

Here, the expression, purification, crystallization and X-ray diffraction data of a family 3 β-glucosidase from the hyperthermophilic bacterium Thermotoga neapolitana are reported.
Keywords: glycoside hydrolase family 3, selenomethionine incorporation, Thermotoga neapolitana, multiple-wavelength anomalous dispersion
Abstract
β-Glucosidases belong to families 1, 3 and 9 of the glycoside hydrolases and act on cello-oligosaccharides. Family 1 and 3 enzymes are retaining and are reported to have transglycosylation activity, which can be used to produce oligosaccharides and glycoconjugates. Family 3 enzymes are less well characterized than their family 1 homologues and to date only two crystal structures have been solved. Here, the expression, purification, crystallization and X-ray diffraction data of a family 3 β-glucosidase from the hyperthermophilic bacterium Thermotoga neapolitana are reported. Crystals of selenomethionine-substituted protein have also been grown. The crystals belong to space group C2221, with unit-cell parameters a = 74.9, b = 127.0, c = 175.2 Å. Native data have been collected to 2.4 Å resolution and the structure has been solved to 2.7 Å using the selenomethionine MAD method. Model building and refinement of the structure are under way.
1. Introduction
Glycoside hydrolases (GH) are enzymes that hydrolyze glycosidic bonds between two or more carbohydrates or between a carbohydrate and a noncarbohydrate moiety. Carbohydrates are essential components of biomass, which is estimated to be produced in a quantity of about 60 Gt y−1 (Cox et al., 2000 ▶) and which contains an array of structural and storage polysaccharides. To utilize these raw materials, microorganisms produce a wide variety of carbohydrate-hydrolyzing and carbohydrate-modifying glycoside hydrolases. These enzymes can also be used as specific catalysts in industrial applications, e.g. in the food and feed industries, the paper and pulp, starch and textile industries and in newly emerging ‘green’ processes (Turner et al., 2006 ▶, 2007 ▶), taking advantage of their specificity in selective preparations of carbohydrate-containing raw materials.
Based on sequence similarities, GH have to date been classified into 108 separate families (Coutinho & Henrissat, 1999 ▶). β-Glucosidases (EC 3.2.1.21) play a role in the carbohydrate metabolism of many organisms by acting on the β-glycosidic linkages of cello-oligosaccharides containing β-d-1,4-glycosidic bonds. These enzymes are classified into three GH families: GH1, GH3 and GH9. Both GH1 and GH3 are families with a retaining mechanism and are dominated by enzymes acting on oligosaccharide substrates, while the GH9 family has an inverting mechanism and mostly contains endoglucanases. Retaining enzymes utilize a double-displacement mechanism with retention of configuration at the anomeric carbon of the sugar ring and often display transglycosylation abilities, which can be of interest for applications focusing on the synthesis of oligosaccharides or related products. The catalysis involves two carboxylate residues located on opposite sides of the sugar plane and the reaction can be divided into two steps: glycosylation, in which a glycosyl-enzyme intermediate is formed, and deglycosylation, in which a water molecule (hydrolysis) or an alcohol (transglycosylation) hydrolyzes the glycosyl-enzyme (McCarter & Withers, 1994 ▶; Sinnott, 1990 ▶). As fold is better conserved than sequence, many GH families have been grouped into structurally related clans (GH A–N; Coutinho & Henrissat, 1999 ▶). Clan A is by far the largest, containing 17 GH families all sharing the (β/α)8-fold, while the other clans (GH B–N) only contain two or three GH families each. GH1 β-glucosidases belong to clan A and have been more thoroughly characterized than the GH3 representatives. Several three-dimensional structures have been solved, some of which are from thermophiles, e.g. a β-glycosidase from Sulfolobus solfataricus (Aguilar et al., 1997 ▶) and a β-glucosidase from Thermotoga maritima (Zechel et al., 2003 ▶).
Enzymes classified as members of GH3 do not belong to any of the known GH clans, indicating a more unusual fold. Generally, knowledge of the function and structure of GH3 enzymes is less abundant, their sequence conservation is relatively low and few enzymes are well characterized. For instance, it is presently still impossible to locate the acid/base catalytic groups based on sequence homology, as even this region has low sequence conservation. Only two crystal structures have been solved to date of GH3 enzymes: a β-1,3-1,4-d-glucan exohydrolase (EC 3.2.1.58) from Hordeum vulgare (barley; Varghese et al., 1999 ▶; PDB code 1ex1) and a β-hexosaminidase (EC 3.2.1.52) from Vibrio cholera (New York Structural Genomics Consortium, unpublished work; PDB codes 1tr9 and 1y65).
