

MAP2569c from M. avium subsp. paratuberculosis, a putative glycosyltransferase implicated in mycobacterial cell-wall biosynthesis, was cloned, expressed, purified and crystallized. X-ray diffraction data were collected to 1.8 Å resolution.
Keywords: glycosyltransferases, GT-A fold, GT2 family, Mycobacterium
Abstract
Glycosidic bond formation is a ubiquitous enzyme-catalysed reaction. This glycosyltransferase-mediated process is responsible for the biosynthesis of innumerable oligosaccharides and glycoconjugates and is often organism- or cell-specific. However, despite the abundance of genomic information on glycosyltransferases (GTs), there is a lack of structural data for this versatile class of enzymes. Here, the cloning, expression, purification and crystallization of an essential 329-amino-acid (34.8 kDa) putative GT of the classic GT-A fold implicated in mycobacterial cell-wall biosynthesis are reported. Crystals of MAP2569c from Mycobacterium avium subsp. paratuberculosis were grown in 1.6 M monoammonium dihydrogen phosphate and 0.1 M sodium citrate pH 5.5. A complete data set was collected to 1.8 Å resolution using synchrotron radiation from a crystal belonging to space group P41212.
1. Introduction
Glycosyltransferases (GTs) catalyse the transfer of glycosyl moieties from mostly nucleotide-activated donor substrates to a myriad of specific acceptor substrates. The oligosaccharide and glycoconjugate products of these reactions are ubiquitous in nature.
This remarkable versatility of GTs is reflected in their sequence diversity. The Carbohydrate-Active Enzyme (CAZy) database (http://www.cazy.org/; Campbell et al., 1997 ▶; Coutinho et al., 2003 ▶) lists over 31 000 known and putative GTs in 89 sequence-based families (as of 7 November 2007). GTs are classified as either inverting or retaining depending on whether the stereochemistry of the anomeric carbon is retained or inverted in the product relative to that in the donor substrate. Moreover, despite the sequence diversity of this class of enzymes, the GT structures solved to date adopt one of only two classic folds. The GT-A and GT-B folds are both described as α/β/α sandwiches, with the GT-A fold containing a single nucleotide-binding or Rossmann-like domain (Rossmann et al., 1974 ▶) and the GT-B fold containing two similar Rossmann-like domains. Members of a particular GT family are proposed to share the same inverting or retaining mechanism and GT-A or GT-B fold.
Owing to the frequent integral membrane or membrane-associated nature of GTs, obtaining such proteins in quantities conducive to structural studies has often proven difficult. Consequently, only 29 of the 89 sequence-based GT families have representative structures. Moreover, for the largest such family, the inverting GT2 family, with over 8600 known and putative members, the structure of the spore-coat biosynthesis protein SpsA from Bacillus subtilis (Charnock & Davies, 1999 ▶) remains its sole representative structure.
The distinctive mycobacterial cell wall is critical to the persistence and pathogenicity of disease-causing mycobacteria within their respective hosts and presents a rich source of oligosaccharides and glycoconjugates unique to Mycobacterium and related Corynebacterineae species. However, despite the importance of the cell wall to the biology of these species, there is a paucity of knowledge on the pathways involved in its biosynthesis. Owing to the complex carbohydrate content of the mycobacterial cell wall, GTs are likely to be the most numerous class of enzymes in these pathways (Berg et al., 2007 ▶). Indeed, there are 41 known or putative GTs from the strain M. tuberculosis H37Rv in the CAZy database, with all 16 such GTs proposed to exhibit the GT-A fold belonging the inverting GT2 family (Cole et al., 1998 ▶). Nevertheless, the recently solved structure of the inverting mannosyltransferase PimA, displaying the GT-B fold, is the only available structure of a GT found in mycobacteria (Guerin et al., 2007 ▶).
One of the predicted GT2-family members found in M. tuberculosis H37Rv is an essential protein implicated in M. tuberculosis cell-wall biosynthesis termed Rv1208 (Sassetti et al., 2003 ▶). As such, Rv1208 presents an attractive candidate for structural/functional studies. However, in addition to the challenges typically associated with the production of GTs noted above, M. tuberculosis proteins are notorious for poor expression and/or solubility in the Escherichia coli system (Moreland et al., 2005 ▶; Vincentelli et al., 2004 ▶). To overcome these barriers and increase our chances of obtaining active protein in sufficient quantities for crystallographic analyses, we investigated a widened ‘expression space’ for Rv1208. This involved screening the expression and solubility of Rv1208 in parallel to those of its putative orthologues from other Mycobacterium species, as was previously found to be successful for structural studies on LpqW (Marland et al., 2006 ▶). Through this approach, we discovered high levels of expression and ample solubility for the closest homologue of Rv1208 (83% sequence identity; Wilbur & Lipman, 1983 ▶; Myers & Miller, 1988 ▶; Chenna et al., 2003 ▶), MAP259c from M. avium subsp. paratuberculosis. Crystals of MAP2569c from M. avium subsp. paratuberculosis K-10 (Li et al., 2005 ▶) were obtained that diffracted to 1.8 Å resolution. Details of the cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis are reported here.
