

Structures of BA0252, an alanine racemase from B. anthracis, in the presence and absence of the inhibitor (R)-1-aminoethylphosphonic acid (l-Ala-P) and determined by X-ray crystallography to resolutions of 2.1 and 1.47 Å, respectively, are described.
Keywords: d-alanine, l-alanine, germination, inhibition, pyridoxal 5′-phosphate, reductive methylation
Abstract
Bacillus anthracis, the causative agent of anthrax, has been targeted by the Oxford Protein Production Facility to validate high-throughput protocols within the Structural Proteomics in Europe project. As part of this work, the structures of an alanine racemase (BA0252) in the presence and absence of the inhibitor (R)-1-aminoethylphosphonic acid (l-Ala-P) have determined by X-ray crystallography to resolutions of 2.1 and 1.47 Å, respectively. Difficulties in crystallizing this protein were overcome by the use of reductive methylation. Alanine racemase has attracted much interest as a possible target for anti-anthrax drugs: not only is d-alanine a vital component of the bacterial cell wall, but recent studies also indicate that alanine racemase, which is accessible in the exosporium, plays a key role in inhibition of germination in B. anthracis. These structures confirm the binding mode of l-Ala-P but suggest an unexpected mechanism of inhibition of alanine racemase by this compound and could provide a basis for the design of improved alanine racemase inhibitors with potential as anti-anthrax therapies.
1. Introduction
In order to facilitate the development of methods for the high-throughput production and crystallization of biomedically important proteins, the Oxford Protein Production Facility (OPPF), as part of the Structural Proteomics in Europe (SPINE) project, has targeted proteins from Bacillus anthracis, the causative agent of anthrax (Au et al., 2006 ▶). The genome of the Ames strain of B. anthracis, a large Gram-positive spore-bearing bacterium (Read et al., 2003 ▶), possesses 5.23 megabases of normal chromosomal DNA, which are predicted to contain 5311 open reading frames, along with two plasmids which encode the major virulence factors. Of the proteins encoded by the chromosomal DNA, a total of 359 were targeted for work within the SPINE project (Au et al., 2006 ▶) and these have to date resulted in more than 50 structure determinations.
One of the targets chosen for study at the OPPF was an alanine racemase (BA0252), which interconverts l- and d-alanine using the cofactor pyridoxal 5′-phosphate (PLP; Grishin et al., 1995 ▶). In the absence of alanine PLP is covalently linked to a lysine residue via a Schiff base, but during catalysis this link is broken and a new Schiff base is formed with the alanine substrate (Fig. 1 ▶). d-Alanine, which is important to both Gram-positive and Gram-negative bacteria since it is required for the synthesis of the peptidoglycan layer of their cell walls, is produced by the action of alanine racemase on l-alanine (Redmond et al., 2004 ▶). With few exceptions, e.g. the fungus Tolypocladium niveum, which requires alanine racemase for cyclosporin biosynthesis (Hoffmann et al., 1994 ▶), alanine racemase activity is unique to bacteria. Since the mammalian hosts of B. anthracis do not synthesize peptidoglycans, they possess no homologues of alanine racemase and specific inhibitors could therefore function as antibiotics (Huang, Elmets et al., 2004 ▶). Furthermore, in B. anthracis spore germination is linked to a reduction in the level of d-alanine (Huang, Foster et al., 2004 ▶; Redmond et al., 2004 ▶). Therefore, an inhibitor of alanine racemase activity could trigger premature germination leading to death in suboptimal environments (Huang, Elmets et al., 2004 ▶). Since alanine racemase is expressed in the easily accessible B. anthracis exosporium, it has attracted much attention as a potential drug target (Huang, Elmets et al., 2004 ▶).
Figure 1.

The classical two-base mechanism of the reaction catalyzed by alanine racemase using pyridoxal 5′-phosphate as a cofactor. The reaction is shown in the direction of conversion from l-alanine to d-alanine, in which Tyr270′ abstracts a proton from l-alanine to form a planar intermediate and Lys41 donates a proton to form d-alanine. A more detailed mechanism avoiding the creation of a quinonoid intermediate has been proposed by Watanabe et al. (2002 ▶).