The enzyme crystallized in this work originates from T. neapolitana (Tn) and is a β-glucosidase (Bgl) classified into GH3. The enzyme is abbreviated TnBgl3B, in accordance with the nomenclature proposed by Henrissat et al. (1998 ▶). The gene was isolated from DSM strain 4359 with the purpose of selecting a candidate catalyst for alkyl glucoside synthesis and the enzyme showed promising results in alkylglucoside production by transglycosylation (Turner et al., 2007 ▶). The structure with the highest homology to TnBgl3B, that of the barley enzyme, is only 20% identical overall; hence, structural information on TnBgl3B is essential in order to gain further knowledge on the function of the family 3 enzymes. In addition, TnBgl3B is the first thermostable representative of GH3, which may allow identification of thermostabilizing features.
2. Materials and methods
2.1. Chemicals
All chemicals were pro analysi from Merck Eurolabs (Darmstadt, Germany) unless otherwise stated.
2.2. Expression and purification of native protein
His-tagged TnBgl3B was produced in Escherichia coli strain Tuner (DE3) grown in minimal medium by fed-batch techniques (de Maré et al., 2005 ▶). Extracts from cultures, harvested 4 h after induction with IPTG (isopropyl β-d-1-thiogalactopyranoside), were purified in a two-step procedure. Heat treatment (343 K, 40 min) precipitated E. coli proteins, which were subsequently removed by centrifugation (27 000g, 30 min). The resulting supernatant containing TnBgl3B was loaded onto an immobilized metal-ion affinity chromatography column and purified as described previously (Turner et al., 2007 ▶).
2.3. SeMet incorporation
The bgl3b-containing plasmid was transformed by electroporation into the methionine-auxotrophic E. coli strain B834 (DE3) (Novagen, Madison, WI, USA). Colonies were inoculated into minimal medium with 25 mg ml−1 seleno-l-methionine (SeMet), cultivated at 310 K with shaking for about 2 d and then transferred to a fresh 200 ml culture of minimal medium supplemented with SeMet and grown at 310 K. The cells were induced by the addition of IPTG to a final concentration of 1 mM when the optical density at 620 nm was 0.6 and expression was allowed to proceed for 2 h.
2.4. Purification of SeMet-Bgl3B
The cell culture was harvested by centrifugation at 5000g and 277 K for 5 min. The pellet was dissolved in 7 ml binding buffer (20 mM Tris–HCl, 0.75 M NaCl, 20 mM imidazole pH 7.5) and lysed by ultrasonication in a UP400S instrument (Dr Hielscher, Stuttgart, Germany). Soluble proteins were separated from the cell debris by centrifugation at 80 000g and 277 K for 10 min. The supernatant was passed through a 0.45 µm filter and purified on a 1 ml HiTrap chelating column (GE Healthcare, Sweden). The gel matrix was washed with deionized water before loading with five volumes of 5 mg ml−1 copper sulfate. The column was then washed with deionized water and equilibrated with 10 ml binding buffer. Crude extract was loaded onto the column and unbound proteins were washed off with 10 ml binding buffer. Elution was achieved with a gradient of 20–500 mM imidazole in 20 mM Tris–HCl, 0.75 M NaCl pH 7.5. 20 fractions of 1 ml each were collected. The fractions containing protein as observed in the chromatogram of light absorption at 280 nm were analyzed and used for crystallization. SDS–PAGE according to Laemmli (1970 ▶) was used to analyze the enzyme purity.
2.5. Protein analyses
In the standard assay for measuring β-glucosidase activity, p-nitrophenol is released from p-nitrophenyl-β-d-glucopyranoside (pNPG). 40 µl enzyme solution was added to 96 µl preheated 2.94 mM pNPG dissolved in 20 mM citrate–phosphate buffer pH 5.6 and incubated for 5 min in a QBD2 block heater (Grant, UK) at 358 K. After incubation, the samples were put on ice for 5 min and the absorbance at 405 nm was read using an Ultrospec 1000 spectrophotometer (GE Healthcare, Sweden). One unit corresponds to the amount of enzyme that will release 1 µmol p-nitrophenol per minute under the described conditions.
