

The X-ray structure of human CutA1 was solved in space group P212121, with unit-cell parameters a = 68.69, b = 88.84, c = 125.33 Å and six molecules per asymmetric unit.
Keywords: divalent-cation tolerance homologues, Homo sapiens, trimeric structures
Abstract
The structure of human brain CutA1 (HsCutA1) has been determined using diffraction data to 2.05 Å resolution. HsCutA1 has been implicated in the anchoring of acetylcholinesterase in neuronal cell membranes, while its bacterial homologue Escherichia coli CutA1 is involved in copper tolerance. Additionally, the structure of HsCutA1 bears similarity to that of the signal transduction protein PII, which is involved in regulation of nitrogen metabolism. Although several crystal structures of CutA1 from various sources with different rotation angles and degrees of interaction between trimer interfaces have been reported, the specific functional role of CutA1 is still unclear. In this study, the X-ray structure of HsCutA1 was determined in space group P212121, with unit-cell parameters a = 68.69, b = 88.84, c = 125.33 Å and six molecules per asymmetric unit. HsCutA1 is a trimeric molecule with intertwined antiparallel β-strands; each subunit has a molecular weight of 14.6 kDa and contains 135 amino-acid residues. In order to obtain clues to the possible function of HsCutA1, its crystal structure was compared with those of other CutA1 and PII proteins.
1. Introduction
A significant number of proteins emerging from genome-sequencing efforts continue to remain as hypothetical proteins of unknown function. Since the biological function of proteins is tightly coupled to their folds, three-dimensional structures in combination with information from sequences can have an important impact in inferring the molecular function of proteins (Kim et al., 2003 ▶). In particular, structural similarity to proteins of known structure and function may provide important clues to the possible function of new proteins.
The precise function of the CutA family of proteins is of great importance owing to their wide presence and implicated role in several diseases (Leonard & Salpeter, 1979 ▶; Bush, 2000 ▶). In the brain, human (Homo sapiens) CutA1 (HsCutA1) appears to be necessary for the localization of acetylcholinesterase (AChE) at the cell surface (Perrier et al., 2000 ▶; Navaratnam et al., 2000 ▶). From biochemical analysis, HsCutA1 has been reported to interact with the hydrophobic proline-rich membrane anchor (PRiMA) protein encoding the AChE anchor in mammalian brain (Perrier et al., 2002 ▶). The human CutA1 gene provides three proteins with alternative hydrophobic N-terminal sequences (Perrier et al., 2000 ▶; Navaratnam et al., 2000 ▶; Yang et al., 2007 ▶). The CutA1 homologues in eukaryotes are very well conserved, with the exception of the N-terminal hydrophobic domain. HsCutA1 is also a homologue of the CutA1 proteins from several bacteria, including Escherichia coli CutA1 (EcCutA1), which is found to be involved in tolerance to Cu2+ and other divalent heavy-metal ions (Fong et al., 1995 ▶). Other CutA1 sequences have been obtained by analysis of transcripts and no function is known for the corresponding proteins. The CutA1 structures are quite similar to that of the signal transduction protein PII, indicating a possible function of CutA1 in signal transduction (Arnesano et al., 2003 ▶). Structural characterization of CutA1 could guide future investigations aimed at understanding the physiological role of the protein. In this study, we describe the detailed structure of the HsCutA1 protein determined at 2.05 Å resolution and identify potential sites for interaction and clues to protein function by comparison with known CutA1 and PII structures.
