

The structure of bovine pancreatic RNase A has been determined in complex with 2′-deoxyguanosine-5′-monophosphate (dGMP) at 1.33 Å resolution at room temperature in a previously unreported unit cell belonging to space group P31.
Keywords: RNase A, 2′-deoxyguanosine-5′-monophosphate
Abstract
The structure of bovine pancreatic RNase A has been determined in complex with 2′-deoxyguanosine-5′-monophosphate (dGMP) at 1.33 Å resolution at room temperature in a previously unreported unit cell belonging to space group P31. There are two molecules of nucleotide per enzyme molecule, one of which lies in the active-site cleft in the productive binding mode, whilst the guanine base of the other dGMP occupies the pyrimidine-specific binding site in a nonproductive mode such that it forms hydrogen bonds to the phosphate group of the first dGMP. This is the first RNase A structure containing a guanine base in the B2 binding site. Each dGMP molecule is involved in intermolecular interactions with adjacent RNase A molecules in the lattice and the two nucleotides appear to direct the formation of the crystal lattice. Because GMP may be produced during degradation of RNA, this association could represent an inhibitor complex and thereby affect the observed enzyme kinetics.
1. Introduction
Bovine pancreatic ribonuclease (EC 3.1.27.5), commonly known as RNase A, has been extensively studied by physical-chemical approaches and by X-ray crystallography for nearly 75 years. The chemical and structural properties of RNase A have been thoroughly reviewed by Richards &Wyckoff (1971 ▶), Blackburn & Moore (1982 ▶), Wlodawer (1985 ▶) and Raines (1998 ▶). RNase A was the fourth protein and the third enzyme (after myoglobin, lysozyme and carboxypeptidase) to have its structure solved by X-ray diffraction (Kartha et al., 1967 ▶; Wyckoff et al., 1967 ▶).
RNase A degrades ribonucleic acid by cleaving selective phosphodiester bonds along its length. The substrate must be single-stranded and the enzyme has a high specificity for a pyrimidine base on the 3′ side of the phosphodiester linkage to be hydrolyzed. Either a pyrimidine or a purine base can be present on the 5′ side. The mechanism of catalysis, which involves a nucleotide 2′,3′-cyclic phosphodiester intermediate, has been thoroughly explored and reviewed by Wlodawer (1985 ▶) and Raines (1998 ▶).
The enzyme consists of a single polypeptide chain of 124 amino acids (Smyth et al., 1963 ▶; Potts et al., 1962 ▶). The secondary structure of the protein, which contains four disulfide bonds, consists primarily of an elongated four-stranded β-sheet, along with three α-helices, each of which is about 2½ turns in length. The extended substrate binds in a long deep cleft passing across the center of the β-sheet, although binding interactions also occur at greater distance from the active site (McPherson, Brayer, Cascio et al., 1986 ▶; McPherson, Brayer & Morrison, 1986 ▶). Three amino-acid residues were identified early on as having particular importance in catalysis: these were His12 and His119, which are involved in general acid–base catalysis, and Lys41, which fixes the phosphate at the active center.
As a basis for the structural descriptions that follow, we note that substrate binding to RNase A has been described in terms of several subsites along the active-site cleft (Raines, 1998 ▶). These include the phosphoryl-binding subsites P0, P1 and P2, where subsite P1 is the active site at which hydrolysis and cleavage occur, and subsites B1, B2 and B3, in which nucleotide–base interactions occur. Subsite B1 is selective for pyrimidine bases and subsites B2 and B3 bind any nucleotide, although B2 has a preference for adenine bases (Raines, 1998 ▶).
We recently reported an investigation of the crystallization of macromolecules in the presence of various small molecules that might serve as reversible linkers between protein molecules during the crystallization process (McPherson & Cudney, 2006 ▶). Subsequent to the crystallization experiments, we investigated the crystal structures of several samples that produced crystals from these experiments (Larson et al., 2007 ▶). In that report, we briefly described the involvement of 2′-deoxyguanosine-5′-monophosphate (dGMP) in the lattice of a new crystal form of RNase A. We now present a full description of the structure of RNase A with dGMP produced by cocrystallization.