2.6. Mass-spectrometric analysis of SeMet incorporation
The molecular masses of the His-tagged proteins (native and SeMet-modified) were determined by electrospray ionization mass spectrometry using a QSTAR hybrid Pulsar i instrument (Applied Biosystems, CA, USA) equipped with a nano-ion source kit (Proxeon, Denmark). The protein samples were desalted using a C4 ZipTip (Millipore, MA, USA). 5 µl of the solution was then mixed with an equal volume of acetonitrile containing 0.1% formic acid before being applied to the nanospray capillary. The nanospray source was set to positive-ion mode with a source voltage of +0.8 kV. The quadrupole system was adjusted to scan between 800–3000 m/z in TOF–MS mode and charge envelopes of the protein variants were obtained from 120 s of data accumulation.
2.7. Crystallization
Purified protein was dialyzed against 20 mM MES buffer pH 6.2 and concentrated to 3–5 mg ml−1. Suitable initial protein concentrations were found using the Pre-Crystallization Screen (PCT, Hampton Research, CA, USA). Initial crystallization conditions were found using the PACT Premier screen (Molecular Dimensions Ltd, UK; Newman et al., 2005 ▶). Crystallization trials were set up in Greiner low-profile 96-well plates using a Mosquito robot (TTP Labtech, UK) at 293 K. Drops consisting of 100 nl protein solution mixed with 100 nl reservoir solution were equilibrated against 80 µl reservoir solution. Parallel trials were set up for the native protein at a concentration of 5 mg ml−1 and for the SeMet protein at 3.1 mg ml−1. Small crystals of both proteins appeared after 4 d in drops where the precipitant was 20% PEG 3350 in 0.1 M bis-Tris propane pH 7.5 and any of 0.2 M NaBr, NaI, KSCN and Na2SO4. The most promising drops were optimized with respect to PEG and salt concentration. The crystals used for data collection grew in hanging drops consisting of 1 µl protein solution and 1 µl reservoir solution equilibrated against 1 ml reservoir solution. The best crystallization conditions for the native protein were 16–20% PEG 3350, 0.1–0.2 M NaI and for the SeMet protein they were 20–24% PEG 3350, 0.1–0.25 M NaI, both in 90 mM bis-Tris propane buffer pH 7.4.
2.8. Data collection
All crystals were cryoprotected with 25% glycerol, 20% PEG 3350, 0.2 M NaI, 90 mM bis-Tris propane pH 7.4 and flash-cooled directly in a liquid-nitrogen stream from an Cryostream cooler (Oxford Cryosystems, UK). The crystals could be soaked in the cryosolution for a period of a few seconds to a few minutes without any observable difference in diffraction quality. Crystals were pre-screened at station I911-5 of the MAX-II synchrotron (Lund, Sweden). Three-wavelength SeMet MAD data to 2.7 Å resolution were collected at station ID29 at the ESRF synchrotron (Grenoble, France) from a crystal measuring approximately 400 × 80 × 20 µm grown in 20% PEG 3350, 0.15 M NaI, 90 mM bis-Tris propane pH 7.4. The oscillation range was 0.7° and 160 images were collected for each wavelength. A native data set to 2.4 Å resolution was collected from a crystal measuring approximately 400 × 200 × 20 µm grown in 16% PEG 3350, 0.1 M KBr, 90 mM bis-Tris propane pH 7.4. The oscillation range was 0.5° and 244 images were collected. The beam size was 50 × 50 µm and the rod-shaped crystals were translated to expose a fresh volume after each wavelength (for the MAD data) and once during each wavelength. Several MAD data sets were collected from separate crystals and that producing the best overall figure of merit for the experimental phases was used for structure determination (Table 1 ▶). Data were integrated and scaled using XDS (Kabsch, 1993 ▶, 2001 ▶). Further data reduction and manipulation used the CCP4 package (Collaborative Computational Project, Number 4, 1994 ▶) driven through the CCP4i interface (Potterton et al., 2003 ▶). The anomalous scattering substructure was solved using autoSHARP (de La Fortelle & Bricogne, 1997 ▶), exploiting SHELXD for Patterson function solution (Usón et al., 2003 ▶). Phases were improved by solvent flipping using SOLOMON (Abrahams & Leslie, 1996 ▶). The optimal solvent content for this process was 47.8%.
Table 1. X-ray data-collection and phasing statistics.