2. Materials and methods
2.1. Expression and purification of HsCutA1
The putative CutA1 from H. sapiens (human brain) used in this study has a molecular weight of 14.7 kDa and consists of 136 amino-acid residues (corresponding to residues 44–179; the accession number for the human CutA1 amino-acid sequence is gi:7341255). We generated an N-terminally truncated version of the HsCutA1 protein as our attempts to express the complete HsCutA1 gene were unsuccessful. A similar short version of the protein that lacks N-terminal residues was copurified with AChE from human brain and encoded as CutA1 (Navaratnam et al., 2000 ▶). Synthesized DNA fragments encoding HsCutA1 (Met44–Pro179) were cloned into the NdeI and BamH1 sites of pET-11a vector and Escherichia coli BL21-Codon Plus (DE3)-RIL cells were transformed with the expression vector for protein production. The E. coli cells were grown without IPTG induction at 310 K in Luria–Bertani medium containing 50 µg ml−1 ampicillin for 20 h. The cells were harvested by centrifugation at 9100g for 4 min at 277 K and subsequently suspended in 20 mM Tris–HCl pH 8.0 containing 0.5 M NaCl and 5 mM 2-mercaptoethanol; they were finally disrupted by sonication and heated at 343 K for 10 min. The cell debris and denatured protein were removed by centrifugation (18 800g for 30 min at 277 K). The supernatant solution was used as the crude extract for purification. The crude extract was desalted by dialysis with 20 mM Tris–HCl pH 8.0 (buffer A) and applied onto a Super Q Toyopearl 650M (Tosoh) column equilibrated with buffer A. After elution with a linear gradient of 0.2–0.8 M NaCl, the fraction containing HsCutA1 was concentrated by ultrafiltration (Vivaspin 5K cut) and loaded onto a HiLoad 16/60 Superdex 75 prep-grade column (Amersham Biosciences) equilibrated with buffer A containing 0.2 M NaCl. The fraction containing HsCutA1 was desalted using a HiPrep 26/10 desalting column (GE Healthcare) and applied onto a Resource Q column (GE Healthcare), again equilibrated with buffer A. After elution with a linear gradient of 0.4–1.0 M NaCl, the fraction containing HsCutA1 was collected. The homogeneity and identity of the purified sample were estimated by SDS–PAGE (Laemmli, 1970 ▶). Further characterization using mass spectrometry showed that the protein lacked the N-terminal Met44 residue. The oligomeric state of the protein was studied by an analytical ultracentrifugation experiment in a Beckman XL-A analytical ultracentrifuge (Beckman-Coulter) using an AN-Ti60 rotor. The result corresponded to a molecular weight of 43.2 ± 0.5 kDa, suggesting a trimeric state in solution. Finally, the purified HsCutA1 was concentrated by ultrafiltration to 8 mg ml−1 in buffer A containing 0.2 M NaCl.
2.2. Crystallization
Crystallization trials were carried out using the oil-microbatch method (Chayen et al., 1990 ▶) using Nunc HLA plates at 291 K. Initial screening for crystallization conditions was performed using Crystal Screens I and II, Crystal Screen Cryo and Crystal Screen Lite from Hampton Research (Jancarik & Kim, 1991 ▶) and Wizard I and II and Wizard Cryo I and II from Emerald BioSystems. Equal volumes of protein solution (1.0 µl) and precipitant solution (1.0 µl) were mixed. The crystallization drop was overlaid with a 1:1 mixture of silicone and paraffin oils, allowing slow evaporation of the water in the drop. The best diffracting crystals were obtained using condition No. 10 of Hampton Research Crystal Screen Cryo [25%(w/v) PEG 4000, 0.17 M ammonium acetate, 0.085 M sodium acetate pH 4.6, 15%(v/v) glycerol]. Single crystals with sharp edges and typical dimensions of 0.2 × 0.2 × 0.1 mm appeared within a week of setup.
2.3. Data collection and structure determination
X-ray diffraction-intensity data were collected on SPring-8 beamline BL26B1 using a Jupiter 210 detector (Ueno et al., 2006 ▶). A crystal was scooped up in a nylon loop and flash-cooled in a cold nitrogen stream at 100 K without the addition of cryoprotectant. A total of 180 frames were collected with 1° oscillation and 25 s exposure time per image. The wavelength of the synchrotron radiation was 1.0 Å and the crystal-to-detector distance was 160 mm. The diffraction data were integrated and scaled to 2.05 Å resolution using DENZO and SCALEPACK as implemented in the HKL-2000 program package (Otwinowski & Minor, 1997 ▶). Autoindexing and data reduction indicated that the crystal belonged to space group P212121, with unit-cell parameters a = 68.69, b = 88.84, c = 125.33 Å. There are six molecules in the asymmetric unit, giving a Matthews coefficient of 2.2 Å3 Da−1 and an approximate solvent content of 43% (Matthews, 1968 ▶). The structure of HsCutA1 was determined using the molecular-replacement method with the software MOLREP from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶; Vagin & Teplyakov, 1997 ▶); a trimer of human CutA1 (PDB code 1xk8, unpublished structure at 2.7 Å resolution) was used as the model in the rotation and translation function. The structure was then refined and water molecules were added using REFMAC5 (Collaborative Computational Project, Number 4, 1994 ▶; Murshudov et al., 1997 ▶) and CNS (Brünger et al., 1998 ▶). The program QUANTA (Accelrys, San Diego, California, USA) was used for molecular rebuilding and visualization of the structure. The stereochemical quality was assessed using the program PROCHECK (Laskowski et al., 1993 ▶). The final refinement of coordinates using the 37.81–2.05 Å resolution reflections resulted in R work = 22.1% and R free = 26.5%. The data-collection and refinement statistics and details of the final model are given in Table 1 ▶.