2. Materials and methods
2.1. Crystallization and data collection
Crystals were obtained in two weeks by the sitting-drop method (McPherson, 1999 ▶) using a reservoir consisting of 0.6 ml 30% PEG 3350 (in 0.1 M HEPES buffer pH 7) and sample drops composed of 2 µl protein stock solution (10–40 mg ml−1 in 0.1 M HEPES buffer pH 7) and 2 µl of a reagent mix containing dGMP, cholesterol, thymine and oxamic acid (each component had a concentration of approximately 0.1 M in 15% PEG 3350 and 0.1 M HEPES buffer pH 7) as previously described (McPherson & Cudney, 2006 ▶).
A single crystal (300 × 300 × 100 µm) was used for the X-ray diffraction experiment. It was mounted in a quartz capillary on an R-AXIS diffraction apparatus utilizing a Cu Kα rotating anode in an RU-200 X-ray generator equipped with Osmic mirrors and image-plate detectors. Data were collected at room temperature (∼295 K) using a 1.0° oscillation and 10 min exposure time per frame. A total of 123 frames of data were collected, all of which were used in data reduction. The intensity data were reduced to structure amplitudes using the program d*TREK (Pflugrath, 1999 ▶). A total of 27 155 unique reflections were obtained from 90 413 observations to a resolution limit of 1.33 Å, resulting in an R merge of 0.049. In order to have the highest data-to-parameter ratio for anisotropic refinement, all data were used in refinement even though the highest resolution shell was only 25% complete. The next highest resolution shell (1.38–1.44 Å) was 72% complete and the overall completeness was 89.3% to 1.33 Å. Table 1 ▶ lists data-processing statistics.
Table 1. Data-processing, refinement and model statistics.
Values in parentheses are for the highest resolution shell.
| Crystal data | |
| Space group | P31 |
| Z | 3 |
| Unit-cell parameters (Å) | a = b = 55.08, c = 39.20 |
| Data-processing statistics | |
| Resolution (Å) | 27.54–1.33 (1.38–1.33) |
| No. of reflections | 27155 |
| Redundancy | 3.33 (1.49) |
| 〈I/σ(I)〉 | 13.7 (0.8) |
| Completeness | 89.3 (24.9) |
| Rmerge† | 0.049 (0.491) |
| Structure-refinement statistics | |
| Resolution (Å) | 1.33 |
| No. of reflections (Fo = 0 removed) | 24138 |
| R/Rfree‡ (all data) | 0.119/0.177 |
| R/Rfree‡ [F > 4σ(F)] | 0.110/0.167 |
| Reflections in test set (%) | 11.1 |
| Least-squares parameters | 10604 |
| No. of restraints | 13218 |
| Model statistics | |
| Protein atoms (full/partial) | 908/84 |
| dGMP atoms (full/partial) | 46/0 |
| Water atoms (full/partial) | 80/57 |
| Geometry: r.m.s. deviations from ideal values (Å) | |
| Bonds | 0.012 |
| Angle distances | 0.032 |
| Planar groups | 0.030 |
| DELU (Å2) | 0.004 |
| SIMU (Å2) | 0.067 |
| ISOR (Å2) | 0.092 |
| Average isotropic B factors (Å2) | |
| Protein (full/partial) | 37.7/38.6 |
| dGMP (full/partial) | 34.1/— |
| Water (full/partial) | 54.9/54.6 |
| Ramachandran plot | |
| Most favored region | 102 [88.7%] |
| Allowed region | 13 [11.3%] |
| Generously allowed and disallowed regions | 0 [0.0%] |
R
merge = 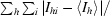
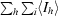 , where I
hi is the ith used observation of unique reflection h and 〈I
h〉 is the mean intensity of unique reflection h.
, where I
hi is the ith used observation of unique reflection h and 〈I
h〉 is the mean intensity of unique reflection h.
R = 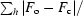
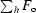 , where F
o and F
c are the observed and calculated structure-factor amplitudes.
, where F
o and F
c are the observed and calculated structure-factor amplitudes.
