

An N-terminal fragment of S. cerevisiae Hsp104 has been crystallized. This is the first report of the crystallization of a eukaryotic member of the Hsp100 family of molecular chaperones.
Keywords: molecular chaperones, AAA+ ATPase, disaggregases, prion-remodeling factors
Abstract
A 40 kDa N-terminal fragment of Saccharomyces cerevisiae Hsp104 was crystallized in two different crystal forms. Native 1 diffracted to 2.6 Å resolution and belonged to space group P212121, with unit-cell parameters a = 66.6, b = 75.8, c = 235.7 Å. Native 2 diffracted to 2.9 Å resolution and belonged to space group P6122 or P6522, with unit-cell parameters a = 179.1, b = 179.1, c = 69.7 Å. This is the first report of the crystallization of a eukaryotic member of the Hsp100 family of molecular chaperones.
1. Introduction
Hsp104 is essential to the yeast stress response (Sanchez & Lindquist, 1990 ▶; Sanchez et al., 1992 ▶) and belongs to the Clp/Hsp100 family of AAA+ ATPases. Like its bacterial ortholog ClpB, Hsp104 forms a homohexameric ring structure (Parsell, Kowal & Lindquist, 1994 ▶) and, together with the yeast Hsp70 chaperone system, rescues proteins from a previously aggregated state (Parsell, Kowal, Singer et al., 1994 ▶; Glover & Lindquist, 1998 ▶). In addition to its role in stress response, Hsp104 also governs the inheritance and maintenance of [PSI+], a yeast prion that increases translational readthrough of nonsense codons (Chernoff et al., 1995 ▶; Patino et al., 1996 ▶; Paushkin et al., 1996 ▶). Perplexingly, overexpression or deletion of Hsp104 cures cells of [PSI+] (Chernoff et al., 1995 ▶), perhaps suggesting that Hsp104 is a prion-remodeling factor. This ability to promote prion fiber assembly and disassembly is not shared with ClpB (Inoue et al., 2004 ▶; Shorter & Lindquist, 2004 ▶), suggesting functional and mechanistic differences between bacterial and fungal members of this family, which might be reflected in their three-dimensional structure.
We have previously reported the crystallization and crystal structure determination of full-length ClpB from Thermus thermophilus (Lee, Hisayoshi et al., 2003 ▶; Lee, Sowa et al., 2003 ▶). ClpB is a multi-domain protein consisting of an N-terminal domain and two AAA+ domains in tandem. Each AAA+ domain consists of a D1/2-large and a D1/2-small domain which are required for ATP binding (Li & Sha, 2002 ▶; Lee, Sowa et al., 2003 ▶). Unlike other Hsp100 proteins, ClpB/Hsp104 possesses an M-domain that is essential for chaperone activity and is inserted into the D1-small domain. While the structure of ClpB provided the much-needed stereochemical framework to further investigate the function of ClpB in vitro and in vivo (Weibezahn et al., 2004 ▶; Haslberger et al., 2007 ▶), the structural and mechanistic basis of the Hsp104-catalyzed prion-remodeling activity remains unknown. Determining the crystal structure of Hsp104 is challenging owing to the lack of well diffracting crystals of the full-length protein. Consequently, we have adopted a ‘divide-and-conquer’ approach by generating smaller Hsp104 fragments that are more amenable to high-resolution structural analysis. Here, we report the overexpression, purification and crystallization of a 40 kDa N-terminal fragment comprising the N-terminal and the D1-large domains of Saccharomyces cerevisiae Hsp104.
2. Materials and methods
2.1. Expression and purification
Escherichia coli BL21 (DE3)-CodonPlus-RIL cells (Stratagene) were transformed with an expression plasmid (pProEX-HTb; Invitrogen) encoding a 40 kDa N-terminal fragment of S. cerevisiae Hsp104 (amino-acid residues 1–360; Cashikar et al., 2002 ▶). Cells were grown at 310 K to mid-log phase in Luria–Bertani medium, induced with 1 mM isopropyl β-d-thiogalactopyranoside (IPTG) and grown at 310 K for a further 4 h.