Table 1. Summary of data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Crystal data | |
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 68.69, b = 88.84, c = 125.34 |
| Subunits per ASU | 6 |
| Data collection and refinement | |
| Temperature (K) | 100 |
| Wavelength (Å) | 1.0 |
| Resolution range (Å) | 40.0–2.05 (2.12–2.05) |
| No. of unique reflections | 45947 (3778) |
| Redundancy | 6.6 (5.8) |
| Completeness (%) | 94.8 (79.2) |
| Average I/σ(I) | 12.5 (2.3) |
| Rmerge† (%) | 6.9 (37.5) |
| Rwork‡/Rfree§ | 0.221/0.265 |
| No. of protein atoms | 5027 |
| No. of water molecules | 252 |
| R.m.s.d. bond lengths (Å) | 0.007 |
| R.m.s.d. bond angles (°) | 1.30 |
| Mean B factor (Å2) | 51.1 |
| Mean B factor, main chains (Å2) | 47.9 |
| Mean B factor, side chains (Å2) | 53.4 |
| Mean B factor, waters (Å2) | 57.5 |
| Ramachandran plot (%) | |
| Most favoured | 96.0 |
| Additionally allowed | 4.0 |
| PDB code | 2zfh |
R
merge = 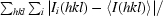
 , where Ii(hkl) and 〈I(hkl)〉 are the observed intensity of measurement i and the mean intensity of the reflection with indices hkl, respectively.
, where Ii(hkl) and 〈I(hkl)〉 are the observed intensity of measurement i and the mean intensity of the reflection with indices hkl, respectively.
R
work = 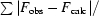
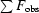 , where F
obs and F
calc are the observed and calculated structure factors, respectively.
, where F
obs and F
calc are the observed and calculated structure factors, respectively.
R free is the R factor for a subset of 5% of the reflections that were omitted from refinement.
The structures were superimposed on each other using the program LSQKAB from the CCP4 package (Collaborative Computational Project, Number 4, 1994 ▶; Kabsch, 1976 ▶). The figures illustrating these structures were prepared with the program PyMOL (DeLano, 2002 ▶). Sequence alignments were generated using ClustalX (Jeanmougin et al., 1998 ▶). Electrostatic surface potentials were calculated using the vacuum electrostatics feature of PyMOL (DeLano, 2002 ▶).
3. Results and discussion
3.1. Overall structure
The final model contains six HsCutA1 polypeptide chains in the crystal asymmetric unit, which consists of 643 amino-acid residues and 252 water molecules, and has a reasonable structure with excellent stereochemistry (Table 1 ▶). Chains A and C consist of residues 61–169 and 63–168, respectively, while the other chains consist of residues 62–168. The N-terminal (45–60) and C-terminal (170–179) residues in the HsCutA1 protein could not be resolved; the interpretable electron density of the HsCutA1 crystals starts at residue Ser61 and terminates at Glu169. HsCutA1 is functional as a trimer and the orthorhombic crystal form contains two such trimers per asymmetric unit (Fig. 1 ▶). Analogous to other CutA1 structures, the HsCutA1 subunit adopts a ferredoxin-like fold made up of a double βαβ motif in which the antiparallel β-sheet packs against antiparallel α-helices and contains an additional short strand (β5) and a C-terminal helix (α3) (Fig. 1 ▶ a). The β2 and β3 strands and the loop between them (the β2-β3 loop) form an extended β-hairpin. The six subunits in the asymmetric unit and the two trimers formed by them are very similar. The root-mean-square deviations (r.m.s.d.s) of the Cα atoms between any pair of subunits are 0.23–0.26 Å and that between the trimers is 0.3 Å. Superposition of the HsCutA1 subunits shows different conformations in the turn region of the β-hairpins (Fig. 2 ▶). This fact, together with the fact that the B factors of the residues residing in the turn region of the β-hairpin are almost twice as high as the mean B-factor value, indicates a high degree of conformational flexibility. Thus, comparison of copies (subunits) of the protein provides some information about the way the protein conformation can vary in protein solution. Strand β2 in HsCutA1 has a kink at its centre that may be caused by the presence of Pro101 in the middle of the β2 strand (Fig. 2 ▶). It is well known that proline tends to accommodate less readily in extended regions of β-sheet structures because of its cyclic structure (MacArthur & Thornton, 1991 ▶; Li et al., 1996 ▶; Reiersen & Rees, 2001 ▶). In HsCutA1, the kink-type deformation of β2 close to Pro101 allows the formation of hydrogen-bond arrangements that stabilize the β2 strand. Overlays of the six subunits show conformational variation of the α2-β4 loop residues His141 and Pro142 in subunit D only (Fig. 2 ▶), which can be explained by the way the molecules pack in the crystal.