2. Materials and methods
2.1. Cloning
The Gateway (Invitrogen) method was used to clone the Rv1208 gene, as well as those encoding its close putative orthologues from M. avium subsp. paratuberculosis and M. smegmatis, for expression in Escherichia coli. The MAP2569c gene was PCR-amplified from M. avium subsp. paratuberculosis K-10 genomic DNA using primers A (5′-CACCATGACGACGTCCGACCTGGT-3′) and B (5′-TCAGCGGGGCCGGATCGCCT-3′) and Proofstart DNA Polymerase (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The PCR product was purified using the UltraClean 15 DNA Purification Kit (Mo Bio Laboratories, California, USA) and cloned into the pENTR/D-TOPO entry vector using the pENTR/D-TOPO Cloning Kit (Invitrogen) to create the pENTR/D-TOPO entry clone. A positive clone was identified by digestion with PvuII and sequenced using T7 promoter and terminator primers. The MAP2569c gene was then transferred from the pENTR/D-TOPO entry clone to a modified version of the pDEST17 (N-terminal His6 tag) destination vector using LR Clonase (Invitrogen) according to the manufacturer’s instructions. The resulting expression construct encoded a 329-amino-acid (34.8 kDa) protein with a vector-derived 25-amino-acid leader sequence (H6GENLYFQGAAITSLYKKAG) containing a tobacco etch virus (TEV) protease-cleavage site and an N-terminal His6 tag, allowing the target protein to be released from the affinity tag prior to crystallization if necessary, and was used to transform E. coli BL21 (DE3).
2.2. Protein expression and purification
All protein expression was performed according to the autoinduction protocols developed by Studier (2005 ▶). Initial expression and solubility screening of Rv1208 was conducted in parallel to those of its close sequence homologues from M. avium subsp. paratuberculosis and M. smegmatis. Briefly, transformed BL21 cells were grown at 303 K overnight in 2 ml Overnight Express Instant TB Medium (Novagen, Wisconsin, USA) containing ampicillin (100 µg ml−1). The cells were lysed by the addition of PopCulture (Novagen) and the crude extracts were added to nickel-Sepharose (Ni–NTA, Qiagen) using a Freedom EVO liquid-handling robotic workstation (Tecan). The Ni–NTA-bound material was washed and the correctly folded recombinant His6-tagged proteins were then eluted with 400 mM imidazole and detected by SDS–PAGE. This initial screening indicated that the expression constructs for the orthologues of Rv1208 from M. avium subsp. paratuberculosis, termed MAP2569c, and from M. smegmatis, but not for Rv1208 itself, were suitable for crystallographic studies (Fig. 1 ▶). However, subsequent large-scale trials revealed that far greater active yields could be obtained for MAP259c than for its orthologue from M. smegmatis.
Figure 1.
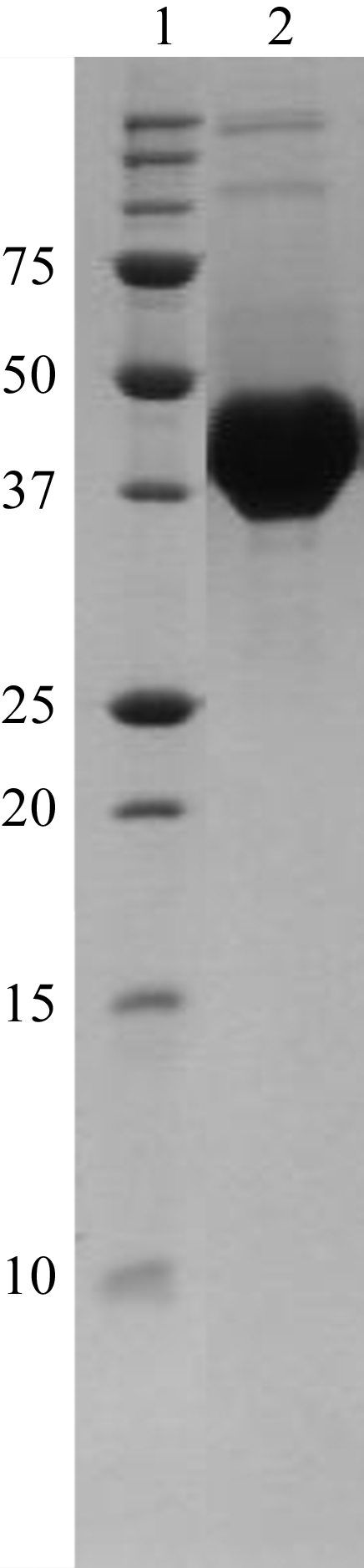
Nonreducing SDS–PAGE of MAP2569c purified using immobilized nickel-affinity, size-exclusion and ion-exchange chromatography (lane 2). The sizes of the molecular-weight markers (lane 1) are indicated. The gel was stained using Coomassie Brilliant Blue.