2.2. Structure solution and refinement
The crystals analyzed here were grown in Experiment III of the ‘silver bullets’ approach to crystallizing proteins described by McPherson & Cudney (2006 ▶). Analysis showed that of the mother-liquor components, only dGMP was incorporated into the crystal and there were not one but two molecules of dGMP per RNase A molecule. There were no indications that any of the other components were present in the crystals. The two dGMP molecules appear to be responsible for the formation of this unique trigonal crystal form.
The crystal grew with lattice symmetry corresponding to the trigonal space group P31, with unit-cell parameters a = b = 55.08, c = 39.20 Å and Z = 3. A search of RNase A structures in the Protein Data Bank (PDB; Berman et al., 2000 ▶) revealed that this is a new space group and unit cell for this enzyme. Therefore, it was necessary to solve the structure using molecular replacement (MR). PDB entry 1rno (Federov et al., 1996 ▶), after removing water and sulfate ions, was used as the search model in the MR routines of the program CNS (Brünger et al., 1998 ▶). The MR solution using 4 Å data gave a correlation coefficient of 0.56 for space group P31 compared with 0.26 for space group P32, verifying the choice of the former space group. Initial refinement using CNS yielded R = 0.283. F o − F c and 2F o − F c maps revealed two molecules of dGMP per molecule of RNase A. The protein model was rebuilt and the two dGMP molecules were built into density and added to the model. All rebuilding was performed using the program O (Jones & Kjeldgaard, 1994 ▶). Continued refinement with CNS produced an R factor of 0.236 (R free = 0.260), at which time waters were successively added to the model. A final CNS refinement of the model containing 153 water molecules gave R = 0.205 (R free = 0.224) at 1.33 Å resolution. Refinement was continued using SHELX-97 (Sheldrick & Schneider, 1997 ▶) with H atoms in riding positions and all non-H atoms treated anisotropically. Water molecules from the CNS model that were inconsistent with those found using SHELWAT were removed from the model and additional waters were added subject to the criteria that they had reasonable intermolecular contacts with the protein and/or other water molecules, that they were in density in 2F o − F c maps contoured at the 0.5σ level and that their B factors did not exceed 80 Å2 at either full or half occupancy. Refinement statistics are listed in Table 1 ▶. All figures were rendered with the program PyMOL (DeLano, 2002 ▶). Ramachandran statistics were produced with PROCHECK (Laskowski et al., 1993 ▶).
3. Results
3.1. The model
The final refined model contains residues 1–124 of the enzyme, two dGMP molecules and 137 water molecules and produced an R factor of 0.119 (R free = 0.177) and a goodness of fit of 1.433 for all data with F o > 0. The side chains of residues Ser15, Thr17, Ser22, Asn24, Asp53, Ser59, Asn62, Gln74, Ser77, Glu86, Gln101 and Asn103 exhibit discrete disorder, with the occupancies of the major components in the range 0.52–0.75. The side chains of residues Lys1, Lys37 and Lys91 were included in the model despite a lack of well defined electron density in 2F o − F c and F o − F c maps. There are 80 fully occupied water sites and 57 partially occupied sites. Other model statistics are given in Table 1 ▶.
3.2. The nucleotides and their interactions
As shown in Fig. 1 ▶, both dGMP molecules are bound in what is commonly described as the active-site cleft of the enzyme. The active-site cleft has traditionally been divided into the purine-binding pocket (subsites P1 and B2) and the pyrimidine-binding pocket (subsite B1), with hydrolysis effected at the phosphate (subsite P1) which joins the nucleotides by His12 and His119. The reaction yields oligonucleotide products having a pyrimidine nucleotide with a 3′-phosphate group. In this crystal, one dGMP (dGMP300) is observed in the purine site with the 5′-phosphate in its expected location, as has been seen in many previous complexes analyzed by X-ray crystallography (Wlodawer, 1985 ▶; Gilliland, 1997 ▶). There was nothing exceptional in this canonical mode of substrate binding.
Figure 1.