Figure 1.

Structure of HsCutA1. (a) Ribbon diagram of a HsCutA1 protomer, which consists of five β-strands and three α-helices. The ribbon model is coloured according to the sequence by a rainbow colour ramp from blue at the N-terminus to red at the C-terminus. (b) Ribbon diagram of trimeric HsCutA1 viewed along of the threefold axis of symmetry vertical to the plane of the paper. The conserved residues are shown in stick models and are mainly positioned around the putative active-site clefts. (c) Hexameric structure of HsCutA1 as found in the crystal asymmetric unit represented as ribbons; each protomer is shown in a different colour.
Figure 2.

Superposition of the six subunits in the asymmetric unit of the HsCutA1 crystal. Circled area 1, kink in the β2-strand at the position of Pro101 and Gln102. Circled area 2, β-hairpins of the protomers; these are flexible in solution and can adopt various conformations upon crystal packing. Circled area 3, packing peculiarity of His141 and Pro142 in the D subunit (coloured yellow).
In the HsCutA1 subunit the conserved residues are mainly gathered into two regions. A loop region between α2 and β4, spatially close to the β-hairpin turn, encompasses the conserved residues His141, Pro142 and Tyr143. The other highly conserved amino acids Cys96 and Tyr107 in β2, Glu118, Lys124 and Thr125 in β3, Trp109 and Gly111 in the β2-β3 loop and Tyr160 and Trp163 in α3 are clustered at the other end of the scaffold. The tertiary fold assembles such that the two conserved regions in the subunits are brought together, thus forming three potential active sites in the clefts at the trimer interfaces (Fig. 1 ▶ b). The β-hairpin acts as an oligomerization site, favouring intersubunit hydrogen-bond formation. The subunits interact by hydrogen-bond pairs between the N-terminal half of strand β2 from one subunit and the C-terminal half of strand β2 from another subunit and between strand β4 from one subunit and the short strand β5 from another subunit (Table 2 ▶). The HsCutA1 trimer is well stabilized electrostatically as negative and positive regions from different subunits are in contact.
Table 2. Hydrogen-bonding interactions between subunits of the HsCutA1 trimer.
The maximum distance cutoff between contact atoms is 3.5 Å.
| Protein atoms | |||
|---|---|---|---|
| Subunit A | Subunit B | Subunit C | Distance (Å) |
| Arg86 NE | Glu115 OE2 | 2.84 | |
| Val97 N | Ile106 O | 2.86 | |
| Val97 O | Ile106 N | 2.78 | |
| Asn98 ND2 | Ser105 OG | 2.82 | |
| Leu99 N | Thr104 O | 2.67 | |
| Leu99 O | Thr104 N | 3.12 | |
| Lys124 NZ | Glu147 OE1 | 2.78 | |
| Glu154 N | Ala150 O | 3.02 | |
| Gln155 O | Ala150 N | 3.10 | |
| Gln155 NE2 | Ser127 O | 2.99 | |
| Asn157 N | Val148 O | 2.81 | |
| Asn157 OD1 | Arg138 NE | 3.30 | |
| Asn157 OD1 | Arg138 NH2 | 2.98 | |
| Asn157 ND2 | Ala146 N | 3.77 | |
| Asn157 ND2 | Ala146 C | 3.84 | |
| Asn157 ND2 | Ala146 O | 2.83 | |
| Tyr160 OH | Glu147 OE1 | 3.19 | |
| Thr104 N | Leu99 O | 3.13 | |
| Thr104 O | Leu99 N | 2.73 | |
| Ser105 OG | Asn98 OD1 | 3.09 | |
| Ile106 N | Val97 O | 2.79 | |
| Ile106 O | Val97 N | 2.90 | |
| Glu115 OE2 | Arg86 NE | 2.87 | |
| Ser127 O | Gln155 NE2 | 2.87 | |
| Ser127 OG | Gln155 NE2 | 3.37 | |
| Arg138 NH1 | Asn157 OD1 | 2.50 | |
| Ala146 O | Asn157 ND2 | 3.10 | |
| Glu147 OE1 | Tyr160 OH | 3.46 | |
| Val148 O | Asn157 N | 2.89 | |
| Ala150 N | Gln155 O | 2.95 | |
| Ala150 O | Glu154 N | 3.07 | |
| Arg86 NH1 | Glu115 OE2 | 2.57 | |
| Val97 N | Ile106 O | 2.80 | |
| Val97 O | Ile106 N | 2.83 | |
| Asn98 OD1 | Ser105 OG | 2.86 | |
| Leu99 N | Thr104 O | 2.83 | |
| Leu99 O | Thr104 N | 3.07 | |
| Lys124 NZ | Glu147 OE2 | 3.32 | |
| Glu154 N | Ala150 O | 2.99 | |
| Gln155 O | Ala150 N | 2.76 | |
| Gln155 NE2 | Ser127 O | 3.13 | |
| Asn157 N | Val148 O | 2.79 | |
