

The first crystals and the 2.8 Å X-ray structure of full-length recombinant human butyrylcholinesterase are reported.
Keywords: BChE, recombinant butyrylcholinesterase, tetramerization domain
Abstract
Human butyrylcholinesterase (BChE) has been shown to function as an endogenous scavenger of diverse poisons. BChE is a 340 kDa tetrameric glycoprotein that is present in human serum at a concentration of 5 mg l−1. The well documented therapeutic effects of BChE on cocaine toxicity and organophosphorus agent poisoning has increased the need for effective methods of producing recombinant therapeutic BChE. In order to be therapeutically useful, BChE must have a long circulatory residence time or associate as tetramers. Full-length recombinant BChE produced in Chinese hamster ovary (CHO) cells or human embryonic kidney cells has been shown to associate as monomers, with a shorter circulatory residence time than the naturally occurring tetrameric serum protein. Based on the preceding observation as well as the need to develop novel methodologies to facilitate the mass production of therapeutic recombinant BChE, studies have been initiated to determine the structural basis of tetramer formation. Towards these ends, full-length monomeric recombinant BChE has been crystallized for the first time. A 2.8 Å X-ray structure was solved in space group P4212, with unit-cell parameters a = b = 156, c = 146 Å.
1. Introduction
Human butyrylcholinesterase (BChE; EC 3.1.1.8) is a tetrameric glycoprotein comprised of four identical subunits, each with a molecular weight of 85 kDa and containing 574 amino acids and nine N-linked glycans (Lockridge et al., 1987 ▶). The C-terminal 40 amino acids constitute the tetramerization domain (Blong et al., 1997 ▶; Altamirano & Lockridge, 1999a ▶,b ▶). The BChE in human serum has a concentration of 5 mg l−1 and a half-life of 12 days (Ostergaard et al., 1988 ▶). BChE is synthesized by the liver and is present in highest amounts in serum, intestine, liver and lung (Jbilo et al., 1994 ▶).
BChE appears to function in the detoxification of ingested or inhaled poisons (Jbilo et al., 1994 ▶). BChE is reactive towards a wider variety of poisons than acetylcholinesterase (AChE) and in this way serves to protect AChE from inhibition. BChE also appears to have a function in the hydrolysis of acetylcholine, as deduced from studies of AChE-knockout mice, which are believed to survive to adulthood because of the activity of BChE in hydrolyzing acetylcholine (Li et al., 2000 ▶; Xie et al., 2000 ▶; Duysen et al., 2002 ▶; Mesulam et al., 2002 ▶).
Prior to this study no structures of full-length BChE have been determined; however, several structures of truncated BChE have been reported (Nachon, Asojo et al., 2005 ▶; Nachon, Nicolet et al., 2005 ▶; Nicolet et al., 2003 ▶). Truncated BChE lacks the tetramerization domain and is 100% monomeric in solution and in all reported crystal forms. Truncated BChE was engineered because of the difficulty in obtaining crystals of full-length BChE (Nachon et al., 2002 ▶).
Full-length BChE can be produced in Chinese hamster ovary (CHO) cells and human embryonic kidney cells. The circulatory residence time of cholinesterases is regulated by post-translational factors such as sialylation and tetramerization (Saxena et al., 1997 ▶, 1998 ▶; Kronman et al., 1995 ▶, 2000 ▶; Chitlaru et al., 1998 ▶, 2001 ▶). Recombinant full-length human BChE secreted by CHO cells is glycosylated and has a molar ratio of sialic acid to galactose residues of 1.0, indicating complete sialylation (Saxena et al., 1998 ▶). Despite its complete sialylation, recombinant full-length BChE has a circulatory half-life of only 2 min in rodents, as opposed to native BChE, which has a circulatory half-life of 56 h in rodents (Saxena et al., 1998 ▶; Duysen et al., 2002 ▶). However, milligram quantities of recombinant human BChE tetramers can be produced by co-expressing BChE with proline-rich peptides such as PRAD (Duysen et al., 2002 ▶) in CHO cells. This yields recombinant BChE that is 70% tetramer and has a significantly improved circulatory residence time. Despite this improvement, the residence time remains significantly lower than that of serum BChE, which is principally tetrameric (98%). This suggests that the circulatory half-life of BChE is dependent on the formation of tetramers.