dGMP200 and dGMP300 are shown as bound in the active-site cleft of a molecule of bovine pancreatic RNase A. Nucleotide-binding subsites are labeled B1 (pyrimidine-specific), P1 (hydrolytic site) and B2 (nucleotide base binding site). The electron density from a 2F o − F c map from which the dGMP molecules have been omitted, contoured at 1.5σ, is superimposed.
The second dGMP (dGMP200) is not bound in a manner consistent with enzymatic catalysis and indeed it cannot be, as it is a purine and not a pyrimidine nucleotide. This dGMP blocks and to some extent fills the pyrimidine-binding pocket with its purine ring, as shown in Fig. 2 ▶(a), but the orientation is otherwise unconventional and represents some abortive or nonproductive mode of binding. The position of the base and sugar are reversed from the positions that would be occupied by a pyrimidine ribonucleotide. The guanine moiety makes two hydrogen bonds to the phosphate group of dGMP300, which sits at the catalytic center, through its amino group N6 and ring N atom N1. At the other end of the abortively bound and fully extended dGMP, its 5′-phosphate reaches out of the binding cleft and makes contact with another RNase A molecule in the lattice which is related by the 31 axis. There, it forms bonds with the side chains of Asn24 and Gln28 of the second protein molecule. Thus, the order of bonded components in the active-site cleft, 3′ to 5′, is base–sugar–phosphate–base–sugar–phosphate in a more-or-less linear arrangement, as seen in Fig. 3 ▶.
Figure 2.

(a) Detailed hydrogen-bonding interactions of dGMP200 with two molecules of RNase A, as well as with dGMP300. (b) The corresponding interactions of dGMP300. The ring-to-ring separation is approximately 3.5 Å between the guanine ring and Tyr115.
Figure 3.

Two nucleotides, dGMP200 in yellow and dGMP300 in red, link three RNase A molecules together in the crystal lattice, shown in cartoon representation, from left to right, in green, gray and yellow. Some amino-acid residues important to binding of the nucleotides are emphasized. There are 14 hydrogen-bonding interactions between the nucleotides and the three protein molecules.
dGMP300 is also not without some interest. Although it is bound as expected in the purine-binding site, it is also in close contact with another symmetry-related RNase A molecule in the lattice. This contact is in the form of a ring-upon-ring stacking interaction between the guanine base and the side-chain ring of Tyr115 of the adjacent protein molecule. Details of this interaction are seen in Fig. 2 ▶(b).
Deoxyribonucleotides prefer the sugar to assume a 2′-endo conformation when part of double-helical DNA structure, but can take on 2′-endo or 3′-endo conformations when they exist as free nucleotides (Altona & Sundaralingam, 1972 ▶). However, ribonucleotides are, with few exceptions, always in the 3′-endo conformation. Both of the nucleotides in this structure have 2′-endo sugar conformations. This suggests that, at least for the purine-binding site, the sugar conformation is of little consequence.
3.3. Intermolecular interactions
As seen in Fig. 3 ▶, at the 5′ end of the catalytic cleft the phosphate of the improperly bound dGMP is associated with Asn24 and Gln28 of a 31 screw-related RNase A molecule (at x = 2/3, y = 1/3), as well as with Arg85 of the RNase A providing the binding cleft. Thus, the phosphate of dGMP200 serves as an intermolecular bridge in the lattice along with five direct interprotein hydrogen bonds between these symmetry-related protein molecules, two of which are part of the extension of the β-strand structure of the primary protein molecule. At the opposite 3′ end of the cleft, the guanine ring of dGMP300 in the B2 binding site simultaneously stacks flush atop Tyr115 of another 31 screw-related RNase A molecule (at x = 1/3, y = 2/3), thereby providing a second intermolecular bridge. There are an additional six direct hydrogen bonds between these two protein molecules. Thus, the enzyme molecule is part of two helical configurations about two screw axes, in one of which the protein molecules are connected through the phosphate of dGMP200 (and other intermolecular interactions) and in the other through the ring stacking of the guanine ring of dGMP300 (and other intermolecular interactions). There is only one other direct intermolecular interaction between protein molecules not associated by these 31 screw axes.