For large-scale expression and purification, transformed BL21 cells were used to inoculate a starter culture grown at 298 K overnight in a complex/modified version of the non-inducing medium PG (Studier, 2005 ▶), here termed LBA-0.8 [5 g l−1 yeast extract, 10 g l−1 tryptone, 1 g l−1 NaCl, 2 mM MgSO4, 0.5% glucose, 25 mM (NH4)2SO4, 50 mM KH2PO4, 50 mM Na2HPO4] containing ampicillin (100 µg ml−1). The addition of 0.5% glucose to the starter culture medium contributed to the repression of the T7 RNA polymerase via the catabolite repression/inducer exclusion mechanism. The LBA-0.8 starter culture was used at a dilution of 1:100 to inoculate a complex/modified version of the inducing medium PA-5052 (Studier, 2005 ▶), here termed LBA-5052 [5 g l−1 yeast extract, 10 g l−1 tryptone, 1 g l−1 NaCl, 2 mM MgSO4, 25 mM (NH4)2SO4, 50 mM KH2PO4, 50 mM Na2HPO4, 0.5% glycerol, 0.05% glucose, 0.2% α-lactose] containing ampicillin (100 µg ml−1). The expression culture was grown at 298 K for 24 h and the cells were harvested by centrifugation at 6000g for 20 min at 277 K. The cell pellets were resuspended in lysis buffer (20 mM Tris–HCl pH 8.0, 150 mM NaCl, 0.2 mM phenylmethylsulfonyl fluoride, 10 mM β-mercaptoethanol) and homogenized by passage through a French press (Avestin, Ottawa, Canada). The cell lysates were centrifuged at 10 000g for 15 min at 277 K and the soluble fraction was applied by gravity onto a 2 ml column of the nickel-affinity chromatography medium Ni Sepharose High Performance (Amersham Biosciences) in equilibration buffer (20 mM imidazole, 20 mM Tris–HCl pH 8.0, 30 mM NaCl). The column was washed with ten column volumes of equilibration buffer and MAP2569c was eluted in 250 mM imidazole, 20 mM Tris–HCl pH 8.0, 300 mM NaCl. The eluted material was supplemented with 2 mM ethylenediaminetetraacetic acid and the enriched His6-tagged protein was further purified by size-exclusion chromatography on a calibrated 16/60 Superdex 200 prep-grade column (Amersham Pharmacia, Uppsala, Sweden) in 20 mM Tris–HCl pH 8.0, 15 mM NaCl. The eluate containing MAP2569c was diluted 1:2 in 20 mM Tris–HCl pH 8.0 and applied onto a Mono Q 5/50 GL anion-exchange column (Amersham Biosciences) in 20 mM Tris–HCl pH 8.0 at 1 ml min−1. The column was washed with ten column volumes of 20 mM Tris–HCl pH 8.0 and MAP2569c was eluted using a linear gradient of 0–600 mM NaCl in 18 ml 20 mM Tris–HCl pH 8.0 at 0.3 ml min−1. The resulting sample showed a single band when analysed by nonreducing SDS–PAGE stained with Coomassie Brilliant Blue (Fig. 1 ▶). N-terminal sequencing indicated that the vector-derived 25-amino-acid leader sequence and the first four amino-acid residues of the protein were removed during purification. The purified MAP2569c was concentrated to 3 mg ml−1 in 20 mM Tris–HCl pH 8.0, 150 mM NaCl using a Centricon YM-10 (Millipore) and stored at 277 K for use in crystallization trials.