A higher percentage of BChE tetramerization appears to correlate with increased circulatory residence time and consequently possible therapeutic use. As a result of this observation and of the need to develop novel methodologies to facilitate the mass production of therapeutic recombinant BChE, studies are ongoing to determine the structural basis of tetramer formation. Towards these ends, we report here the first crystals and X-ray structure of full-length recombinant BChE.
2. Materials and methods
2.1. Recombinant expression and purification of BChE
Full-length human BChE, consisting of 574 amino acids and six Asn-linked glycans on Asn57, Asn106, Asn241, Asn256, Asn341 and Asn485, was expressed in CHO cells. The four glycosylation sites at Asn17, Asn455, Asn481 and Asn486 were deleted by mutating Asn to Gln. Asn485 is not glycosylated in wild-type BChE, but is glycosylated in the 4-sugars-off mutant used in these studies. The locations of the glycosylation sites in the full-length recombinant BChE were the same as in the truncated BChE, the crystal structure of which has previously been solved (Nachon et al., 2002 ▶; Nicolet et al., 2003 ▶). The accession number for the human BChE amino-acid sequence is gi:116353.
The mammalian expression plasmid contained rat glutamine synthetase to allow the selection of transfected cells in 25 µM methionine sulfoximine. CHO cells were grown in 2 l roller bottles and the culture medium was collected over a period of six months until 36 l had accumulated. The serum-free and glutamine-free culture medium (Ultraculture, BioWhittaker, Walkersville, MD, USA; catalogue No. 12-725B) had a total of 210 000 units of BChE activity, which corresponds to about 300 mg BChE. BChE was purified by affinity chromatography on procainamide-Sepharose eluted with NaCl, followed by ion-exchange chromatography on Whatman DE52 in a gravity-flow column and finally ion-exchange chromatography on a 10 × 100 mm Protein-Pak anion-exchange column (DEAE 8HR 1000 Å 8 µm, Part No. 35650; Waters/Millipore, Milford, MA, USA) on a Waters HPLC. The yield was 54 mg pure BChE.
2.2. Crystallization
The concentration of the pure BChE protein solution used for crystallization experiments was 8 mg ml−1 in a solution comprised of 50 mM Tris–HCl and 10 mM MES pH 8.0. Crystals were grown at 293 K by vapor diffusion in sitting drops. Crystallization drops were prepared by mixing 5 µl protein solution with 1.5 µl reservoir solution. Crystals or crystalline precipitate were obtained from reservoir solutions containing high concentrations of at least 1.6 M ammonium sulfate and moderate amounts of other additives. Additives included 0.2 M diammonium phosphate, 0.2 M ammonium iodide and 0.5 M lithium chloride. The largest crystals, which were less than 0.1 mm on the smallest face, were obtained within three weeks. Two crystal morphologies were obtained. Smaller chunk-shaped crystals were obtained at acidic pH using either 0.1 M MES or Tris buffer at pH 6.5 (Fig. 1 ▶ a), while larger plate-like crystals were obtained from solutions at greater than pH 7.5 (Fig. 1 ▶ b). The crystals were cryoprotected by 5 min soaks in solutions containing 15% glycerol and equivalent amounts of precipitants as mother liquor. The crystals were then flash-cooled directly in a stream of N2 prior to data collection at 100 K. Cocrystallization attempts were also carried out with a tetramerization peptide (PPPPPPPPPPPPPPPLP). This 17-mer peptide was chemically synthesized by GenScript (http://www.genescript.com) and confirmed by HPLC to be greater than 95% pure. The polyproline-rich peptide was added to pure BChE in attempts to induce monomers to assemble into tetramers.
