

A thermostable esterase (EstA) from Thermotoga maritima was cloned and purified. Crystals of EstA and its selenomethionine derivative were grown and diffract to beyond 2.6 Å resolution at 100 K using synchrotron radiation.
Keywords: esterases, Thermotoga maritima
Abstract
A predicted esterase (EstA) with an unusual new domain from the hyperthermophilic bacterium Thermotoga maritima has been cloned and overexpressed in Escherichia coli. The purified protein was crystallized by the hanging-drop vapour-diffusion technique in the presence of lithium sulfate and polyethylene glycol 8000. Selenomethionine-substituted EstA crystals were obtained under the same conditions and three different-wavelength data sets were collected to 2.6 Å resolution. The crystal belongs to space group H32, with unit-cell parameters a = b = 130.2, c = 306.2 Å. There are two molecules in the asymmetric unit, with a V M of 2.9 Å3 Da−1 and 58% solvent content.
1. Introduction
Esterases and lipases catalyse the hydrolysis of ester bonds, resulting in the formation of an alcohol and a carboxylic acid. Both enzyme types share structural characteristics, including an α/β-hydrolase fold and a conserved catalytic triad that is usually composed of a serine, an aspartate and a histidine residue (Jaeger et al., 1999 ▶). Esterases differ from lipases in that they show a preference for short-chain acyl esters (shorter than ten C atoms; Jaeger et al., 1999 ▶). Ester-hydrolyzing enzymes are attractive biocatalysts because they have cofactor-independent activity, high regiospecificity and stereospecificity and are generally rather stable and active in organic solvents. There is a special interest from industry in esterases of thermophilic origin since these enzymes are both stable in organic solvents and function at elevated temperatures (Atomi & Imanaka, 2004 ▶).
The hyperthermophilic anaerobic bacterium Thermotoga maritima was isolated from geothermally heated marine sediments at Vulcano, Italy. The organism grows optimally between 328 and 363 K and can metabolize many simple and complex carbohydrates, including glucose, cellulose and starch (Nelson et al., 1999 ▶). During bioinformatic analysis of the genome of T. maritima, several open reading frames potentially encoding new thermostable esterases were found, including one (estA, TM0033) that was annotated as a hypothetical protein. EstA was produced in Escherichia coli and found to exhibit esterase activity with a preference for esters of short-chain fatty acids. It consists of 395 amino-acid residues and has a predicted molecular weight of 44.7 kDa. A BLAST search of EstA at NCBI (http://www.ncbi.nlm.nih.gov/BLAST/; Altschul et al., 1997 ▶) reveals highest similarity to other hypothetical proteins and a few predicted peptidases and carboxylesterases. An alignment with the obtained homologous sequences revealed EstA to be composed of two domains. The C-terminal domain of EstA contains a predicted α/β-hydrolase fold and includes the characteristic conserved Ser-Asp-His catalytic triad. However, the amino-acid sequence of the N-terminal domain has no homology to known proteins in the database and this domain therefore has unknown function and structure. It is tempting to speculate that the N-terminal domain might be involved in substrate binding and recognition. Therefore, determination of the structure of EstA may provide insight into the function of this new N-terminal domain and reveal the molecular basis of substrate recognition and catalysis by this enzyme.
This paper describes the cloning, purification, crystallization and preliminary X-ray analysis of T. maritima EstA as a first step in the structure determination of this new esterase.
2. Cloning, expression and purification
The gene encoding EstA was amplified by PCR, without the predicted signal peptide (the first 16 amino acids) and without its stop codon (fused to a His6 tag), using chromosomal DNA of T. maritima as a template and the two primers BG1962 (5′-GCGCCATGGAGGATGTTACTGTGAAGAGTG-3′) and BG1963 (5′-GCGCTCGAGTCTACTTTGTTCAAACAGCCAC-3′), introducing NcoI and XhoI restriction sites. The generated PCR product was digested by NcoI and XhoI and the product was purified and ligated into pET-24d digested with the same restriction enzymes, resulting in the plasmid pWUR350. The construct was designed with a hexahistidine tag engineered at the C-terminus of the enzyme to facilitate purification. E. coli BL21(DE3)/pSJS1244 was transformed with pWUR350.