| Asn157 OD1 | Arg138 NH1 | 3.04 | |
| Asn157 ND2 | Ala146 O | 2.84 | |
| Tyr160 OH | Glu147 OE2 | 2.75 | |
Electrostatic calculations conducted on the trimer show large negatively charged patches in and around the trimer-interface clefts (Fig. 3 ▶ a). A positively charged area of the β-hairpin turn (Trp109–Lys112) is placed near the negatively charged clefts. The charged nature of the clefts creates a potential docking site for ligands and the discrete charge distribution around the clefts suggests that electrostatics play a very important role in guiding the substrate molecules to their correct docking sites. The surface of one side of the HsCutA1 trimer is almost nonpolar, with a negative cavity formed mainly by residue Glu154 (Fig. 3 ▶ c), whereas the other face is entirely charged (Fig. 3 ▶ b). In the hexamer the nonpolar face of the HsCutA1 trimer interacts with the same face of a second trimer and the negatively charged face is exposed to the solvent. A dimer of trimers is formed in which the second trimer is rotated by 25° around the ternary axis with respect to the first trimer. The buried interface between the two trimers is about 2470 Å2, whereas the total interacting surface between subunits in a trimer is 6840 Å2, indicating that the interactions within the trimer are more extensive than those between the two trimers.
Figure 3.

Electrostatic surface representation of the HsCutA1 trimer. Red represents negative charge and blue positive. (a) Side view of the protein, showing the putative active-site cleft. (b) The view from one side of the threefold axis of symmetry vertical to the plane of the paper. (c) The view rotated 180° around the threefold axis from the orientation shown in (b). In the hexamer the trimer interacts using this face.
3.2. Comparison with other CutA1 proteins
In all CutA1 structures the ferredoxin-like subunits assemble into trimers involving a long β-hairpin. After superposition of Cα atoms, the r.m.s.d. between the present coordinates and those of human CutA1 refined at 2.7 Å (PDB code 1xk8; unpublished results) are 0.29 and 0.45 Å for the trimer and hexamer, respectively. The overall structure of HsCutA1 resembles that of rat CutA1 (Arnesano et al., 2003 ▶). They both belong to the same space group with very similar unit-cell parameters, six subunits in the asymmetric unit and the same rotation angle between the trimer interfaces around the ternary axis with similar charge interactions. The r.m.s.d. for Cα atoms between HsCutA1 and rat CutA1 are 0.43 and 0.53 Å for the trimers and hexamers, respectively, which confirm a high degree of homology between them. The trimers in the asymmetric unit of EcCutA1 (PDB code 1naq; Arnesano et al., 2003 ▶) also interact through nonpolar faces, but the rotation angle of the trimers around the ternary axis is different compared with that in HsCutA1. Although the CutA1 trimers interact in a different way in the crystal, a rigid trimeric core structure appears to be common to the CutA1 proteins. Superposition of CutA1 trimers from different sources shows that the main differences are localized in the turn area of the β-hairpins (Fig. 4 ▶ b); another peculiarity is the observation of a conformational kink in the β2 strand when a proline residue is present in the sequence of β2 (Fig. 4 ▶). CutA1 from Pyrococcus horikoshii (PhCutA1) and CutA1 from Thermotoga maritima (TmCutA1), both of which do not have a proline in β2, do not show any kink. This effect in the hyperthermostable PhCutA1 (Tanaka et al., 2006 ▶) could indicate a possible role of the conformation of β2 in protein thermostability. Therefore, it would be worth observing the alteration in the conformation of β2 and the consequent effect on protein stability induced by the mutation or deletion of Pro101 in HsCutA1.