Figure 1.

The two crystal morphologies of full-length monomeric BChE. (a) Form I crystals are smaller (less than 0.2 mm on the largest face) low-resolution crystals obtained in solutions buffered at acidic pH, while (b) form II crystals are larger crystals (greater than 0.8 mm on the largest face) that diffract to higher resolution.
2.3. Data collection and structure determination
X-ray diffraction data extending to 4.6 and 2.8 Å were collected from crystal forms I (acidic buffered crystals) and II (basic pH crystals), respectively. Both data sets were collected using an R-AXIS IV++ detector (Rigaku Research). The X-ray source was a Rigaku Micromax-007 rotating-anode generator operating at 40 kV and 20 mA with an Osmic double-focused mirror system. A complete data set for crystal form I was collected from a single crystal using a crystal-to-detector distance of 200 mm and an exposure time of 60 min per 1.0° oscillation. The data set from crystal form II was also collected from a single crystal using a crystal-to-detector distance of 150 mm and an exposure time of 20 min per 0.5° oscillation. Both X-ray data sets were processed using the HKL-2000 program suite (Otwinowski & Minor, 1997 ▶). Crystal form I had a monomer in the asymmetric unit and belonged to space group I422, with unit-cell parameters a = b = 155, c = 130 Å. These unit-cell parameters are slightly larger than those of the truncated form of BChE, which was cloned without all 40 C-terminus residues and had unit-cell parameters a = b = 154, c = 127 Å. Crystal form II belonged to space group P4212, with unit-cell parameters a = b = 156, c = 146 Å, and had a dimer in the asymmetric unit. At resolution limits below 4.5 Å, crystal form II can be integrated and scaled in space group I422. In addition, crystal form I can also be integrated and scaled in space group P4212, with similar unit-cell parameters to those of crystal form II. This suggests that despite their differences in morphology, both crystals actually belong to the same space group P4212 and it is the absence of higher resolution data that allows the assignment of space group I422 to crystal form I.
The 2.8 Å structure was solved by molecular replacement with Phaser (McCoy et al., 2005 ▶; Storoni et al., 2004 ▶; Read, 2001 ▶) using a polyalanine monomer of PDB entry 1xlw (Nachon, Asojo et al., 2005 ▶) stripped of ligands and waters as the search model. The molecular-replacement solution had a dimer in the asymmetric unit and had a solvent content of 62%, corresponding to a Matthews coefficient (V M) of 3.24 Å3 Da−1 (Matthews, 1968 ▶). The molecular-replacement solution model had R factors in the low-40% range and free R factors in the mid- to high-40% range. Most of the amino-acid residues of the truncated model were clearly visible in a 2F o − F c OMIT electron-density map calculated from molecular-replacement phases. The final model was obtained through iterative cycles of manual model building in O (Jones et al., 1991 ▶) followed by structure refinement using a maximum-likelihood refinement procedure and NCS phase restraints in the program REFMAC5 (Pannu et al., 1998 ▶; Murshudov et al., 1999 ▶, 1997 ▶) and was refined to an R factor of 22.4% and a free R factor of 29.7%. Data and model statistics are shown in Table 1 ▶.
Table 1. Crystallographic data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| PDB code | 2pm8 |
| Resolution (Å) | 50–2.8 (2.9–2.8) |
| Rmerge† (%) | 11.6 (52.9) |
| Completeness (%) | 95.0 (95.0) |
| Redundancy | 4.9 (3.0) |
| I/σ(I) | 5.6 (1.6) |
| Refinement statistics | |
| R factor‡ (%) | 22.5 (30.2) |
| Rfree§ (%) | 29.2 (36.2) |
| Mean B factor (Å2) | 56.4 |
| R.m.s. deviation from ideal | |
| Bond length (Å) | 0.012 |
| Bond angle (°) | 1.536 |
| Chiral (Å3) | 0.101 |
| Ramachandran plot | |
| Most favored regions (%) | 80.0 |
| Additional allowed regions (%) | 18.9 |
| Generously allowed regions (%) | 0.5 |
| Disallowed regions (%) | 0.5 |
R
merge = 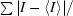
 , where I is the observed intensity and 〈I〉 is the average intensity obtained from multiple observations of symmetry-related reflections after rejections.