4. Discussion
4.1. Importance of dGMP for crystal lattice formation
Although a multitude of RNase A and RNase A–nucleotide complex crystal forms have been grown and analyzed by X-ray diffraction analysis, this particular form is unique and has not been seen previously. The reason seems clear from the structure. Although there are numerous lattice contacts, the contacts involving the bound nucleotides must play an important role in directing the organization of the molecules and the formation of direct protein–protein interactions. Because of the hydrogen bonds formed between the guanine of dGMP200 and the phosphate of dGMP300, it is unlikely that a similar series of interactions would be formed using any other nucleotide. It is not clear that the absence of a hydroxyl at the 2′ position is relevant in this complex and GMP might have served as well.
It would appear to us that in spite of the unusual disposition of dGMP200 with respect to the B1 binding site, it must form this interaction with individual protein molecules in solution prior to crystallization. The six hydrogen bonds involving dGMP200, two to Arg85, two to Thr45 and two to the phosphate group of the dGMP300 (see Fig. 2 ▶ a), all suggest that it is tightly bound, although improperly bound in an enzymatic sense.
This analysis of the complex between RNase A and dGMP shows that the particular chemical groups presented by the guanosine moiety of GMP or dGMP are uniquely able to form multiple hydrogen bonds with the phosphate group at the active center. This allows it to associate nonproductively with RNase A in a manner that must, to at least some extent, inhibit its enzymatic activity. Because GMP can be produced during the normal course of RNA hydrolysis, this example of selective product inhibition should affect the observed kinetics of the reaction and the apparent K m of the enzyme for substrate.
4.2. The B1 binding site
Several structures of guanosine-containing dinucleotide complexes have been reported [PDB codes 1rnd, 1rnc (Aguilar et al., 1992 ▶), 1rca (Listgarten et al., 1995 ▶) and 1eow (Vitagliano et al., 2000 ▶)], all of which contain the guanine base in the B1 binding site rather than the B2 binding site. Each of these structures was crystallized in the presence of SO4 2− or PO4 2− ions which occupy the P1 binding site and apparently could not be displaced by the dinucleotides. These structures suggest that although the B1 binding site is selective for pyrimidine nucleotides for hydrolytic activity, the site is more selective for guanine than pyrimidine bases when the P1 site is already occupied by SO4 2− or PO4 2− groups to which the guanine can form two additional hydrogen bonds. In these particular structures, the pyrimidine moieties are disordered.
4.3. The B2 binding site
To our knowledge, the structure presented here is the first with a guanine base in the B2 binding site. Several structures of RNase A complexed with adenosine-containing nucleotides have been reported [PDB codes 1rcn (Fontecilla-Camps et al., 1994 ▶), 1z6s (Hatzopoulos et al., 2005 ▶), 1o0f, 1o0h (Leonidas et al., 2003 ▶), 1rpg (Zegers et al., 1994 ▶), 1jn4 (Jardine et al., 2001 ▶), 1qhc (Leonidas et al., 1999 ▶) and 1afk (Leonidas et al., 1997 ▶)]. The adenine ring in those structures containing a 5′-monophosphate group (1rcn, 1z6s, 1o0f and 1rpg) bind such that Asn71 forms hydrogen bonds to the N6 and N1 atoms of the adenine ring (see Fig. 4 ▶ a). Where the adenosine has a 5′-diphosphate group (1jn4, 1qhc, 1afk and 1o0h), the ring is oriented such that Asn71 forms hydrogen bonds with the N6 and N7 atoms of the adenine ring. Perhaps the most interesting of these structures is 1rcn, in which RNase A is complexed with the pentanucleotide d(ApTpApApG), the 2′-deoxy analog of a natural substrate. The terminal guanosine monophosphate group is disordered in this structure. The thymidine is in the B1 site as expected and the next two adenosine nucleotides occupy the B2 and B3 sites. As noted above, Asn71 anchors the adenine ring in the B2 site through hydrogen bonds to atoms N1 and N6. The adenine ring in the B3 site is loosely stacked on the adenine ring in the B2 site and is also hydrogen bonded to Gln69 through atom N1. In our structure (Fig. 4 ▶ b), dGMP300 in the B2 site is hydrogen bonded to Gln69 through atom O6 and to Glu111 through the amino atom N2, but only interacts with Asn71 through a water molecule.