2.3. Crystallization, X-ray diffraction data collection and analysis
All crystallization experiments were performed at 293 K. Initial experiments involved screening Crystal Screen, Index Screen and PEG/Ion Screen (Hampton Research) conditions using a HoneyBee nanolitre-dispensing robotic workstation (Genome Solutions). Equal volumes of protein and precipitant solution (100 nl each) were mixed in sitting-drop vapour-diffusion experiments in 96-well Intelliplates (Hampton Research). A number of promising conditions were identified and manually reproduced and optimized in terms of pH, precipitant concentration, drop ratio and volume, and protein concentration using the hanging-drop vapour-diffusion technique in 24-well Linbro plates (Hampton Research). The best crystals were obtained by mixing 1 µl 6 mg ml−1 protein solution and 1 µl reservoir buffer containing 1.6 M monoammonium dihydrogen phosphate as the precipitant and 0.1 M sodium citrate pH 5.5. The colourless tetragonal crystals grew overnight to maximum dimensions of 0.3 × 0.3 × 0.1 mm (Fig. 2 ▶). In preparation for X-ray diffraction analysis, crystals were soaked in reservoir buffer containing 5, 10 and 15% 2-methyl-2,4-pentanediol as cryoprotectant for 5 min (in each solution) before flash-cooling in liquid nitrogen.
Figure 2.

Diffraction-quality crystal of MAP2569c.
X-ray diffraction data were collected at 100 K on BioCARS beamline 14-BM-C at the Advanced Photon Source, Chicago, USA using a Quantum 315 charge-coupled device detector. A complete data set to 1.8 Å resolution comprising 117 images (0.5° oscillation, 2 s exposure per oscillation) was collected from a single crystal and was processed and analysed using HKL-2000 (Otwinowski & Minor, 1997 ▶; Table 1 ▶).
Table 1. Crystal and data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Temperature (K) | 100 |
| X-ray source | APS, BioCARS, 14-BM-C |
| Detector | ADSC Quantum 315 |
| Wavelength (Å) | 1.0000 |
| Space group, crystal system | P41212, tetragonal |
| Unit-cell parameters (Å) | a = b = 86.6, c = 104.3 |
| Matthews coefficient VM (Å3 Da−1) | 2.7 |
| Solvent content (%) | 53.5 |
| Molecules per ASU | 1 |
| Resolution range (Å) | 100–1.80 (1.86–1.80) |
| Mosaicity (°) | 0.3 |
| Total observations | 172279 |
| Unique observations | 37229 |
| Multiplicity | 4.6 (4.7) |
| Data completeness (%) | 99.1 (100) |
| 〈I/σ(I)〉 | 43.8 (2.6) |
| Rmerge† (%) | 3.0 (50.5) |
R
merge = 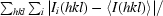
 , where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
, where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
3. Results and discussion
The autoinduction methods developed by Studier (2005 ▶) allow high-density cultures (i.e. grown to saturation) and thus high levels of expression of target proteins. Indeed, although approximately 90% of the recombinant MAP259c obtained from BL21 expression in the complex inducing medium used here was insoluble, the active yields attained for MAP2569c were approximately 1.6 mg l−1.
The parallel multiple-orthologue expression and solubility-screening approach adopted here allowed the identification of a suitable homologue of an essential M. tuberculosis protein of interest for crystallographic analyses. Although there are no obvious (sequence-based) clues as to why the orthologue of Rv1208 from M. avium subsp. paratuberculosis, termed MAP2569c, exhibited far superior expression and solubility in the E. coli system than its close homologues from M. tuberculosis and M. smegmatis, this phenomenon has proven useful for investigating M. tuberculosis biology though structural studies. Moreover, owing to the high sequence identity of MAP2569c to Rv1208, MAP259c is expected to adopt the same structure and thus perform the same important function as its essential homologue from the tuberculosis bacillus.
MAP2569c was successfully enriched using nickel-affinity chromatography and was approximately 80% pure following this step as estimated by SDS–PAGE. A polypeptide of approximately 37 kDa was observed that matched the predicted size of the recombinant MAP2569c (37.6 kDa), including the TEV protease-cleavage site, N-terminal His6 tag and additional amino acids encoded by the modified version of the pDEST17 destination vector (Invitrogen) used here. MAP2569c eluted from the gel-filtration column in monomeric form and was further purified by anion-exchange chromatography and concentrated to 6 mg ml−1 for optimized crystallization experiments. The crystals of MAP2569c grown here belong to the tetragonal space group P41212, with unit-cell parameters a = b = 86.6, c = 104.3 Å and one molecule per asymmetric unit, corresponding to a Matthews coefficient of 2.7 Å3 Da−1 and a solvent content of 54%. As MAP2569c shows low sequence identity (13%) to the sole structural representative of the GT2 family, SpsA (Charnock & Davies, 1999 ▶), the structure of MAP2569c will be pursued through the multiple anomalous dispersion method.
