

The C-terminal soluble domain of the catalytic subunit (STT3) of the oligosaccharyltransferase from P. furiosus was purified and crystallized. A native crystal and a SeMet derivative have been analyzed using X-ray diffraction.
Keywords: N-glycosylation, oligosaccharyltransferase, STT3
Abstract
Oligosaccharyltransferase catalyzes the transfer of preassembled oligosaccharides onto asparagine residues in nascent polypeptide chains. The STT3 subunit is thought to bear the catalytic site. The C-terminal domain of the STT3 protein of Pyrococcus furiosus was expressed in Escherichia coli cells. STT3 protein prepared from two different sources, the soluble fraction and the inclusion bodies, produced crystals that diffracted to 2.7 Å. During crystallization screening, cocrystals of P. furiosus STT3 with an E. coli 50S ribosomal protein, L7/L12, were accidentally obtained. This cross-species interaction is not biologically relevant, but may be used to design a built-in polypeptide substrate for the STT3 crystals.
1. Introduction
Asparagine-linked glycosylation (N-glycosylation) is widespread in eukaryotic, archaeal and some eubacterial organisms (Szymanski et al., 2003 ▶; Upreti et al., 2003 ▶; Szymanski & Wren, 2005 ▶; Kelleher & Gilmore, 2006 ▶) and is essential for the structural and functional integrity of many secretory and membrane proteins. Oligosaccharyltransferase (OST) catalyzes the transfer of preassembled oligosaccharides on lipid carriers onto asparagine residues in the sequon Asn-X-Thr/Ser, where X can be any residue except Pro, in nascent polypeptide chains (Bause & Hettkamp, 1979 ▶; Gavel & von Heijne, 1990 ▶).
The OST enzyme is a multisubunit protein complex residing in the endoplasmic reticulum membrane in eukaryotic cells (Knauer & Lehle, 1999 ▶). In contrast, the OST of the Gram-negative eubacterium Campylobacter jejuni is composed of one subunit (Szymanski et al., 2003 ▶). Several lines of biochemical and genetic evidence have indicated that the STT3 protein is the catalytic subunit of the OST enzyme in eukaryotes and C. jejuni (Yan & Lennarz, 2002 ▶; Nilsson et al., 2003 ▶; Karamyshev et al., 2005 ▶; Feldman et al., 2005 ▶; Glover et al., 2005 ▶). The inactivation of the stt3 gene in the archaeon Methanococcus voltae resulted in the underglycosylation of flagellin and S-layer proteins (Chaban et al., 2006 ▶). This experiment suggested that the STT3 protein also functions as the catalytic subunit of the OST enzyme in archaea. The STT3 protein in all three domains of life consists of an N-terminal half segment, containing 10–13 predicted transmembrane helices (TMs), and a C-terminal globular domain (Zufferey et al., 1995 ▶; Knauer & Lehle, 1999 ▶). The C-terminal domain contains a highly conserved five-residue motif, Trp-Trp-Asp-Tyr-Gly, and is thought to bear the catalytic site.
Here, we report the expression, crystallization and preliminary crystallographic analysis of the C-terminal domain of the STT3 protein from the thermophilic archaeon Pyrococcus furiosus.
2. Materials and methods
2.1. Expression of the soluble domain of P. furiosus STT3 protein
The soluble domain (residues 471–967) of the STT3 protein (sSTT3) from P. furiosus (accession No. PF0156, Q8U4D2_PYRFU) was produced in Escherichia coli BL21-CodonPlus (DE3)-RIL cells (Stratagene) using the NcoI and BamHI restriction sites of the pET21d vector (Invitrogen). The transformed E. coli cells were cultured at 310 K in LB media containing 50 mg l−1 ampicillin. After the OD600 had reached 0.6, the culture was supplemented with 0.5 mM IPTG for induction. The cells were cultured at 310 K for 4 h or at 289 K overnight for the production of the sSTT3 protein in inclusion bodies or as soluble protein, respectively. After the cells had been harvested by centrifugation, the cell pellet was resuspended in buffer A (50 mM Tris–HCl pH 8.0, 150 mM NaCl and 2 mM EDTA) supplemented with 4 mM MgCl2 and 10 U ml−1 Benzonase (Merck). The cells were then disrupted by sonication. The resultant solution was separated into supernatant and pellet fractions by centrifugation at 10 000g for 30 min.