Figure 4.

Relative positions of the purine rings in (a) structure 1rcn, (b) the present structure and (c) structure 1z6d after superposition of the Cα atoms of the active-site residues His12, Thr45, His119 and Phe120. The active-site residues in the background are displayed as line drawings to establish a point of reference for the positioning of the purine rings in each structure. The interactions for each purine ring are indicated by dashed lines. Residue Asn71 is fairly constantly disposed with respect to the active-site residues, while Gln69 and Glu111 move to accommodate the various base rings of the bound nucleotides at the B2 site. The displacement is approximately in the plane of the adenine ring with a magnitude of ∼2 Å for the guanine ring and of ∼4 Å for the hypoxanthine ring. This demonstrates how the enzyme can accommodate a variety of bases in the B2 site.
The guanosine-5’-monophosphate analog inosinic acid (IMP) provides the only other example of a non-adenine base in the B2 binding site (PDB code 1z6d; Hatzopoulos et al., 2005 ▶). Although IMP has atom O6 with which it could interact with Gln69, analogous to dGMP300, it does not. Instead, the anchoring interaction is between atom N1 of the ring and Glu111 (Fig. 4 ▶ c). Again, interaction with Asn71 is only through a water molecule. When the Cα atoms of the active-site residues His12, Thr45, His119 and Phe120 are superimposed (as seen in Fig. 4 ▶), the relative positions of the adenine (1rcn), guanine (our structure) and hypoxanthine (1z6d) rings are such that it appears as though the base ring slides across the imidazole ring of His119 with the guanine ring displaced ∼2 Å from the adenine position and the hypoxanthine ring displaced ∼4 Å owing to the change in interactions. IMP is also present in site B1 in a nonproductive binding mode in exactly the same manner as exhibited in all the nonproductively bound guanosine structures, except that hypoxanthine can only form a single hydrogen bond with the phosphate group in the P1 site.
4.4. Arg85 involvement
Of all the structures with a purine base at the B1 binding site, the structure presented here is the only one showing a strong interaction between Arg85 and the phosphate group. In the structure presented here, Asp83 forms a salt bridge with Arg85 through atoms N∊ and Nη2. Despite the proximity of Arg85 to Asp83 and to the phosphate groups of structures containing a purine nucleotide in binding site B1, the Arg85 residue does not interact with either of these moieties.
Supplementary Material
PDB reference: RNase A–dGMP complex, 2qca, r2qcasf
Acknowledgments
This work was supported by NIH grant GM074899 for the establishment of the Center for High Throughput Structural Biology. The authors thank Steve Mayclin for his assistance in preparing the figures.
References
- Aguilar, C. F., Thomas, P. J., Mills, A., Moss, D. S. & Palmer, R. A. (1992). J. Mol. Biol.224, 265–267. [DOI] [PubMed]
- Altona, C. & Sundaralingam, M. (1972). J. Am. Chem. Soc.94, 8205–8212. [DOI] [PubMed]
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000). Nucleic Acids Res.28, 235–242. [DOI] [PMC free article] [PubMed]
- Blackburn, P. & Moore, S. (1982). The Enzymes, edited by P. D. Boyer, 3rd ed., Vol. 15, pp. 317–340. New York: Academic Press.
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- DeLano, W. L. (2002). The PyMOL Molecular Visualization System. DeLano Scientific, San Carlos, CA, USA. http://www.pymol.org.
- Federov, A. A., Joseph-McCarthy, D., Federov, E., Sirakova, D., Graf, I. & Almo, S. C. (1996). Biochemistry, 35, 15962–15979. [DOI] [PubMed]
- Fontecilla-Camps, J. C., de Llorens, R., le Du, M.-H. & Cuchillo, C. M. (1994). J. Biol. Chem.269, 21526–21531. [DOI] [PubMed]
- Gilliland, G. L. (1997). Ribonucleases: Structures and Functions, edited by G. D’Alessio & J. F. Riordan, pp. 305–341. New York: Academic Press.