The first structure of an alanine racemase, from B. stearothermophilus (now Geobacillus stearothermophilus; PDB code 1sft; Shaw et al., 1997 ▶), has been followed by several more from B. stearothermophilus as well as from Pseudomonas aeruginosa, Mycobacterium tuberculosis and Streptomyces lavendulae, including structures of inhibitor-bound enzymes. The most relevant of these structures to the current report are those of B. stearothermophilus alanine racemase with the same inhibitor discussed here (PDB code 1bd0; Stamper et al., 1998 ▶) and with two close analogues of the inhibitor (PDB codes 1l6f and 1l6g; Watanabe et al., 2002 ▶). In all cases the enzyme comprises two domains: the larger N-terminal domain forms a TIM fold and the smaller C-terminal domain is mainly composed of β-structure. The enzyme appears to be active as a homodimer and two catalytic bases, Lys39 and Tyr265 (B. stearothermophilus numbering), are essential for activity. The lysine is also responsible for the formation of a Schiff base with PLP.
There are several well known natural inhibitors of alanine racemase, such as d-cycloserine (Seromycin, a clinically approved antibiotic for use against tuberculosis, but also used for treating anxiety disorders and autism) and O-carbamoyl-d-serine (Neuhaus, 1967 ▶). Many alanine homologues have also been shown to exhibit inhibitory activity, including β-chloroalanine and β-fluoroalanine (Badet et al., 1984 ▶), 1-aminocyclopropane phosphonate (Erion & Walsh, 1987 ▶), alanine phosphonate (Copié et al., 1988 ▶) and β,β,β-trifluoroalanine (Faraci & Walsh, 1989 ▶). However, they often lack specificity and inhibit other PLP-containing enzymes (Shaw et al., 1997 ▶).
Here, we report the first crystal structures of the potential anti-anthrax drug target alanine racemase from B. anthracis, both with covalently bound PLP at 1.47 Å resolution and with PLP and (R)-1-aminoethylphosphonic acid (l-Ala-P) at 2.1 Å resolution. Together, these structures provide new insight into the mode of inhibition of alanine racemase by l-Ala-P. Moreover, the structure determination is methodologically interesting as an example of the successful use of quantitative reductive methylation to overcome difficulties in crystallizing B. anthracis alanine racemase (Walter et al., 2006 ▶), which had defeated previous attempts at structural analysis by us and others.
2. Materials and methods
Cloning, expression and protein purification employed standard OPPF pipeline protocols as described elsewhere (Ren et al., 2005 ▶; Alzari et al., 2006 ▶). Briefly, the alanine racemase gene (BA0252) was amplified from genomic B. anthracis Ames strain DNA by PCR with forward primer AAGTTCTGTTTCAGGGCCCGATGGAAGAAGCACCATTTTATCG and reverse primer ATGGTCTAGAAAGCTTTACTATATATCGTTCAAATAATTAATTAC. The PCR product was cloned into pOPINB, a modified pET28a vector (Berrow et al., 2007 ▶), using the In-Fusion method (Clontech Takara Bio Europe), which resulted in the overexpressed protein incorporating a 3C protease-cleavable N-terminal hexahistidine purification tag (the added protein sequence was MGSSHHHHHHSSGLEVLFQ↓GP, cleaved as indicated by the arrow to leave two residues preceding the natural N-terminus in the final protein, although these residues are not resolved in our structures and our numbering reflects the natural sequence). Following small-scale trials and sequencing, overexpression of the protein was autoinduced by Overnight Express Autoinduction System 1 (Novagen) using Escherichia coli Rosetta pLysS cells. The mixture of Overnight Express autoinduction solution (20 ml solution 1, 50 ml solution 2 and 1 ml solution 3) and 1 l Luria Broth medium with start-up culture was incubated for 4.5 h at 310 K followed by 18 h at 298 K. Cells were harvested by centrifugation at 6000g for 30 min, washed with PBS buffer and then re-centrifuged at 6000g for 30 min with the final pellet stored at 193 K. For purification, the pellet was thawed in an ice bath and resuspended in lysis buffer containing 50 mM Tris–HCl, 500 mM NaCl, 20 mM imidazole, 0.2%(v/v) Tween, DNase and protease inhibitor [one cocktail tablet (Complete; Roche) in 30 ml buffer] at pH 7.5. After 30 min rocking in the ice bath, the cell extract was lysed in a cell disruptor (Constant Systems Ltd) at 207 MPa. After centrifugation at 30 000g for 45 min, the supernatant was loaded onto a 1 ml His-trap nickel column (GE Healthcare), washed with 5 ml 50 mM Tris–HCl, 500 mM NaCl, 20 mM imidazole and eluted with 5 ml 50 mM Tris–HCl, 500 mM NaCl, 500 mM imidazole. The elutant was loaded onto a gel-filtration column (HiLoad 16/60 Superdex 200; GE Healthcare) with 20 mM Tris–HCl pH 7.5, 200 mM NaCl buffer. The yield of purified protein was of the order of 40 mg per litre of culture. To cleave the purification tag, 800 µl of 0.5 mg ml−1 3C protease was added and the mixture was incubated with stirring overnight at 277 K. The His6-tagged protease was then removed by passage over a nickel column.