Owing to the countless combinations of donor and acceptor substrate and the seemingly limitless range of possible regiochemical and stereochemical linkages in products for GTs, these enzymes are often organism- or cell-specific and this presents potential drug targets in aetiological agents, including M. tuberculosis and other Mycobacterium species. Consequently, the crystallographic characterization of MAP2569c is not only likely to provide the first structure of a GT of the GT-A fold found in mycobacteria, but will also provide a structural template for putative orthologues of MAP2569c that may present such promising targets in future.
Acknowledgments
We thank the BioCARS staff for their assistance in data collection at the Advanced Photon Source, Chicago, USA. This work was supported by the Australian Research Council (ARC) Centre of Excellence in Structural and Functional Microbial Genomics and the National Health and Medical Research Council (NHMRC) of Australia. JR is an ARC Federation Fellow and TB is an NHMRC Career Development Award Fellow.
References
- Berg, S., Kaur, D., Jackson, M. & Brennan, P. J. (2007). Glycobiology, 17, 35R–56R. [DOI] [PubMed]
- Campbell, J. A., Davies, G. J., Bulone, V. & Henrissat, B. (1997). Biochem. J.326, 929–939. [DOI] [PMC free article] [PubMed]
- Charnock, S. J. & Davies, G. J. (1999). Biochemistry, 38, 6380–6385. [DOI] [PubMed]
- Chenna, R., Sugawara, H., Koike, T., Lopez, R., Gibson, T. J., Higgins, D. G. & Thompson, J. D. (2003). Nucleic Acids Res.31, 3497–3500. [DOI] [PMC free article] [PubMed]
- Cole, S. T. et al. (1998). Nature (London), 393, 537–544.
- Coutinho, P. M., Deleury, E., Davies, G. J. & Henrissat, B. (2003). J. Mol. Biol.328, 307–317. [DOI] [PubMed]
- Guerin, M. E., Kordulakova, J., Schaeffer, F., Svetlikova, Z., Buschiazzo, A., Giganti, D., Gicquel, B., Mikusova, K., Jackson, M. & Alzari, P. M. (2007). J. Biol. Chem.282, 20705–20714. [DOI] [PubMed]
- Li, L., Bannantine, J. P., Zhang, Q., Amonsin, A., May, B. J., Alt, D., Banerji, N., Kanjilal, S. & Kapur, V. (2005). Proc. Natl Acad. Sci. USA, 102, 12344–12349. [DOI] [PMC free article] [PubMed]
- Marland, Z., Beddoe, T., Zaker-Tabrizi, L., Lucet, I. S., Brammananth, R., Whisstock, J. C., Wilce, M. C., Coppel, R. L., Crellin, P. K. & Rossjohn, J. (2006). J. Mol. Biol.359, 983–997. [DOI] [PubMed]
- Moreland, N., Ashton, R., Baker, H. M., Ivanovic, I., Patterson, S., Arcus, V. L., Baker, E. N. & Lott, J. S. (2005). Acta Cryst. D61, 1378–1385. [DOI] [PubMed]
- Myers, E. W. & Miller, W. (1988). Comput. Appl. Biosci.4, 11–17. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Rossmann, M. G., Moras, D. & Olsen, K. W. (1974). Nature (London), 250, 194–199. [DOI] [PubMed]
- Sassetti, C. M., Boyd, D. H. & Rubin, E. J. (2003). Mol. Microbiol.48, 77–84. [DOI] [PubMed]
- Studier, F. W. (2005). Protein Expr. Purif.41, 207–234. [DOI] [PubMed]
- Vincentelli, R., Canaan, S., Campanacci, V., Valencia, C., Maurin, D., Frassinetti, F., Scappucini-Calvo, L., Bourne, Y., Cambillau, C. & Bignon, C. (2004). Protein Sci.13, 2782–2792. [DOI] [PMC free article] [PubMed]
- Wilbur, W. J. & Lipman, D. J. (1983). Proc. Natl Acad. Sci. USA, 80, 726–730. [DOI] [PMC free article] [PubMed]