, where I is the observed intensity and 〈I〉 is the average intensity obtained from multiple observations of symmetry-related reflections after rejections.
R factor = 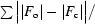
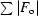 , where F
o are observed and F
c are calculated structure-factor amplitudes.
, where F
o are observed and F
c are calculated structure-factor amplitudes.
The R free set uses a randomly chosen 5% of reflections.
3. Results and discussion
We have solved a 2.8 Å structure of full-length recombinant BChE by molecular replacement. There are two monomers in the asymmetric unit (Fig. 2 ▶). The dimer is oriented such that the active-site gorge openings of the two monomers are on opposite sides. The main chains of the monomers overlay well, with an r.m.s. deviation of 0.319 Å. In addition, each monomer is similar to the monomer of truncated BChE; main-chain residues, side-chain residues and even sugars overlay. The r.m.s. deviation for an overlay of the main chains of each monomer with the main chains of a typical truncated BChE 530-stop structure (PDB code 1xlw) are 0.350 and 0.390 Å, respectively. The main differences between the 530-stop truncated structure and the monomers of the full-length structure lie in the presence of additional residues in the model. Firstly, the main chains of residues 378 and 379, which are consistently disordered in all the 530-stop truncated structures, are ordered in the recombinant full-length structures. Secondly, in the C-terminus there is additional electron density into which residues 530–534 can be built (Fig. 3 ▶); these residues are absent in all the reported 530-stop truncated structures, which only have unambiguous density to residue 529. The tetramerization domain (residues 535–574) is disordered and such carboxy-terminus disorder was also observed in the structure of Electrophorus electricus acetylcholinesterase (Bourne et al., 1999 ▶).
Figure 2.

Cartoon of the BChE dimer showing that each monomer has the typical fold. Each monomer is colored from blue at the N-terminus to red at the C-terminus.
Figure 3.

A 2F o − F c electron-density map at 1σ indicates the presence of residues 530–534. The electron-density map is in aquamarine.
The crystals do not diffract any better at the synchrotron beamline 14-BM-C at APS Chicago than at the rotating-anode home source. It is likely that the inherent disorder in the tetramerization domain may contribute to the difficulty in growing better diffracting crystals. Within the crystallographic unit cell, there is very limited interaction between the C-terminal tetramerization domains of the monomers that make up the crystallographic dimer.
The structure of a synthetic peptide representing the tetramerization domain of the homologous acetylcholinesterase in complex with a truncated tetramer-assembling peptide PRAD has been reported (Dvir et al., 2004 ▶). While the sequence of PRAD is CCLLTPPPPPLFPPPFF (Bon et al., 1997 ▶), the crystals were grown using a synthetic peptide that lacked the first two cysteines. Furthermore, the tetramerization domain could not be crystallized in the absence of the PRAD. This suggests a need for the tetramerization peptide for ordered formation of the tetrameric domain. The crystal structure of the acetylcholinesterase peptide structure shows tightly packed helices wrapped around PRAD (Fig. 4 ▶ b). The PRAD peptide makes numerous stacking interactions with the tryptophans of the tetramerization-domain peptide. The conserved tryptophans occur every seventh residue and are oriented toward the PRAD peptide. The prolines of PRAD interact with the tryptophans of the tetramerization domain (Fig. 4 ▶ b). The model shows that one PRAD peptide interleaves with the four tetramerization domains of four cholinesterase subunits (Fig. 4 ▶ b). These four helices of the tetramerization domain are parallel and form a superhelix. The PRAD molecule is antiparallel, adopting a left-handed polyproline type II conformation (Dvir et al., 2004 ▶). It is important to point out that there are three conserved Trp residues in the tetramerization domains of both AChE and BChE (Fig. 5 ▶). Interestingly, the electron density of the low-resolution structure of E. electricus acetylcholinesterase reveals a similar four-helix bundle of the tetramerization domain (Bourne et al., 1999 ▶) akin to that observed in the acetylcholinesterase peptide–PRAD complex structure. Such a packing of the tetramerization domain is impossible for our structure of recombinant full-length BChE (Fig. 4 ▶ a). Instead, the crystal packing does not allow interactions between any four C-termini (Fig. 4 ▶ a).