Extensive crystallization trials with the cleaved but otherwise unmodified protein were conducted using the sitting-drop method at 294 K with the standard OPPF nanolitre-scale procedures and screens (Walter et al., 2003 ▶, 2005 ▶). However, no crystals were produced in 768 trials at each of two concentrations both with and without added PLP. In an attempt to obtain crystals, the enzyme was subjected to a reductive-methylation procedure. A full discussion of this technique, including its application to alanine racemase, can be found in Walter et al. (2006 ▶) and thus the procedure is only summarized here. The buffer was changed to 50 mM HEPES and 250 mM NaCl at pH 7.5 by centrifugation at 2800g using a 10 kDa molecular-weight cutoff (MWCO) concentrator (Vivascience). The protein was then diluted to a concentration of 1.0 mg ml−1. 20 µl 1 M dimethylamine–borane complex (ABC) solution and 40 µl 1 M formaldehyde solution (36.5%) were added to each 1 ml of protein solution. After incubation at 277 K for 2 h, 20 µl 1 M ABC solution and 40 µl 1 M formaldehyde solution were again added. After a further 2 h incubation at 277 K, the solution was supplemented with 10 µl 1 M ABC solution followed by 12 h incubation at 277 K. The buffer was changed back to 20 mM Tris–HCl and 200 mM NaCl pH 7.5 using a 10 kDa MWCO concentrator (by centrifugation at 2800g) and re-purified by gel filtration. Mass spectra were obtained following the standard OPPF protocols (Nettleship et al., 2008 ▶).
The crystallization trials were repeated with the methylated enzyme at different concentrations. Good-quality crystals were obtained with a protein concentration of 60 mg ml−1 using 0.2 M magnesium chloride, 25%(w/v) polyethylene glycol 3350 and 0.1 M bis-Tris pH 6.5 as the reservoir solution. Crystals formed quickly and continued to grow for about 5 d, reaching final dimensions of approximately 750 × 100 × 100 µm. In order to bind the inhibitor, some of the crystals were soaked for 45 min in a solution of mother liquor mixed with 10 mM l-Ala-P prior to data collection.