2.2. Protein preparation by refolding from inclusion bodies
The pellet of the sonicated lysate contained sSTT3 in inclusion bodies. The inclusion bodies were washed three times by resuspension and centrifugation in buffer A containing 1% Triton X-100 and then three times in buffer A without Triton X-100. The washed inclusion bodies were dissolved in buffer A containing 6 M urea and the insoluble materials were removed by centrifugation. The supernatant was loaded onto an anion-exchange Q-Sepharose FF column (GE Healthcare). The sSTT3 protein was eluted as a single peak with a linear gradient of NaCl in the presence of 6 M urea and the fractions containing sSTT3 were collected. Refolding was carried out by direct injection into a gel-filtration Superdex 200 column (GE Healthcare) pre-equilibrated with buffer A without urea. The fractions containing the refolded sSTT3 protein were combined and concentrated to 10 mg ml−1 by ultrafiltration (10 kDa cutoff, Amicon Ultra, Millipore) in 10 mM Tris–HCl pH 8.0.
2.3. Protein preparation by heat treatment of the supernatant
The supernatant of the sonicated lysate contained soluble sSTT3. After the supernatant had been heated to 353 K for 15 min, the denatured E. coli proteins were removed by centrifugation at 16 000g for 15 min. The resultant supernatant was diluted with buffer (50 mM Tris–HCl pH 8.0, 2 mM EDTA) and loaded onto a Resource Q anion-exchange column (GE Healthcare). The sSTT3 protein was eluted with a linear gradient of NaCl. The fractions containing sSTT3 were combined and concentrated by ultrafiltration. The protein was further purified by gel-filtration chromatography on a Superdex 200 column (GE Healthcare) pre-equilibrated with buffer A. The fractions at the top of the peak (peak-top) containing sSTT3 were combined and concentrated to 10 mg ml−1 by ultrafiltration in 10 mM Tris–HCl pH 8.0. The fractions at the tailing edge of the peak were also collected and processed in the same manner.
A selenomethionyl (SeMet) derivative of sSTT3 was expressed in minimal medium containing seleno-l-methionine at a concentration of 25 mg l−1. SeMet sSTT3 was purified by the same procedure as described for the peak-top fraction of the supernatant sSTT3 and the incorporation of selenium was confirmed by MALDI–TOF mass spectrometry.
2.4. Crystallization
Initial screening was carried out using the sitting-drop vapour-diffusion method in 96-well Intelli-plates (Art Robbins Instruments) and optimization was performed using the hanging-drop vapour-diffusion method in 24-well VDX greased plates (Hampton Research). Sitting drops were set up by mixing equal volumes (0.2 µl each) of the protein solution and the reservoir solution using an automated dispenser (Hydra II Plus One system; Apogent Discoveries), whereas hanging drops were prepared manually by mixing 0.7 µl protein solution and 0.7 µl reservoir solution. Each sitting drop was placed over 0.1 ml reservoir solution and each hanging drop was placed over 0.4 ml reservoir solution.
3. Results and discussion
3.1. Preparation
The C-terminal 497-residue fragment of the STT3 protein (sSTT3) from P. furiosus was expressed in E. coli cells. The majority of the expressed sSTT3 resided in the pellet, but the supernatant also contained a small but significant amount of sSTT3. The insoluble sSTT3 contained in the pellet fraction was purified by anion-exchange chromatography in the presence of urea and was refolded by direct injection into a gel-filtration column pre-equilibrated without urea. The majority of the sSTT3 protein eluted as a single peak (upper chromatogram in Fig. 1 ▶). By assuming a monomeric form (55.9 kDa), the point corresponding to the peak-top elution volume is on the calibration curve (inset in Fig. 1 ▶), suggesting successful refolding as the monomeric form. The typical yield of sSTT3 from the pellet was 30 mg per litre of culture. On the other hand, the supernatant fraction was heated at 353 K. The thermostable sSTT3 remained in the solution, but the majority of the E. coli proteins precipitated. After centrifugation, the sSTT3 was recovered in the supernatant and was further purified by anion-exchange and gel-filtration chromatography. The sSTT3 protein eluted as a single peak in the gel-filtration chromatography at the same elution volume as the refolded sSTT3, but with a tailing peak (lower chromatogram in Fig. 1 ▶). The peak-top fractions contained pure sSTT3. The typical yield of sSTT3 from the supernatant was 2 mg per litre of culture. The fractions at the tailing edge were also collected. All protein stock solutions used for crystallization contained 10 mg ml−1 protein in 10 mM Tris–HCl pH 8.0.
Figure 1.