The cell pellet was suspended in buffer A consisting of 20 mM Tris–HCl pH 8.0, 150 mM NaCl and 20 mM imidazole and lysed by passage through a microfluidizer processor. His6-TEV-Hsp104 (1–360) was purified from the cleared lysate by application onto an Ni–NTA agarose (Qiagen) column, washing in buffer A and elution with a linear gradient of 20–800 mM imidazole in Tris-buffered saline over ten column volumes. After elution, the polyhistidine tag was cleaved off with TEV protease. The sample was then dialyzed against buffer A and reapplied onto Ni–NTA agarose to remove the liberated His tag, the TEV protease and any uncleaved His-tagged protein. The cleaved Hsp104 fragment consists of a short leader peptide of sequence Gly-Ala-Met-Gly-Ser-Gly followed by amino acids 1–360 of S. cerevisiae Hsp104. This construct is hereafter referred to as Hsp104 (1–360). Hsp104 (1–360) was further purified by negative binding to DEAE Sepharose (GE Healthcare) followed by ion-exchange chromatography on a Mono-S column (GE Healthcare). The protein was eluted from the Mono-S column with a linear gradient of 0–300 mM NaCl in 50 mM Tris–HCl pH 7.6 and 10 mM MgCl2 over 12 column volumes. After purification, the protein was judged to be greater than 98% pure by SDS–PAGE and Coomassie Blue staining (data not shown). Purified Hsp104 (1–360) was concentrated to ∼20 mg ml−1 without buffer exchange and was stored at 193 K in 50 mM Tris–HCl pH 7.6, 10 mM MgCl2 and approximately 100 mM NaCl. The protein concentration was estimated by measuring the absorption at 280 nm in 6.0 M guanidine–HCl, 20 mM potassium phosphate buffer pH 6.5 using a molar extinction coefficient of 11 520 M −1 cm−1.
2.2. Crystallization
Hsp104 (1–360) was crystallized at 277 K by hanging-drop vapor diffusion using VDX plates (Hampton Research). All crystallization experiments were set up against a 1 ml reservoir. Crystal form 1 (native 1) belonged to the primitive orthorhombic space group and was obtained by mixing 2 µl protein sample (20 mg ml−1) with an equal volume of well solution consisting of 25% PEG 4000, 50 mM Tris–HCl pH 8.5 and 20 mM ammonium citrate. Crystals appeared after two weeks and reached maximum dimensions of 0.4 × 0.05 × 0.05 mm after two months (Fig. 1 ▶). Crystal form 2 (native 2) belonged to the primitive hexagonal space group and was obtained by mixing 2 µl protein sample (19 mg ml−1) with 1 µl well solution consisting of 33% PEG 400, 50 mM HEPES–HCl pH 7.2 and 150 mM sodium formate. Crystals appeared after one week and reached maximum dimensions of 0.2 × 0.1 × 0.1 mm after two months (Fig. 2 ▶).
Figure 1.

Crystals of Hsp104 (1–360) belonging to the primitive orthorhombic space group (native 1).
Figure 2.

Crystals of Hsp104 (1–360) belonging to the primitive hexagonal space group (native 2).
2.3. Data collection
Crystals were harvested into a cryoprotectant solution consisting of 20%(w/v) glycerol, 25% PEG 4000, 50 mM Tris–HCl pH 8.5 and 20 mM ammonium citrate (native 1) or well solution only (native 2). For data collection, crystals were suspended in small nylon loops at the end of Hampton mounting pins (Hampton Research) and flash-frozen by plunging into liquid nitrogen.
All data were processed and scaled using the HKL software package (Otwinowski & Minor, 1997 ▶) and analyzed using CNSsolve v.1.1 (Brünger et al., 1998 ▶).
3. Results
Crystals of the 40 kDa N-terminal fragment diffracted to 2.6 Å resolution (native 1) and 2.9 Å resolution (native 2) using synchrotron radiation (Table 1 ▶). Complete native data sets were collected at the SBC ID19 beamline (native 1) and the NSLS X25 beamline (native 2) and were used for preliminary crystallographic analysis. The calculated Matthews coefficient (V M = 2.49 Å3 Da−1; Matthews, 1968 ▶) suggests the presence of three molecules in the asymmetric unit for native 1 and either one molecule (V M = 3.91 Å3 Da−1) or possibly two molecules (V M = 1.96 Å3 Da−1) in the asymmetric unit for native 2. However, no noncrystallographic symmetry was detected in the self-rotation function calculated for each crystal form (data not shown).
Table 1. Statistics of the native data sets for Hsp104 (1–360).
Values in parentheses are for the highest resolution shell.
| Native 1 | Native 2 | |
|---|---|---|
| Source | SBC ID19 | NSLS X25 |
| Wavelength (Å) | 1.04 | 1.10 |
| Detector | SBC-2 CCD | Brandeis B4 CCD |
| Temperature (K) | 100 | 100 |
| Space group | P212121 | P6122 or P6522 |
| Unit-cell lengths (Å) | a = 66.6, b = 75.8, c = 235.7 | a = 179.1, b = 179.1, c = 69.7 |
| Unit-cell angles (°) | α = 90, β = 90, γ = 90 | α = 90, β = 90, γ = 120 |
| Matthews coefficient (Å3 Da−1) | 2.49 | 3.91 or 1.96 |
| Solvent content (%) | 50.6 | 68.5 or 37.2 |
| No. of molecules in the ASU | 3 | 1 or 2 |
| Resolution (Å) | 46.0–2.6 (2.7–2.6) | 41.5–2.9 (3.0–2.9) |
| No. of observed reflections | 238759 | 206446 |
| No. of unique reflections | 36873 | 13869 |
| Data completeness (%) | 97.3 (80.3) | 91.9 (48.4) |
| Redundancy | 6.5 (3.1) | 14.9 (1.3) |
| Rsym† (%) | 0.058 (0.346) | 0.076 (0.289) |
| I/σ(I) | 16.5 (4.5) | 17.7 (3.3) |
R
sym = 
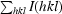 , where 〈I(hkl)〉 is the mean of the symmetry-equivalent reflections of I(hkl).