Crystals were mounted in a nylon-fibre loop, cryoprotected in a mixture containing 25% glycerol and 75% reservoir solution and flashed-cooled to 100 K in a nitrogen-gas stream. All diffraction images were collected on BM14, the UK beamline, at the European Synchrotron Radiation Facility, Grenoble on a MAR 225 CCD detector. For the data set without inhibitor the detector was placed 168 mm from the crystal and 936 images were collected using 0.3° oscillations and exposure times of 10 s. For the data set with the inhibitor the detector was placed 243 mm from the crystal and 180 images were collected using 1° oscillations and exposure times of 30 s (Table 1 ▶). Both sets of images were indexed, integrated and scaled using HKL-2000 (Otwinowski & Minor, 1997 ▶), resulting in the data sets described in Table 1 ▶. The structure without inhibitor was solved by molecular replacement from a B. stearothermophilus alanine racemase structure (PDB code 1sft; 57% identity; Shaw et al., 1997 ▶) using MOLREP (Vagin & Teplyakov, 1997 ▶) from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶). The sequence of the model was manually corrected using Coot (Emsley & Cowtan, 2004 ▶). The model was improved by rounds of manual rebuilding in Coot interleaved with refinement using REFMAC (Murshudov et al., 1999 ▶) using modified residue definitions for the 18 (out of 20) lysine residues on each chain assumed to be methylated, a modified residue definition for the Lys41–PLP adduct and including riding H-atom positions in the refinement. The final model, after checking with MolProbity (Lovell et al., 2003 ▶) and refined to a resolution of 1.47 Å, has a crystallographic R factor of 0.168 (the free R factor was 0.199), stereochemistry characterized by root-mean-square (r.m.s.) deviations from target bond lengths and angles of 0.010 Å and 1.4°, respectively, and 97.3% of residues in favoured Ramachandran regions (Table 1 ▶). The inhibitor-bound structure was solved from this refined model by molecular replacement using MOLREP. The structure was refined to a resolution of 2.1 Å (Table 1 ▶) in a manner analogous to that outlined above except that NCS restraints were applied and Lys41 was no longer linked to the PLP. Instead, the Schiff base formed between PLP and the inhibitor was modelled by simultaneously refining with overlapping PLP–l-Ala-P and PLP–d-Ala-P moieties each having their occupancy set to 0.5. Structural conclusions were checked against three further models: one refined in the absence of any PLP moiety, one with only PLP–l-Ala-P at full occupancy and one with only PLP–d-Ala-P at full occupancy.
Table 1. Crystallographic data-collection, processing and refinement statistics.
Values in parentheses are for the outer resolution shell indicated in each section.
| Without inhibitor | With inhibitor | |
|---|---|---|
| Experiment type | Single wavelength | Single wavelength |
| Beamline | BM14 (ESRF) | BM14 (ESRF) |
| Wavelength (Å) | 0.979 | 0.979 |
| Data processing | ||
| Resolution limits (Å) | 50–1.47 (1.52–1.47) | 50–2.10 (2.18–2.10) |
| Unique reflections | 116056 (9318) | 48124 (4647) |
| Space group | P212121 | P212121 |
| Unit-cell parameters (Å) | a = 57.6, b = 88.4, c = 139.0 | a = 59.7, b = 96.5, c = 140.7 |
| Completeness (%) | 96.3 (78.3) | 99.9 (98.7) |
| Multiplicity | 9.9 (4.6) | 6.9 (4.1) |
| Mosaicity for image batches (°) | 0.5–0.7 | 0.3–0.6 |
| I/σ(I) | 18.6 (1.2) | 7.1 (1.2) |
| Rmerge† | 0.106 (0.954) | 0.230 (0.878) |
| Refinement statistics | ||
| Resolution limits (Å) | 50–1.47 (1.51–1.47) | 50–2.10 (2.16–2.10) |
| Rcryst‡ | 0.168 (0.292) | 0.188 (0.261) |
| Free Rcryst‡ (random 5%) | 0.199 (0.367) | 0.239 (0.324) |
| No. of non-H atoms§ | ||
| Protein | 6278 | 6196 |
| Cofactor and inhibitor | 37 | 94 |
| Water | 1020 | 871 |
| Stereochemical parameters | ||
| R.m.s. bond-length deviation (Å) | 0.010 | 0.008 |
| R.m.s. bond-angle deviation (°) | 1.4 | 1.2 |
| Mean B value (Å2) | 10.3 | 14.3 |
| Residues in favoured Ramachandran regions¶ (%) | 97.3 | 97.1 |
R
merge = 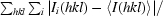
 .
.
R
cryst = 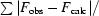
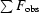 .
.
In the absence of inhibitor each PLP cofactor forms a Schiff base with Lys41, whereas in the presence of inhibitor each PLP cofactor is modelled forming a Schiff base with the racemized inhibitor (50% occupancy for each enantiomer). The cofactor atom count includes four (without inhibitor) or three (with inhibitor) magnesium ions and three chloride ions, which are visible in the structure but not considered to be functionally important. The atoms of PLP–L-Ala-P and PLP–D-Ala-P with occupancies of 0.5 are counted separately.