Values in parentheses are for the highest resolution shell.
| SeMet | ||||
|---|---|---|---|---|
| Peak | Inflection point | High-energy remote | Native | |
| Data collection | ||||
| Unit-cell parameters | a = 74.9, b = 127.0, c = 175.2 | a = 74.9, b = 127.0, c = 175.2 | a = 74.9, b = 127.0, c = 175.2 | a = 74.9, b = 127.2, c = 175.2 |
| Wavelength (Å) | 0.97909 | 0.97924 | 0.97565 | 0.97906 |
| Resolution (Å) | 24–2.7 (2.87–2.7) | 24–2.7 (2.87–2.7) | 19–2.9 (3.07–2.9) | 40–2.4 (2.5–2.4) |
| Rmerge† (%) | 6.3 (43.7) | 6.4 (41.1) | 7.9 (46.3) | 6.4 (45.3) |
| Total observations | 102212 | 99797 | 82094 | 143480 |
| Unique reflections | 43310 | 42644 | 36227 | 32799 |
| Average redundancy‡ | 2.4 | 2.3 | 2.3 | 4.4 |
| Completeness (%) | 97.6 (97.4) | 97.2 (96.0) | 97.3 (97.5) | 98.9 (98.2) |
| 〈I/σ(I)〉 | 10.7 (2.1) | 11.0 (2.1) | 9.9 (1.8) | 16.7 (3.7) |
| Snorm/Sano§ | 1.16 (1.04) | 1.15 (1.03) | 1.07 (1.04) | 1.02 (1.02) |
| Phasing (to 2.7 Å) | ||||
| Phasing power (dispersive/anomalous) | —/0.813 | 0.071/0.806 | 0.218/0.588 | |
| RCullis¶ (dispersive/anomalous) | —/0.862 | 0.678/0.868 | 0.876/0.963 | |
| Mean figure of merit (centric/acentric) | 0.168/0.358 | |||
R
merge(I) = 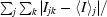
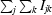 , where I
jk are the k individual observations of each reflection j and 〈I〉j is the value after weighted averaging.
, where I
jk are the k individual observations of each reflection j and 〈I〉j is the value after weighted averaging.
Friedel mates are treated as separate reflections for the MAD data set.
S norm/S ano = 〈σ(I)〉 assuming Friedel’s law to be true/〈σ(I)〉 assuming Friedel’s law to be false.
R
cullis = 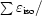
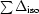 for acentric reflections and
for acentric reflections and 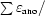
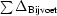 for anomalous differences, where ∊iso and ∊ano are the isomorphous and anomalous lack of closure, respectively, Δiso is the isomorphous difference and ΔBijvoet is the Bijvoet difference.
for anomalous differences, where ∊iso and ∊ano are the isomorphous and anomalous lack of closure, respectively, Δiso is the isomorphous difference and ΔBijvoet is the Bijvoet difference.
3. Results and discussion
3.1. Overall sequence
Glycoside hydrolase family 3 includes 873 gene sequences, of which almost all are bacterial (631) or eukaryotic (236). Despite the large number of sequences, rather few GH3 enzymes have been biochemically characterized. Only one, that from H. vulgare, has been characterized at the structural level (Varghese et al., 1999 ▶; Hrmova et al., 2004 ▶, 2005 ▶).
GH family 3 also includes thermostable enzymes and in this work we have crystallized a β-glucosidase originating from the hyperthermophile T. neapolitana (DSM strain 4359). The amino-acid sequence of TnBgl3B is 96% identical to a β-glucosidase isolated from a different strain of the same species and the major difference is a nonconserved stretch close to the N-terminus (Zverlov et al., 1997 ▶). The TnBgl3B sequence was aligned against a number of other sequences from the GH3 family (Fig. 1 ▶). The overall sequence identity with the structurally determined barley enzyme (HvBgl) is only 20%. Distinct phylogenetic clusters of enzymes within the family have been identified, with six major branches (Harvey et al., 2000 ▶). TnBgl3B is located in cluster 5, together with enzymes from T. maritima (AE001690), Prevotella ruminicola (U35425), Clostridium thermocellum (X15644), C. stercorarium (Z94045) and Ruminicoccus albus (U92808). TnBgl3B is relatively distant from the H. vulgare enzyme, which is found in cluster 1 together with representatives from a number of other plants. Alignments also showed a difference in the length of the sequence between these two enzymes, with the thermostable TnBgl3B having a C-terminal extension of approximately 115 amino acids, indicating a difference in the number of domains. This finding is also supported by cluster analysis (Harvey et al., 2000 ▶).