A single colony was used to inoculate 4 ml Luria–Bertani medium containing kanamycin and spectinomycin (both 50 µg ml−1) and incubated overnight at 310 K with shaking. The preculture was used to inoculate (1:1000) 2 l of the same medium and growth was continued for 8 h (an OD600 above 2.0 was reached). Subsequently, the culture was induced by adding IPTG (isopropyl β-d-1-thiogalactopyranoside) to a final concentration of 0.5 mM. The culture was incubated for a further 16 h at 310 K.
Cells were harvested by centrifugation at 10 000g for 10 min. The cell pellet was resuspended in 30 ml buffer (50 mM Tris–HCl pH 7.5, 300 mM NaCl, 10 mM imidazole). The cells were disrupted by passage twice through a French press at 110 MPa. The crude cell extract was treated with DNAse I at room temperature for 30 min and subsequently centrifuged at 43 000g for 30 min in order to remove cell debris. The supernatant was heated at 343 K for 30 min and then centrifuged to remove the precipitated proteins. The supernatant was filtered and applied onto a nickel-chelating column packed with 20 ml Ni–NTA His-Bind Resin (Novagen) and equilibrated in 50 mM Tris–HCl buffer pH 7.8 containing 300 mM NaCl. The column was washed with 20 mM imidazole in the same buffer and proteins were subsequently eluted with a linear gradient of 20–500 mM imidazole in 50 mM Tris–HCl pH 7.5, 300 mM NaCl. Fractions containing esterase activity were pooled and applied onto a Hi-Prep desalting column (Amersham Biosciences) equilibrated with 50 mM disodium phosphate buffer pH 7.5. The homogeneity of the protein was checked by SDS–PAGE and activity staining of the SDS–PAGE gels using α-naphtyl acetate, as described previously (Levisson et al., 2007 ▶). The protein concentration was determined at 280 nm using a NanoDrop ND-1000 Spectrophotometer (NanoDrop). For the preparation of selenomethionine-substituted EstA (SeMet-EstA), the overproducing strain was grown as described previously (Akerboom et al., 2003 ▶) and the enzyme was purified as described above. EstA and SeMet-EstA were assayed for esterase activity using p-nitrophenyl-valerate as a substrate (data not shown).
3. Crystallization
The purified protein was dialyzed against 10 mM potassium phosphate buffer pH 7.5 and concentrated to 15 mg ml−1. Crystallization was performed using the hanging-drop vapour-diffusion method at room temperature. Initial crystallization conditions were screened using Crystal Screen and Crystal Screen II (Hampton Research). Drops consisting of equal volumes (1 µl) of protein and reservoir solution were equilibrated over 500 µl reservoirs. Crystals of EstA were obtained using a reservoir solution consisting of 1.0 M lithium sulfate monohydrate, 2%(w/v) polyethylene glycol 8000 (Crystal Screen condition No. 49). SeMet-EstA crystals were obtained under the same conditions (Fig. 1 ▶).
Figure 1.

(a) Crystals of EstA. (b) Crystals of SeMet-EstA.
4. Data collection and preliminary X-ray analysis
For cryoprotection, crystals were soaked for a few seconds in reservoir solution containing 20%(w/v) glycerol. The crystals were mounted in a cryoloop and subsequently flash-frozen in liquid nitrogen. X-ray data were collected at 100 K on beamline ID29 at ESRF, Grenoble. A native data set was collected to 2.6 Å resolution. The crystal belongs to space group H32, with unit-cell parameters a = b = 130.2, c = 306.2 Å. There are two molecules in the asymmetric unit, with a V M of 2.9 Å3 Da−1 and 58% solvent content (Matthews, 1968 ▶). Crystals of SeMet-EstA showed a well defined Se K absorption edge on fluorescence scanning. A single SeMet-EstA crystal was used for MAD data collection at peak (0.9791 Å), inflection (0.9793 Å) and remote (0.9557 Å) wavelengths to 2.6 Å resolution. All data were indexed and integrated using MOSFLM (Leslie, 1992 ▶) and scaled with anisotropic scaling corrections using SCALA (Evans, 1993 ▶) within the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶). Processing statistics for the native and SeMet data sets are shown in Table 1 ▶.