Figure 4.

(a) Sequence alignment of HsCutA1 with other selected CutA1 proteins for which three-dimensional structures are available. The secondary-structural elements shown above the sequences and the numbering of the residues refers to HsCutA1. Completely conserved residues are highlighted by white characters on a red background. Helices (α-helices) are represented by squiggles, β-strands are shown as arrows and β-turns are marked TT. This sequence alignment was created using the following sequences from the PDB: RnCutA1 (CutA1 from Rattus norvegicus; PDB code 1osc), TtCutA1 (CutA1 from Thermus thermophilus; 1nza), EcCutA1 (CutA1 from E. coli; 1naq), TmCutA1 (CutA1 from Thermotoga maritima; 1kr4; Savchenko et al., 2003 ▶), PhCutA1 (CutA1 from P. horikoshi; 1v9b). (b) Superposition of the trimers of selected CutA1 structures: HsCutA1 (green), RnCutA1 (blue), TtCutA1 (yellow), EcCutA1 (magenta), TmCutA1 (cyan) and PhCutA1 (orange). Circled area 1, the β2-strand kink observed in HsCutA1, RnCutA1, TtCutA1 and EcCutA1 but not in TmCutA1 and PhCutA1. Circled area 2, the β-hairpin turn; the largest differences in the superposition of the CutA1 trimers are found in the flexible β-hairpin turns.
3.3. Comparison with PII proteins
Despite extremely limited sequence conservation, the CutA1 architecture is similar to those of several well characterized PII signal transduction proteins: GlnB (Cheah et al., 1994 ▶; Carr et al., 1996 ▶; Xu et al., 1998 ▶, 2003 ▶), GlnK (Sakai et al., 2005 ▶; Yildiz et al., 2007 ▶) and the PII-like domains of YqfO (Godsey et al., 2007 ▶) and of the hypothetical protein SA1388 (Saikatendu et al., 2006 ▶). The major structural differences are the presence of a C-terminal β-strand in PII, which replaces helix α3 of CutA1; in PII there is also a large loop between strands β2 and β3 that protrudes into the solvent, while in CutA1 strands β2 and β3 are longer and form a β-hairpin (Fig. 5 ▶). The X-ray studies show that the β2-β3 loop of PII adopts various conformations upon binding of the various ligands and thus modulates protein–protein interactions (Xu et al., 1998 ▶; Llácer et al., 2007 ▶; Mizuno et al., 2007 ▶; Yildiz et al., 2007 ▶). In the YqfO and SA1388 proteins, the trimeric PII-like domains have been reported to act as a signal sensor to regulate the still-unknown catalytic activity of the more conserved domains (Godsey et al., 2007 ▶; Saikatendu et al., 2006 ▶). Although the PII, bacterial CutA1 and mammalian CutA1 proteins have been found to be involved in diverse functions, the evolutionarily conserved trimeric assembly points to a similar mechanism of action. Acting as a signal transduction protein, HsCutA1 may serve as a sensor/modulator of the redox state of thiol groups in the PRiMA–AChE complex (Arnesano et al., 2003 ▶). For this functionally crucial role, HsCutA1 may use the thiol group of the conserved Cys96 placed at the N-terminal end of β2 very close to the cleft of the trimer interface. In the interface clefts of several PII structures, ATP or ADP was found in an orientation with the base in the deepest part of the active-site cleft and the phosphates poking out toward the solvent (Xu et al., 1998 ▶; Schwarzenbacher et al., 2004 ▶; Sakai et al., 2005 ▶; Mizuno et al., 2007 ▶; Yildiz et al., 2007 ▶). Binding of nucleotides stabilizes a conformation of the protruding β2-β3 loop of PII that favours its distal region insertion into the interdomain crevice of the interacting protein. A mercury ion or mercuribenzoic acid were found in the analogous clefts of EcCutA1 (Arnesano et al., 2003 ▶). Most of the conserved residues in HsCutA1 are placed in regions corresponding to the clefts at the trimer interfaces (Fig. 1 ▶ b), allowing them to be considered as putative active-site residues. Our attempts to obtain the HsCutA1 structure in the presence of divalent metals or nucleotides have as yet been unsuccessful. The clefts of HsCutA1 contain many Asp and Glu residues that provide negative charge, while the clefts in PII are positively charged. From the lack of conservation in charge and binding residues between HsCutA1 and PII, it is difficult to predict that nucleotides will be the natural ligand for HsCutA1. Attempts to model a nucleotide in the HsCutA1 cleft led to inescapable steric clashes with protein atoms. However, considering the binding flexibility of the cleft that is displayed in proteins with this fold and the presence of the aromatic rings of Trp163, Hys141, Tyr143, Trp109 in the immediate vicinity of the HsCutA1 clefts, nucleotides should not be ruled out as possible natural ligands. It is more likely that the negatively charged clefts of HsCutA1 may function as channels to target cationic effectors, for example metal ions.