, where 〈I(hkl)〉 is the mean of the symmetry-equivalent reflections of I(hkl).
Our attempts to solve the structure of Hsp104 (1–360) by molecular replacement using either CNSsolve v.1.1 (Brünger et al., 1998 ▶) or the CCP4 suite of programs (Collaborative Computational Project, Number 4, 1994 ▶) with an in silico-generated ClpB fragment consisting of the N-terminal and D1-large domains of T. thermophilus ClpB (PDB code 1qvr; Lee, Sowa et al., 2003 ▶) as a search model were unsuccessful, even when using either domain alone. This is surprising as yeast Hsp104 and T. thermophilus ClpB share a sequence identity of 42% over the first 360 amino-acid residues. We speculate that the inability to find a molecular-replacement solution may be caused by an inherent interdomain movement of the Hsp104 (1–360) construct. Hence, we propose to solve the crystal structure of Hsp104 (1–360) by the multiple-wavelength anomalous diffraction technique. The value of this structure derives from the fact that crystals not only diffract X-rays to high resolution, but are likely to contain an intact N-terminal domain of S. cerevisiae Hsp104, for which no structural information is currently available.
Acknowledgments
We thank D. A. Hattendorf and S. L. Lindquist for providing the pProEX-HTb/His6-TEV-Hsp104 (1–360) expression construct, the staff of MacCHESS (A-1), NSLS (X25) and APS-SBC (ID-19 and BM-19) for access to and help with their respective synchrotron X-ray sources, A. B. Biter for critical reading of the manuscript and members of the Tsai and Lindquist laboratories for helpful discussions. SL acknowledges support from the Department of Defense (W81XWH-04-1-0033) and the American Heart Association Texas Affiliate (0665082Y). FTFT is supported by the National Institutes of Health and the Robert A. Welch Foundation (Q-1530).
References
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Cashikar, A. G., Schirmer, E. C., Hattendorf, D. A., Glover, J. R., Ramakrishnan, M. S., Ware, D. M. & Lindquist, S. L. (2002). Mol. Cell, 9, 751–760. [DOI] [PubMed]
- Chernoff, Y. O., Lindquist, S. L., Ono, B., Inge-Vechtomov, S. G. & Liebman, S. W. (1995). Science, 268, 880–884. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Glover, J. R. & Lindquist, S. (1998). Cell, 94, 73–82. [DOI] [PubMed]
- Haslberger, T., Weibezahn, J., Zahn, R., Lee, S., Tsai, F. T. F., Bukau, B. & Mogk, A. (2007). Mol. Cell, 25, 247–260. [DOI] [PubMed]
- Inoue, Y., Taguchi, H., Kishimoto, A. & Yoshida, M. (2004). J. Biol. Chem.279, 52319–52323. [DOI] [PubMed]
- Lee, S., Hisayoshi, M., Yoshida, M. & Tsai, F. T. F. (2003). Acta Cryst. D59, 2334–2336. [DOI] [PubMed]
- Lee, S., Sowa, M. E., Watanabe, Y., Sigler, P. B., Chiu, W., Yoshida, M. & Tsai, F. T. F. (2003). Cell, 115, 229–240. [DOI] [PubMed]
- Li, J. & Sha, B. (2002). J. Mol. Biol.318, 1127–1137. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Parsell, D. A., Kowal, A. S. & Lindquist, S. (1994). J. Biol. Chem.269, 4480–4487. [PubMed]
- Parsell, D. A., Kowal, A. S., Singer, M. A. & Lindquist, S. (1994). Nature (London), 372, 475–478. [DOI] [PubMed]
- Patino, M. M., Liu, J.-J., Glover, J. R. & Lindquist, S. (1996). Science, 273, 622–626. [DOI] [PubMed]
- Paushkin, S. V., Kushnirov, V. V., Smirnov, V. N. & Ter-Avanesyan, M. D. (1996). EMBO J.15, 3127–3134. [PMC free article] [PubMed]
- Sanchez, Y. & Lindquist, S. (1990). Science, 248, 1112–1115. [DOI] [PubMed]
- Sanchez, Y., Taulien, J., Borkovich, K. A. & Lindquist, S. (1992). EMBO J.11, 2357–2364. [DOI] [PMC free article] [PubMed]
- Shorter, J. & Lindquist, S. (2004). Science, 304, 1793–1797. [DOI] [PubMed]
- Weibezahn, J., Tessarz, P., Schlieker, C., Zahn, R., Maglica, Z., Lee, S., Zentgraf, H., Weber-Ban, E. U., Dougan, D. A., Tsai, F. T. F., Mogk, A. & Bukau, B. (2004). Cell, 119, 653–665. [DOI] [PubMed]