- Hatzopoulos, G. N., Leonidas, D. D., Kardakaris, R., Kobe, J. & Oikonomakos, N. G. (2005). FEBS J.272, 3988–4001. [DOI] [PubMed]
- Jardine, A. M., Leonidas, D. D., Jenkins, J. L., Park, C., Raines, R. T., Acharya, K. R. & Shapiro, R. (2001). Biochemistry, 40, 10262–10272. [DOI] [PubMed]
- Jones, T. A. & Kjeldgaard, M. (1994). O – The Manual, v.5.10. Uppsala, Sweden: Uppsala University Press.
- Kartha, G., Bello, J. & Harker, D. (1967). Nature (London), 213, 862–865. [DOI] [PubMed]
- Larson, S. B., Day, J. S., Cudney, R. & McPherson, A. (2007). Acta Cryst. D63, 310–318. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Leonidas, D. D., Chavali, G. B., Oikonomakos, N. G., Chrysina, E. D., Kosmopoulou, M. N., Vlassi, M., Frankling, C. & Acharya, K. R. (2003). Protein Sci.12, 2559–2574. [DOI] [PMC free article] [PubMed]
- Leonidas, D. D., Shapiro, R., Irons, L. I., Russo, N. & Acharya, K. R. (1997). Biochemistry, 36, 5578–5588. [DOI] [PubMed]
- Leonidas, D. D., Shapiro, R., Irons, L. I., Russo, N. & Acharya, K. R. (1999). Biochemistry, 38, 10287–10297. [DOI] [PubMed]
- Listgarten, J. N., Maes, D., Wyns, L., Aguilar, C. F. & Palmer, R. A. (1995). Acta Cryst. D51, 767–771. [DOI] [PubMed]
- McPherson, A. (1999). Crystallization of Biological Macromolecules. New York: Cold Spring Harbor Laboratory Press.
- McPherson, A., Brayer, G., Cascio, D. & Williams, R. (1986). Science, 232, 765–768. [DOI] [PubMed]
- McPherson, A., Brayer, G. D. & Morrison, R. D. (1986). J. Mol. Biol.189, 305–327. [DOI] [PubMed]
- McPherson, A. & Cudney, R. (2006). J. Struct. Biol.156, 387–406. [DOI] [PubMed]
- Pflugrath, J. W. (1999). Acta Cryst. D55, 1718–1725. [DOI] [PubMed]
- Potts, J. T., Berger, A., Cooke, J. & Anfinsen, C. B. (1962). J. Biol. Chem.237, 1851–1855. [PubMed]
- Raines, R. T. (1998). Chem. Rev.98, 1045–1065. [DOI] [PubMed]
- Richards, F. M. & Wyckoff, H. W. (1971). The Enzymes, edited by P. D. Boyer, 3rd ed., Vol. 4, pp. 647–806. New York: Academic Press.
- Sheldrick, G. M. & Schneider, T. R. (1997). Methods Enzymol.277, 319–343. [PubMed]
- Smyth, D. G., Stein, W. H. & Moore, S. (1963). J. Biol. Chem.238, 227–234. [PubMed]
- Vitagliano, L., Merlino, A., Zagari, A. & Mazzarella, L. (2000). Protein Sci.9, 1217–1225. [DOI] [PMC free article] [PubMed]
- Wlodawer, A. (1985). Biological Macromolecules and Assemblies, Vol. 2, edited by F. A. Jurnak & A. McPherson, pp. 393–439. New York: John Wiley & Sons.
- Wyckoff, H. W., Doscher, M., Tsernoglou, D., Inagami, T., Johnson, L. N., Hardman, K. D., Allewell, N. M., Kelly, D. M. & Richards, F. M. (1967). J. Mol. Biol.27, 563–578. [DOI] [PubMed]
- Zegers, I., Maes, D., Dao-Thi, M.-H., Poortmans, F., Palmer, R. & Wyns, L. (1994). Protein Sci.3, 2322–2339. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: RNase A–dGMP complex, 2qca, r2qcasf