Comparison of gel-filtration chromatograms of the two different preparations of the C-terminal soluble domain of the STT3 protein from P. furiosus. The upper chromatogram shows the refolded sSTT3 recovered from the pellet fraction and the lower chromatogram shows the sSTT3 recovered from the heat-treated cell lysate. The pooled fractions for crystallization are indicated by horizontal bars. The inset shows the calibration curve for the estimation of apparent molecular weight. The open circle represents the peak-top position of the chromatograms, assuming the molecular weight of the monomeric form (55.9 kDa). a, Void volume; b, albumin, 67 kDa; c, ovalbumin, 43 kDa; d, chymotrypsinogen, 25 kDa; e, ribonuclease A, 13.7 kDa.
3.2. Crystallization
Screenings were carried out at 293 K using Crystal Screens 1 and 2 (Hampton Research). Crystals of sSTT3 purified as the peak-top fractions in the gel filtration from the supernatant fraction (Fig. 1 ▶, sample 2) were obtained with Crystal Screen 1 solution No. 22 (0.1 M Tris–HCl pH 8.5, 30% PEG 4000, 0.2 M sodium acetate) and Crystal Screen 2 solution No. 30 (0.1 M HEPES pH 7.5, 10% PEG 6000, 5% MPD). After optimization, the crystals grew from a hanging drop with a 1:1 volume ratio of 10 mg ml−1 protein stock solution and reservoir solution (0.1 M Tris–HCl pH 9.0, 22% PEG 4000, 0.3 M sodium acetate) at 303 K in 1–3 weeks (Fig. 2 ▶ a). Crystallization at 303 K was essential to obtain large rod-like crystals. The SDS–PAGE of the crystals revealed their purity (Fig. 2 ▶ a, lane C). Crystals of sSTT3 prepared by refolding from the pellet fraction (Fig. 1, sample 1) grew under the same conditions.
Figure 2.

Crystals of P. furiosus sSTT3 protein prepared from the heat-treated cell lysate and SDS–PAGE analysis of the crystals. (a) Crystals obtained from the peak-top fractions (Fig. 1 ▶, sample 2) after two weeks at 303 K; (b) crystals obtained from the tailing-edge fractions (Fig. 1 ▶, sample 3) after 2 d at 293 K. The scale bars represent 0.1 mm. Crystals were transferred to the corresponding mother liquors for washing, mixed with 5× SDS sample buffer, heated for 5 min at 368 K and then subjected to SDS–PAGE (10–20% gradient gel, Daiichi). The gels were stained with SYPRO Orange protein gel stain (Invitrogen) and were photographed with an LAS-3000 multicolour image analyzer (Fuji Film) using a Blue LED (460 nm) illuminator and a Y515-Di filter. Lane M, prestained markers (Precision Plus protein standards, dual colour, Bio-Rad); lane S, protein solution used for crystallization; lane C, washed crystals. The amount of the markers used was extremely small, so that only the two bands prestained with a fluorescent dye are visible. The 12 kDa band was excised from the CBB-stained gel and subjected to in-gel digestion with trypsin and the eluted peptides were loaded onto LC-ESI-MS/MS (LC Q DECA, Finnigan). The protein was identified by a peptide-fingerprint search using the Mascot software (Matrix Science).
Small crystals were obtained when the peak-tailing edge fractions in the gel filtration from the supernatant fraction were used (Fig. 1 ▶, sample 3), but they grew much faster and with a different shape. Optimization resulted in a lower PEG concentration and the addition of Y3+ ion as an additive. Crystals grew from a hanging drop with a 1:1 volume ratio of 10 mg ml−1 protein stock solution and reservoir solution (0.1 M Tris–HCl pH 9.0, 15% PEG 4000, 0.3 M sodium acetate, 5 mM YCl3) at 293 K in 2 d (Fig. 2 ▶ b). EXAFS measurements showed that the Y3+ ion was not incorporated into the crystals. Surprisingly, SDS–PAGE analysis revealed that the washed crystals contained a stoichiometric amount of an unknown protein band with a molecular weight of about 12 kDa in addition to the sSTT3 protein band (Fig. 2 ▶ b, lane C). The protein solution used for crystallization contained some extra protein bands (Fig. 2 ▶ b, lane S), but apart from the 12 kDa protein these contaminating proteins were not incorporated into the crystals. In-gel trypsin-digestion and LC-ESI-MS/MS analysis revealed that the 12 kDa protein was an E. coli 50S ribosomal protein, L7/L12 (entry name RL7_ECOLI). L7/L12 is a ribosomal subunit protein (for recent reviews, see Wahl & Moller, 2002 ▶; Gonzalo & Reboud, 2003 ▶). L7 is the N-terminally acetylated form of L12. L7/L12 is present in each 50S ribosomal subunit in four copies, organized as two dimers, and is necessary for the proper function of the elongation factors EF-G and EF-Tu. Functional counterparts, termed P1/P2 proteins, exist in archaeal and eukaryotic ribosomes but have no sequence homology.