Figure 1.

Amino-acid sequence alignment of glycoside hydrolase family 3. The regions surrounding the putative active-site region are shown. Abbreviations: TnBglB, Thermotoga neapolitana DSM 4359 β-glucosidase 3B; TmBgl, T. maritima MSB8 β-glucosidase; FnGH, Fervidobacterium nodosum glycoside hydrolase; AhBgl, Aeromonas hydrophila β-glucosidase; FspBgl, Flavobacterium sp. MED217 β-glucosidase; PaBgl, Prevotella albensis β-glucosidase; YpBgl, Yersinia pestis Nepal516 β-glucosidase; CtBglB, Clostridium thermocellum β-glucosidase B; CsBgl, C. stercorarium β-glucosidase; PspGlc, Paenibacillus sp. TS12 glucocerebrosidase; AnBgl, Aspergillus niger β-glucosidase; FmBgl, Flavobacterium meningosepticum β-glucosidase; HvBgl, barley β-d-glucan exohydrolase ExoI. Amino acids in bold have been determined experimentally as catalytic amino acids, an inverted triangle denotes the position of the nucleophile and diamonds denote the positions of verified acid/base residues.
3.2. Production and SeMet incorporation
Production of the His-tagged β-glucosidase in E. coli Tuner (DE3) [maximum specific growth rate (μmax) = 0.7 h−1 at 310 K] resulted in an intracellular recombinant protein level corresponding to approximately 15% of the total protein.
The methionine-auxotrophic strain grew slowly (μmax = 0.3 h−1 at 310 K), but the SeMet-substituted TnBgl3B also constituted approximately 15% of the total protein. Both the SeMet-substituted and the native glucosidase were successfully purified to about 90% (Fig. 2 ▶) and the activity of the purified samples was verified (data not shown).
Figure 2.

SDS–PAGE analysis of protein purified by IMAC. (a) SeMet-TnBgl3B, (b) TnBgl3B.
SeMet incorporation was confirmed by nanospray mass spectrometry. Purified samples of His-tagged native and SeMet-TnBgl3B were examined and the average masses were 82 225 and 82 740 Da, respectively. The additional mass of 515 Da corresponds to the difference obtained when selenium replaces sulfur in 11 methionine residues, the total number in the sequence. SeMet therefore occupies all possible methionine sites in the protein.
3.3. Crystallization and data collection
Both native TnBgl3B and SeMet-TnBgl3B crystallized in the PACT Premier Screen and the conditions were further optimized. Crystals belong to space group C2221, with the unit-cell parameters given in Table 1 ▶. There is one TnBgl3B molecule in the asymmetric unit, giving a solvent content of 57%.
The crystals used for data collection were plate-shaped and had dimensions of around 300 × 200 × 20 µm (Fig. 3 ▶). X-ray data were collected to a resolution of 2.7 Å for the SeMet protein and 2.4 Å for the native protein (Fig. 4 ▶ and Table 1 ▶).
Figure 3.

A native TnBgl3B crystal in the X-ray beam at beamline ID29 of the ESRF. The length of the crystal is approximately 400 µm.
Figure 4.

First diffraction image from the native TnBgl3B crystal. The blue circle indicates a resolution of 2.3 Å.
Phasing statistics are presented in Table 1 ▶. The anomalous signal was weak: autoSHARP suggested resolution cutoffs of 3.67 and 4.1 Å for the peak and inflection-point data, respectively, despite a diffraction resolution limit of 2.7 Å. Ten Se positions were found and refined to occupancies of 0.4–0.9. The low occupancies were unexpected, as the incorporation of SeMet had been shown to be 100% by mass spectrometry. However, two different batches of protein were used: one for checking the incorporation of SeMet and one for growing the crystals from which the X-ray data were collected. The same protocol for SeMet incorporation was used, but the reproducibility of incorporation was not tested. Some of the methionine residues may be on the surface and thus less well ordered. Nevertheless, the electron-density maps after solvent flattening were of good quality and manual model building and refinement are in progress.
Acknowledgments
We thank the beamline staff at MAX-lab and the ESRF for assistance with data collection. The B834 (DE3) strain was a kind gift from Dr Claes von Wachenfeldt, Department of Cell and Organism Biology, Lund University. Financial support from The Foundation for Strategic Environmental Research, Mistra to ENK and PT and from the Swedish Research Council (Vetenskapsrådet) to ENK and DL is gratefully acknowledged. ENK also acknowledges the Krapperup Foundation for additional support.