Figure 4.

The crystallographic tetramers of full-length recombinant BChE (a) are not mediated by the tetramerization domain as is the case in the cocrystal structure of the tetramerization domain of acetylcholinesterase with PRAD (b), PDB code 1vzj (Dvir et al., 2004 ▶), which instead has two monomers with C-termini oriented away from the other two monomers.
Figure 5.

Sequence of the 40-residue tetramerization domains of human AChE and BChE.
Our studies also show that while the addition of tetramerization peptide during recombinant expression facilitates the formation of tetrameric protein (Altamirano & Lockridge, 1999a ▶), addition to purified recombinant full-length BChE does not aid the formation of tetrameric crystals. In fact, all cocrystals or soaks of recombinant full-length BChE with the tetramer-assembling peptide yield crystal form II with disordered C-termini and peptide.
Thus, our crystal structures appear to have oligomerized to form nonbiological tetramers. If the crystallographic dimers and tetramers of recombinant BChE are similar to the solution dimers, this may account for the relatively short half-life of the recombinant protein relative to the native protein, as the oligomers are nonbiological and do not allow the C-terminal tetramerization domain to interact. The formation of nonbiological dimers does not affect the activity of recombinant BChE, as the catalytic site is distinct from the tetramerization domain. Recombinant full-length and even truncated BChE remain catalytically active, hydrolyzing a variety of substrates including cocaine. Nonetheless, the short residence time of monomeric recombinant BChE renders it therapeutically ineffective; studies are therefore under way to crystallize full-length tetrameric BChE in order to further our understanding of the structural basis of tetramer formation. There are two possible sources of tetrameric BChE: serum-derived protein and recombinant production via the addition of PRAD during expression. Since the recombinant BChE produced by the addition of PRAD during expression is a mixture of monomers, dimers and tetramers from which it is difficult and costly to isolate sufficient quantities of homogeneous tetramers, our current crystallization efforts are focused on serum-derived BChE, which is 98% tetrameric.
Supplementary Material
PDB reference: butyrylcholinesterase, 2pm8, r2pm8sf
References
- Altamirano, C. V. & Lockridge, O. (1999a). Biochemistry, 38, 13414–13422. [DOI] [PubMed]
- Altamirano, C. V. & Lockridge, O. (1999b). Chem. Biol. Interact.119–120, 53–60. [DOI] [PubMed]
- Blong, R. M., Bedows, E. & Lockridge, O. (1997). Biochem. J.327, 747–757. [DOI] [PMC free article] [PubMed]
- Bon, S., Coussen, F. & Massoulie, J. (1997). J. Biol. Chem.272, 3016–3021. [DOI] [PubMed]
- Bourne, Y., Grassi, J., Bougis, P. E. & Marchot, P. (1999). J. Biol. Chem.274, 30370–30376. [DOI] [PubMed]
- Chitlaru, T., Kronman, C., Velan, B. & Shafferman, A. (2001). Biochem. J.354, 613–625. [DOI] [PMC free article] [PubMed]
