whole body autoregulation in reduced renal mass hypertension. Hypertension. 1992;20(5):659–65. doi: 10.1161/01.hyp.20.5.659. [DOI] [PubMed] [Google Scholar]
- 24.Chao J, et al. Human kallikrein gene delivery attenuates hypertension, cardiac hypertrophy, and renal injury in Dahl salt-sensitive rats. Hum Gene Ther. 1998;9(1):21–31. doi: 10.1089/hum.1998.9.1-21. [DOI] [PubMed] [Google Scholar]
- 25.Schneemann A, et al. Nitric oxide/guanylate cyclase pathways and flow in anterior segment perfusion. Graefe's Archive for Clinical and Experimental Ophthalmology. 2002;V240(11):936–941. doi: 10.1007/s00417-002-0559-7. [DOI] [PubMed] [Google Scholar]
- 26.Wu ZL, et al. Protein tyrosine phosphatases are up-regulated and participate in cell death induced by polyglutamine expansion. J Biol Chem. 2002;277(46):44208–13. doi: 10.1074/jbc.M206890200. [DOI] [PubMed] [Google Scholar]
- 27.Raij L, Azar S, Keane W. Mesangial immune injury, hypertension, and progressive glomerular damage in Dahl rats. Kidney Int. 1984;26(2):137–43. doi: 10.1038/ki.1984.147. [DOI] [PubMed] [Google Scholar]
- 28.Clements JA. The glandular kallikrein family of enzymes: tissue-specific expression and hormonal regulation. Endocr Rev. 1989;10(4):393–419. doi: 10.1210/edrv-10-4-393. [DOI] [PubMed] [Google Scholar]
- 29.Wang DZ, et al. Expression and cellular localization of tissue kallikrein-kinin system in human adrenal gland. Am J Physiol. 1996;271(3 Pt 2):F709–16. doi: 10.1152/ajprenal.1996.271.3.F709. [DOI] [PubMed] [Google Scholar]
- 30.Wolf WC, et al. Localization and expression of tissue kallikrein and kallistatin in human blood vessels. J Histochem Cytochem. 1999;47(2):221–8. doi: 10.1177/002215549904700210. [DOI] [PubMed] [Google Scholar]
- 31.Bhoola KD, Figureroa CD, Figueroa, Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev. 1992;44(1):1–80. [PubMed] [Google Scholar]
- 32.Hall JM. Bradykinin receptors. Gen Pharmacol. 1997;28(1):1–6. doi: 10.1016/s0306-3623(96)00174-7. [DOI] [PubMed] [Google Scholar]
- 33.Siragy HM. Evidence that intrarenal bradykinin plays a role in regulation of renal function. Am J Physiol. 1993;265(4 Pt 1):E648–54. doi: 10.1152/ajpendo.1993.265.4.E648. [DOI] [PubMed] [Google Scholar]
- 34.Madeddu P, et al. Chronic inhibition of bradykinin B2-receptors enhances the slow vasopressor response to angiotensin II. Hypertension. 1994;23(5):646–52. doi: 10.1161/01.hyp.23.5.646. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, et al. Human tissue kallikrein induces hypotension in transgenic mice. Hypertension. 1994;23(2):236–43. doi: 10.1161/01.hyp.23.2.236. [DOI] [PubMed] [Google Scholar]
- 36.Wang D, et al. Enhanced renal function in bradykinin B(2) receptor transgenic mice. Am J Physiol Renal Physiol. 2000;278(3):F484–91. doi: 10.1152/ajprenal.2000.278.3.F484. [DOI] [PubMed] [Google Scholar]
- 37.Oldenburg O, Qin Q, Krieg T, Yang XM, Philipp S, Critz SD, Cohen MV, Downey JM. Bradykinin induces mitochondrial ROS generation via NO, cGMP, PKG, and mitoKATP channel opening and leads to cardioprotection. Am J Physiol Heart Circ Physiol. 2004;286(1):H468–76. doi: 10.1152/ajpheart.00360.2003. [DOI] [PubMed] [Google Scholar]