3.3. Data collection and phase determination
The sSTT3 crystals (Fig. 2 ▶ a) were cryoprotected by increasing the concentration of PEG 4000 to 24% and were cryocooled in a nitrogen-gas stream (100 K). The crystals diffracted to 2.7 Å at BL41XU, SPring-8 (Harima, Japan). Crystals of SeMet sSTT3 grew under the same conditions and were processed in the same manner and diffracted to 3.0 Å. The diffraction data were processed with HKL-2000 (Otwinowski & Minor, 1997 ▶). The crystals belonged to space group P21212, with unit-cell parameters a = 137, b = 266, c = 74 Å. A search for Se-atom sites was carried out with SOLVE (Terwilliger & Berendzen, 1999 ▶), resulting in 16 out of 20 Se sites, assuming four SeMet residues in each molecule. Phases were further improved by density-modification methods using autoSHARP (Vonrhein et al., 2006 ▶), with a mean figure-of-merit value of 0.737. The statistics of data collection and phasing are summarized in Table 1 ▶. The resulting electron-density map allowed tracing of the main chains of the polypeptides. Model building and structure refinement are now in progress.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| SeMet sSTT3 | Native sSTT3 | Native sSTT3 + L7/L12 | |
|---|---|---|---|
| Data collection | |||
| Space group | P21212 | P21212 | C2221 |
| Unit-cell parameters (Å) | |||
| a | 137.34 | 136.93 | 102.23 |
| b | 266.60 | 265.29 | 106.04 |
| c | 73.95 | 74.13 | 289.38 |
| Wavelength (Å) | 0.97915 (peak) | 0.96419 | 1.00000 |
| Resolution range (Å) | 30–3.0 (3.1–3.0) | 40–2.7 (2.8–2.7) | 50–3.35 (3.5–3.35) |
| No. of measured reflections | 610027 | 458643 | 168317 |
| No. of unique reflections | 55387 (5441) | 75226 (7371) | 23023 (2238) |
| Completeness (%) | 100.0 (100.0) | 99.9 (99.4) | 100.0 (100.0) |
| Multiplicity | 11.0 (11.1) | 6.1 (5.8) | 7.3 (7.5) |
| Average I/σ(I) | 7.4 (3.8) | 12.4 (3.5) | 7.8 (4.5) |
| Rmerge† | 0.136 (0.598) | 0.081 (0.443) | 0.127 (0.465) |
| Mosaicity (°) | 0.254 | 0.261 | 0.458 |
| Molecules per AU | 4 | 4 | 2 (sSTT3) + 2 (L7/L12) |
| Phasing statistics | |||
| Se sites (found/all) | 16/20 | ||
| Phasing power (centric/acentric) | 0.00/0.608 | ||
| 〈FOM〉 (after SOLVE/after SHARP) | 0.298/0.737 |
R
merge = 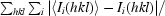
 , where I
i(hkl) is the observed intensity and 〈I
i(hkl)〉 is the average intensity obtained from multiple observations of symmetry-related reflections.
, where I
i(hkl) is the observed intensity and 〈I
i(hkl)〉 is the average intensity obtained from multiple observations of symmetry-related reflections.
The crystals containing sSTT3 and E. coli L7/L12 (Fig. 2 ▶ b) were cryoprotected by transferring them to a mixture of mother liquor and ethylene glycol (8:2 volume ratio) and were cryocooled in a nitrogen-gas stream. The crystals diffracted to 3.4 Å at BL41XU and optimization of the crystallization conditions is under way. The space group was C2221, with unit-cell parameters a = 102, b = 106, c = 289 Å (Table 1 ▶). As the preliminary electron densities indicated extra short α-helices in addition to the sSTT3 molecules, the interactions between sSTT3 and L7/L12 seem to be stoichiometric and specific in the crystals. The interaction probably also occurred weakly in solution, as the E. coli L7/L12 protein coeluted with sSTT3 at an elution volume earlier than that expected for the 24 kDa L7/L12 dimer (Fig. 1 ▶). We do not think that this cross-species interaction is biologically relevant, but L7/L12 might mimic the interaction with the partner protein(s) in situ. Finally, such ‘xenogeneic cocrystals’ may provide chances for unexpected research and applications. For example, L7/L12 will serve as the template for a polypeptide substrate specifically designed for cocrystallization by engrafting an Asn-X-Thr/Ser sequon.