Figure 5.

Ribbon diagram of the trimer of HsCutA1 (coloured in green) overlaid with PII proteins of similar structure. The PII proteins are from Methanococcus jannaschii (PDB code 2j9d; Yildiz et al., 2007 ▶; marine), Synechococcus elongates (2v5h; Llácer et al., 1991 ▶; yellow), Bacillus cereus (2gx8; Godsey et al., 2007 ▶; magenta) and E. coli (2gnk; Xu et al., 1998 ▶; cyan). The molecules of ATP bound to E. coli GlnK (2gnk; Xu et al., 1998 ▶) in the interface clefts are presented as ball-and-stick models.
It can be concluded from the structural data that the functionally important areas of HsCutA1 appear to be the putative active-site clefts and the flexible β-hairpins. Oligomerization of HsCutA1 allows the small (14.6 kDa) protein to form a compact cylinder-shaped structure and offers the three β-hairpins and clefts sites for specific interactions with other proteins and effector molecules. These regions contain conserved residues and the flexible β-hairpin may change conformation on the binding of effectors and/or docking with a receptor. The negatively charged cleft of HsCutA1 reflects the positive charge of the substrate ligands and is readily accessible to solvent. Elucidation of the structure of HsCutA1 might serve as an important initial step towards providing clues to its function. Future work will include determining the natural ligand(s) of this protein in order to illuminate the specific function(s) of HsCutA1. Also, the role of the hydrophobic N-terminal domain, the structure of which has not been solved owing to its flexibility, is as yet unclear.
Supplementary Material
PDB reference: CutA1, 2zfh, r2zfhsf
Acknowledgments
We thank the staff at beamline BL26B1 of SPring-8 for excellent facilities and assistance. This work was supported by the ‘National Project on Protein Structural and Functional Analyses’ funded by the MEXT of Japan.
References
- Arnesano, F., Banci, L., Benvenuti, M., Bertini, I., Calderone, V., Mangani, S. & Viezzoli, M. S. (2003). J. Biol. Chem.278, 45999–46006. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Bush, A. I. (2000). Curr. Opin. Chem. Biol.4, 184–191.
- Carr, P. D., Cheah, E., Suffolk, P. M., Vasudevan, S. G., Dixon, N. E. & Ollis, D. L. (1996). Acta Cryst. D52, 93–104. [DOI] [PubMed]
- Chayen, N. E., Shaw Stewart, P. D., Maeder, D. L. & Blow, D. M. (1990). J. Appl. Cryst.23, 297–302.
- Cheah, E., Carr, P. D., Suffolk, P. M., Vasudevan, S. G., Dixon, N. E. & Ollis, D. L. (1994). Structure, 2, 981–990. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, California, USA.
- Fong, S. T., Camakaris, J. & Lee, B. T. (1995). Mol. Microbiol.15, 1127–1137. [DOI] [PubMed]
- Godsey, M. H., Minasov, G., Shuvalova, L., Brunzelle, J. S., Vorontsov, I. I., Collart, F. R. & Anderson, W. F. (2007). Protein Sci.16, 1285–1293. [DOI] [PMC free article] [PubMed]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411.
- Jeanmougin, F., Thompson, J. D., Gouy, M., Higgins, D. G. & Gibson, T. J. (1998). Trends. Biochem. Sci.23, 403–405. [DOI] [PubMed]
- Kabsch, W. (1976). Acta Cryst. A32, 922–923.