- Chitlaru, T., Kronman, C., Zeevi, M., Kam, M., Harel, A., Ordentlich, A., Velan, B. & Shafferman, A. (1998). Biochem. J.336, 647–658. [DOI] [PMC free article] [PubMed]
- Duysen, E. G., Bartels, C. F. & Lockridge, O. (2002). J. Pharmacol. Exp. Ther.302, 751–758. [DOI] [PubMed]
- Dvir, H., Harel, M., Bon, S., Liu, W. Q., Vidal, M., Garbay, C., Sussman, J. L., Massoulie, J. & Silman, I. (2004). EMBO J.23, 4394–4405. [DOI] [PMC free article] [PubMed]
- Jbilo, O., Bartels, C. F., Chatonnet, A., Toutant, J. P. & Lockridge, O. (1994). Toxicon, 32, 1445–1457. [DOI] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Kronman, C., Chitlaru, T., Elhanany, E., Velan, B. & Shafferman, A. (2000). J. Biol. Chem.275, 29488–29502. [DOI] [PubMed]
- Kronman, C., Velan, B., Marcus, D., Ordentlich, A., Reuveny, S. & Shafferman, A. (1995). Biochem. J.311, 959–967. [DOI] [PMC free article] [PubMed]
- Li, B., Stribley, J. A., Ticu, A., Xie, W., Schopfer, L. M., Hammond, P., Brimijoin, S., Hinrichs, S. H. & Lockridge, O. (2000). J. Neurochem.75, 1320–1331. [DOI] [PubMed]
- Lockridge, O., Bartels, C. F., Vaughan, T. A., Wong, C. K., Norton, S. E. & Johnson, L. L. (1987). J. Biol. Chem.262, 549–557. [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Mesulam, M. M., Guillozet, A., Shaw, P., Levey, A., Duysen, E. G. & Lockridge, O. (2002). Neuroscience, 110, 627–639. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A., Lebedev, A., Wilson, K. S. & Dodson, E. J. (1999). Acta Cryst. D55, 247–255. [DOI] [PubMed]
- Nachon, F., Asojo, O. A., Borgstahl, G. E., Masson, P. & Lockridge, O. (2005). Biochemistry, 44, 1154–1162. [DOI] [PubMed]
- Nachon, F., Nicolet, Y. & Masson, P. (2005). Ann. Pharm. Fr. 63, 194–206. [DOI] [PubMed]
- Nachon, F., Nicolet, Y., Viguie, N., Masson, P., Fontecilla-Camps, J. C. & Lockridge, O. (2002). Eur. J. Biochem.269, 630–637. [DOI] [PubMed]
- Nicolet, Y., Lockridge, O., Masson, P., Fontecilla-Camps, J. C. & Nachon, F. (2003). J. Biol. Chem.278, 41141–41147. [DOI] [PubMed]
- Ostergaard, D., Viby-Mogensen, J., Hanel, H. K. & Skovgaard, L. T. (1988). Acta Anaesthesiol. Scand.32, 266–269. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pannu, N. S., Murshudov, G. N., Dodson, E. J. & Read, R. J. (1998). Acta Cryst. D54, 1285–1294. [DOI] [PubMed]
- Read, R. J. (2001). Acta Cryst. D57, 1373–1382. [DOI] [PubMed]
- Saxena, A., Raveh, L., Ashani, Y. & Doctor, B. P. (1997). Biochemistry, 36, 7481–7489. [DOI] [PubMed]
- Saxena, A., Viragh, C., Frazier, D. S., Kovach, I. M., Maxwell, D. M., Lockridge, O. & Doctor, B. P. (1998). Biochemistry, 37, 15086–15096. [DOI] [PubMed]
- Storoni, L. C., McCoy, A. J. & Read, R. J. (2004). Acta Cryst. D60, 432–438. [DOI] [PubMed]
- Xie, W., Stribley, J. A., Chatonnet, A., Wilder, P. J., Rizzino, A., McComb, R. D., Taylor, P., Hinrichs, S. H. & Lockridge, O. (2000). J. Pharmacol. Exp. Ther.293, 896–902. [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: butyrylcholinesterase, 2pm8, r2pm8sf