Acknowledgments
We thank Dr Yoshizumi Ishino of Kyushu University for providing the P. furiosus cells and Ms Mizuho Oda, Ms Emiko Fujimoto and Ms Masumi Otsu of the Laboratory for Technical Support, Medical Institute of Bioregulation, Kyushu University for the in-gel digestion, LC-ESI-MS/MS analysis and DNA sequencing. The experiments at SPring-8 were approved by the Japan Synchrotron Radiation Research Institute (JASRI) as proposals 2005A0877, 2005B0157, 2006A1739 and 2007A1204. The experiments at the Photon Factory were approved by the High Energy Accelerator Research Organization (KEK) as proposal 2007G208. DK and KM were supported by Grants-in-Aid for Scientific Research in Priority Areas and the National Project on Target Protein Analyses from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan. TO was supported by Research Fellowships from the Japan Society for the Promotion of Science (JSPS) for Young Scientists.
References
- Bause, E. & Hettkamp, H. (1979). FEBS Lett.108, 341–344. [DOI] [PubMed]
- Chaban, B., Voisin, S., Kelly, J., Logan, S. M. & Jarrell, K. F. (2006). Mol. Microbiol.61, 259–268. [DOI] [PubMed]
- Feldman, M. F., Wacker, M., Hernandez, M., Hitchen, P. G., Marolda, C. L., Kowarik, M., Morris, H. R., Dell, A., Valvano, M. A. & Aebi, M. (2005). Proc. Natl Acad. Sci. USA, 102, 3016–3021. [DOI] [PMC free article] [PubMed]
- Gavel, Y. & von Heijne, G. (1990). Protein Eng.3, 433–442. [DOI] [PMC free article] [PubMed]
- Glover, K. J., Weerapana, E., Numao, S. & Imperiali, B. (2005). Chem. Biol.12, 1311–1315. [DOI] [PMC free article] [PubMed]
- Gonzalo, P. & Reboud, J. P. (2003). Biol. Cell, 95, 179–193. [DOI] [PubMed]
- Karamyshev, A. L., Kelleher, D. J., Gilmore, R., Johnson, A. E., von Heijne, G. & Nilsson, I. (2005). J. Biol. Chem.280, 40489–40493. [DOI] [PubMed]
- Kelleher, D. J. & Gilmore, R. (2006). Glycobiology, 16, 47R–62R. [DOI] [PubMed]
- Knauer, R. & Lehle, L. (1999). Biochim. Biophys. Acta, 1426, 259–273. [DOI] [PubMed]
- Nilsson, I., Kelleher, D. J., Miao, Y., Shao, Y., Kreibich, G., Gilmore, R., von Heijne, G. & Johnson, A. E. (2003). J. Cell Biol.161, 715–725. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Szymanski, C. M., Logan, S. M., Linton, D. & Wren, B. W. (2003). Trends Microbiol.11, 233–238. [DOI] [PubMed]
- Szymanski, C. M. & Wren, B. W. (2005). Nature Rev. Microbiol.3, 225–237. [DOI] [PubMed]
- Terwilliger, T. C. & Berendzen, J. (1999). Acta Cryst. D55, 849–861. [DOI] [PMC free article] [PubMed]
- Upreti, R. K., Kumar, M. & Shankar, V. (2003). Proteomics, 3, 363–379. [DOI] [PubMed]
- Vonrhein, C., Blanc, E., Roversi, P. & Bricogne, G. (2006). Methods Mol. Biol.364, 215–230. [DOI] [PubMed]
- Wahl, M. C. & Moller, W. (2002). Curr. Protein Pept. Sci.3, 93–106. [DOI] [PubMed]
- Yan, Q. & Lennarz, W. J. (2002). J. Biol. Chem.277, 47692–47700. [DOI] [PubMed]
- Zufferey, R., Knauer, R., Burda, P., Stagljar, I., te Heesen, S., Lehle, L. & Aebi, M. (1995). EMBO J.14, 4949–4960. [DOI] [PMC free article] [PubMed]