- Kim, S.-H., Shin, D. H., Choi, I. G., Schulze-Gahmen, U., Chen, S. & Kim, R. (2003). J. Struct. Funct. Genomics, 4, 129–135. [DOI] [PubMed]
- Laemmli, U. K. (1970). Nature (London), 227, 680–685. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Leonard, J. P. & Salpeter, M. M. (1979). J. Cell Biol.82, 811–819. [DOI] [PMC free article] [PubMed]
- Li, S. C., Goto, N. K., Williams, K. A. & Deber, C. M. (1996). Proc. Natl Acad. Sci. USA, 93, 6676–6681.
- Llácer, J. L., Contreras, A., Forchhammer, K., Marco-Marín, C., Gil-Ortiz, F., Maldonado, R., Fita, I. & Rubio, V. (2007). Proc. Natl Acad. Sci. USA, 104, 17644–17649. [DOI] [PMC free article] [PubMed]
- MacArthur, M. W. & Thornton, J. W. (1991). J. Mol. Biol.218, 397–412. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Mizuno, Y., Moorhead, G. B. G. & Ng, K. K.-S. (2007). J. Biol. Chem.282, 35733–35740. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Navaratnam, D. S., Fernando, F. S., Priddle, J. D., Giles, K., Clegg, S. M., Pappin, D. J., Craig, I. & Smith, A. D. (2000). J. Neurochem.74, 2146–2153. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Perrier, A. L., Cousin, X., Boschetti, N., Haas, R., Chatel, J. M., Bon, S., Roberts, W. L., Pickett, S. R., Massoulie, J., Rosenberry, T. L. & Krejci, E. (2000). J. Biol. Chem.275, 34260–34265. [DOI] [PubMed]
- Perrier, A. L., Massoulie, J. & Krejci, E. (2002). Neuron, 33, 275–285. [DOI] [PubMed]
- Reiersen, H. & Rees, A. R. (2001). Trends Biochem. Sci.26, 679–684. [DOI] [PubMed]
- Sakai, H., Wang, H., Takemoto-Hori, C., Kaminishi, T., Yamaguchi, H., Kamewari, Y., Terada, T., Kuramitsu, S., Shirouzu, M. & Yokoyama, S. (2005). J. Struct. Biol.149, 99–110. [DOI] [PubMed]
- Saikatendu, K. S., Zhang, X., Kinch, L., Leybourne, M., Grishin, N. V. & Zhang, H. (2006). BMC Struct. Biol.6, 27. [DOI] [PMC free article] [PubMed]
- Savchenko, A., Skarina, T., Evdokimova, E., Watson, J. D., Laskowski, R., Arrowsmith, C. H., Edwards, A. M., Joachimiak, A. & Zhang, R. G. (2003). Proteins, 54,162–165. [DOI] [PMC free article] [PubMed]
- Schwarzenbacher, R. et al. (2004). Proteins, 54, 810–813. [DOI] [PubMed]
- Tanaka, T., Sawano, M., Ogasahara, K., Sakaguchi, Y., Bagautdinov, B., Katoh, E., Kuroishi, C., Shinkai, A., Yokoyama, S. & Yutani, K. (2006). FEBS Lett.580, 4224–4230. [DOI] [PubMed]
- Ueno, G., Kanda, K., Hirose, R., Ida, K., Kumasaka, T. & Yamamoto, Y. (2006). J. Struct. Funct. Genomics, 7, 15–22. [DOI] [PubMed]
- Xu, Y., Carr, P. D., Clancy, P., Garcia-Dominguez, M., Forchhammer, K., Florencio, F., Tandeau de Marsac, N., Vasudevan, S. G. & Ollis, D. L. (2003). Acta Cryst. D59, 2183–2190. [DOI] [PubMed]
- Xu, Y., Cheah, E., Carr, P. D., van Heeswijk, W. C., Westerhoff, H. V., Vasudevan, S. G. & Ollis, D. L. (1998). J. Mol. Biol.202, 149–165. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.
- Yang, J., Yang, H., Yan, L., Yang, L. & Yu, L. (2007). Mol. Biol. Rep. doi:10.1007/s11033-007-9152-9. [DOI] [PubMed]
- Yildiz, Ö., Kalthoff, C., Raunser, S. & Kühlbrandt, W. (2007). EMBO J.26, 589–599. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: CutA1, 2zfh, r2zfhsf