References
- Abrahams, J. P. & Leslie, A. G. W. (1996). Acta Cryst. D52, 30–42. [DOI] [PubMed]
- Aguilar, C. F., Sanderson, I., Moracci, M., Ciaramella, M., Nucci, R., Rossi, M. & Pearl, L. H. (1997). J. Mol. Biol.271, 789–802. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Coutinho, P. M. & Henrissat, B. (1999). Recent Advances in Carbohydrate Bioengineering, edited by H. J. Gilbert, G. Davies, B. Henrissat & B. Svensson, pp. 3–12. Cambridge: Royal Society of Chemistry.
- Cox, P. M., Betts, R. A., Jones, C. D., Spall, S. A. & Totterdell, I. J. (2000). Nature (London), 408, 184–187. [DOI] [PubMed]
- Harvey, A. J., Hrmova, M., De Gori, R., Varghese, J. N. & Fincher, G. B. (2000). Proteins, 41, 257–269. [DOI] [PubMed]
- Henrissat, B., Teeri, T. T. & Warren, R. A. J. (1998). FEBS Lett.425, 352–354. [DOI] [PubMed]
- Hrmova, M., De Gori, R., Smith, B. J., Vasella, A., Varghese, J. N. & Fincher, G. B. (2004). J. Biol. Chem.279, 4970–4980. [DOI] [PubMed]
- Hrmova, M., Streltsov, V. A., Smith, B. J., Vasella, A., Varghese, J. N. & Fincher, G. B. (2005). Biochemistry, 44, 16529–16539. [DOI] [PubMed]
- Kabsch, W. (1993). J. Appl. Cryst.26, 795–800.
- Kabsch, W. (2001). International Tables for Crystallography, Vol. F, edited by M. G. Rossmann & E. Arnold, pp. 730–734. Dordrecht: Kluwer Academic Publishers.
- La Fortelle, E. de & Bricogne, G. (1997). Methods Enzymol.276, 472–494. [DOI] [PubMed]
- Laemmli, U. (1970). Nature (London), 227, 680–685. [DOI] [PubMed]
- McCarter, J. D. & Withers, S. (1994). Curr. Opin. Struct. Biol.4, 885–892. [DOI] [PubMed]
- de Maré, L., Velut, S., Ledung, E., Cimander, C., Norrman, B., Karlsson, E. N., Holst, O. & Hagander, P. (2005). Biotechnol. Lett.27, 983–990. [DOI] [PubMed]
- Newman, J., Egan, D., Walter, T. S., Meged, R., Berry, I., Ben Jelloul, M., Sussman, J. L., Stuart, D. I. & Perrakis, A. (2005). Acta Cryst. D61, 1426–1431. [DOI] [PubMed]
- Potterton, E., Briggs, P., Turkenburg, M. & Dodson, E. (2003). Acta Cryst. D59, 1131–1137. [DOI] [PubMed]
- Sinnott, M. L. (1990). Chem. Rev.90, 1171–1202.
- Turner, C., Turner, P., Jacobson, G., Waldebäck, M., Sjöberg, P., Nordberg Karlsson, E. & Markides, K. (2006). Green Chem.8, 949–959.
- Turner, P., Svensson, D., Adlercreutz, P. & Karlsson, E. N. (2007). J. Biotechnol.130, 67–74. [DOI] [PubMed]
- Usón, I., Schmidt, B., von Bulow, R., Grimme, S., von Figura, K., Dauter, M., Rajashankar, K. R., Dauter, Z. & Sheldrick, G. M. (2003). Acta Cryst. D59, 57–66. [DOI] [PubMed]
- Varghese, J. N., Hrmova, M. & Fincher, G. B. (1999). Structure, 7, 179–190. [DOI] [PubMed]
- Zechel, D. L., Boraston, A. B., Gloster, T., Boraston, C. M., Macdonald, J. M., Tilbrook, D. M. G., Stick, R. V. & Davies, G. (2003). J. Am. Chem. Soc.125, 14313–14323. [DOI] [PubMed]
- Zverlov, V. V., Volkov, I. Y., Velikodvorskaya, T. V. & Schwarz, W. H. (1997). Microbiology, 143, 3537–3542. [DOI] [PubMed]