- 38.Hiyoshi H, Yayama K, Takano M, Okamoto H. Stimulation of Cyclic GMP Production via AT2 and B2 Receptors in the Pressure-Overloaded Aorta After Banding. Hypertension. 2004;43:1258–1263. doi: 10.1161/01.HYP.0000128022.24598.4f. [DOI] [PubMed] [Google Scholar]
- 39.Polte T, Hemmerle A, Berndt G, Grosser N, Abate A, Schroder H. Atrial natriuretic peptide reduces cyclosporin toxicity in renal cells: role of cGMP and heme oxygenase-1. Free Radic Biol Med. 2002;32(1):56–63. doi: 10.1016/s0891-5849(01)00761-4. [DOI] [PubMed] [Google Scholar]
- 40.Clavell AL, B.J. Physiologic and pathophysiologic roles of endothelin in the kidney. Curr Opin Nephrol Hypertens. 1994;3(1):66–72. doi: 10.1097/00041552-199401000-00010. [DOI] [PubMed] [Google Scholar]
- 41.Potter GS, Johnson RJ, Fink GD. Role of endothelin in hypertension of experimental chronic renal failure. Hypertension. 1997;30(6):1578–84. doi: 10.1161/01.hyp.30.6.1578. [DOI] [PubMed] [Google Scholar]
- 42.Neuhofer W, Pittrow D. Role of endothelin and endothelin receptor antagonists in renal disease. Eur J Clin Invest. 2006;36(Suppl 3):78–88. doi: 10.1111/j.1365-2362.2006.01689.x. [DOI] [PubMed] [Google Scholar]
- 43.Dumont Y, D'Amours M, Lebel M, Lariviere R. Blood pressure-independent effect of angiotensin AT1 receptor blockade on renal endothelin-1 production in hypertensive uremic rats. Journal of Hypertension. 2001;19(8):9. doi: 10.1097/00004872-200108000-00017. [DOI] [PubMed] [Google Scholar]
- 44.Jose PA, Eisner GM, Felder RA. Renal dopamine receptors in health and hypertension. Pharmacol Ther. 1998;80(2):149–82. doi: 10.1016/s0163-7258(98)00027-8. [DOI] [PubMed] [Google Scholar]
- 45.Wong BP, Wong NL. Renal hypersensitivity to vasopressin in congestive heart failure. Cardiology. 1998;90(1):20–7. doi: 10.1159/000006811. [DOI] [PubMed] [Google Scholar]
- 46.Tahara A, T.J., Tomura Y, Kusayama T, Momose K, Taniguchi N, Suzuki T, Yatsu T, Shibasaki M. Binding and signal transduction characteristics of the nonpeptide vasopressin V1A receptor-selective antagonist YM218 in cultured rat mesangial cells. Pharmacology. 2006;78(2):10. doi: 10.1159/000095698. [DOI] [PubMed] [Google Scholar]
- 47.Ghosh PM, et al. Arginine vasopressin stimulates mesangial cell proliferation by activating the epidermal growth factor receptor. Am J Physiol Renal Physiol. 2001;280(6):F972–979. doi: 10.1152/ajprenal.2001.280.6.F972. [DOI] [PubMed] [Google Scholar]
- 48.Ide JM, et al. Prophylaxis of iodinated contrast media-induced nephropathy: a pharmacological point of view. Invest Radiol. 2004;39(3):155–70. doi: 10.1097/01.rli.0000101483.60710.2c. [DOI] [PubMed] [Google Scholar]
- 49.Fan JM, N.Y., Hill PA, Nikolic-Paterson DJ, Mu W, Atkins RC, Lan HY. Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int. 1999;56(4):1455–67. doi: 10.1046/j.1523-1755.1999.00656.x. [DOI] [PubMed] [Google Scholar]
- 50.Li JH, W.W., Huang XR, Oldfield M, Schmidt AM, Cooper ME, Lan HY. Advanced glycation end products induce tubular epithelial-myofibroblast transition through the RAGE-ERK1/2 MAP kinase signaling pathway. Am J Pathol. 2004;164(4):1389–97. doi: 10.1016/S0002-9440(10)63225-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.HECQUET C, T.F., MARCIC BM, ERDOS EG. Human Bradykinin B2 Receptor Is Activated by Kallikrein and Other Serine Proteases. Mol Pharmacol. 2000;58(4):828–836. doi: 10.1124/mol.58.4.828. [DOI] [PubMed] [Google Scholar]