Table 1. Data-collection and processing statistics for EstA.
Values in parentheses are for the highest resolution shell.
| Native | SeMet peak | SeMet inflection | SeMet remote | |
|---|---|---|---|---|
| Wavelength (Å) | 1.0000 | 0.9791 | 0.9793 | 0.9757 |
| Resolution limits (Å) | 50.0–2.6 (2.74–2.6) | 50.0–2.6 (2.74–2.6) | 50.0–2.6 (2.74–2.6) | 50.0–2.6 (2.74–2.6) |
| Space group | H32 | H32 | H32 | H32 |
| Unit-cell parameters (Å) | ||||
| a = b | 130.2 | 131.0 | 131.0 | 131.0 |
| c | 306.2 | 306.8 | 306.8 | 306.8 |
| Observed reflections | 173066 | 353388 | 358408 | 358161 |
| Unique reflections | 31079 | 31295 | 31587 | 31457 |
| Completeness (%) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) |
| Rmerge† | 0.087 (0.430) | 0.086 (0.498) | 0.082 (0.412) | 0.075 (0.437) |
| 〈I/σ(I)〉 | 14.2 (3.6) | 22.3 (4.6) | 22.1 (4.4) | 24.8 (5.0) |
| Redundancy | 5.6 (5.7) | 11.4 (11.7) | 11.3 (11.4) | 11.4 (11.3) |
R
merge = 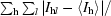
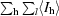 , where I
l is the lth observation of reflection h and 〈I
h〉 is the weighted average intensity for all observations l of reflection h.
, where I
l is the lth observation of reflection h and 〈I
h〉 is the weighted average intensity for all observations l of reflection h.
Structure determination is currently ongoing. However, an initial model shows that there are indeed two domains present in EstA: a domain resembling the α/β-hydrolase fold and a domain resembling the immunoglobulin fold. It is speculated that the α/β-hydrolase domain is the catalytically active domain and that the immunoglobulin domain might be involved in substrate binding or cell adhesion. Structure refinement combined with biochemical analysis is expected to provide improved insights into the function of this esterase.
Acknowledgments
We would like to thank Andrew McCarthy for his assistance at ESRF beamline ID29. This research was financially supported by the Graduate School VLAG and an NWO-Vici grant to JvdO (Wageningen University).
References
- Akerboom, J., Turnbull, A. P., Hargreaves, D., Fisher, M., de Geus, D., Sedelnikova, S. E., Berrisford, J. M., Baker, P. J., Verhees, C. H., van der Oost, J. & Rice, D. W. (2003). Acta Cryst. D59, 1822–1823. [DOI] [PubMed]
- Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997). Nucleic Acids Res.25, 3389–3402. [DOI] [PMC free article] [PubMed]
- Atomi, H. & Imanaka, T. (2004). Tetrahedron Asymm.15, 2729–2735.
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Evans, P. R. (1993). Proceedings of the CCP4 Study Weekend. Data Collection and Processing, edited by L. Sawyer, N. Isaacs & S. Bailey, pp. 114–122. Warrington: Daresbury Laboratory.
- Jaeger, K. E., Dijkstra, B. W. & Reetz, M. T. (1999). Annu. Rev. Microbiol.53, 315–351. [DOI] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Levisson, M., van der Oost, J. & Kengen, S. W. (2007). FEBS J.274, 2832–2842. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Nelson, K. E. et al. (1999). Nature (London), 399, 323–329.