As defined by MolProbity (Lovell et al., 2003 ▶).
Structure superpositions were calculated using the program SHP (Stuart et al., 1979 ▶). Sequence alignments were prepared using ESPript (Gouet et al., 1999 ▶) and structure figures were prepared using BobScript (Esnouf, 1997 ▶) and RASTER3D (Merritt & Bacon, 1997 ▶).
3. Results and discussion
3.1. Reductive methylation
Of the 21 potential methylation sites in B. anthracis alanine racemase (20 lysine residues and the N-terminal nitrogen), mass-spectroscopic analysis of the methylated product suggests that only 19 are actually methylated (Fig. 2 ▶). Prior to methylation (Fig. 2 ▶ a) the main peak at 43 814 Da corresponds to unmodified alanine racemase (theoretical weight 43 816 Da) and a small peak at 44 044 Da (230 Da heavier) corresponds to the adduct between alanine racemase and PLP. After methylation (Fig. 2 ▶ b) the corresponding peaks have shifted to 44 354 and 44 584 Da, respectively, an increase of 540 Da (the theoretical increase for 19 dimethylation sites is 532 Da).
Figure 2.

Mass-spectrometric analysis of B. anthracis alanine racemase: (a) before and (b) after reductive methylation.
The structure revealed two clearly unmethylated lysine residues: Lys41 is involved in forming the Schiff base with PLP, while Lys260 is buried inside the protein and forms salt-bridge interactions. It is assumed that the other lysine residues and the N-terminus are all completely methylated, although several are disordered in both structures. It is noteworthy that the Lys41–PLP Schiff base is sufficiently stable that some survived the denaturing conditions employed for mass spectroscopy (Fig. 2 ▶).
3.2. Overall structure
In both crystal structures there are two protein molecules in the asymmetric unit with continuous chain traces for residues 3–389 (without inhibitor) or 4–389 (with inhibitor). The chains in each structure form a homodimer with a combined interface area of ∼3600 Å2 (calculated using PISA; Krissinel & Henrick, 2007 ▶) and the active sites are formed on this dimer interface. This suggests that, as with other alanine racemases, the dimer is the biologically relevant form (Fig. 3 ▶ a). In each structure, the dimer subunits have very similar structures with r.m.s. deviations between equivalent Cα positions of <0.30 Å. The change in the unit-cell b dimension on soaking with l-Ala-P (Table 1 ▶) appears to arise primarily from a rotation of the complete dimer by ∼10° about an axis roughly parallel to the c axis rather than from any significant internal rearrangement. Given this similarity, the discussion focuses on the A chain in each structure.
Figure 3.

The structure of B. anthracis alanine racemase. (a) Overall structure of the alanine racemase dimer showing the subunits in light and dark blue and indicating the positions of the active sites by red sticks for the Lys41–PLP adduct. (b) Secondary structure of the monomer (coloured from blue at the N-terminus to red at the C-terminus). For clarity, the C-terminal domain (top) is moved up and rotated 90° about a vertical axis with respect to the N-terminal domain (bottom). Secondary-structural elements are labelled. (c) A sequence alignment based on structure (calculated using SHP) between the alanine racemases of B. anthracis and B. stearothermophilus (PDB code 1sft; Shaw et al., 1997 ▶). The residue numbering and secondary-structural assignments refer to the B. anthracis sequence. Lys41, which forms aSchiff base with PLP, is indicated by a red asterisk. Residues in the B. anthracis structure that interact with PLP (any interatomic distance less than 4.0 Å) are indicated by green triangles, while the additional residues that interact with PLP–l-Ala-P/PLP–d-Ala-P are indicated by blue circles.
The alanine racemase monomer is composed of two domains: an N-terminal (α/β)8 TIM-barrel domain (residues 16–248; bottom of Fig. 3 ▶ b) and a C-terminal domain which includes the extreme N-terminus (residues 3–15 and 249–389; top of Fig. 3 ▶ b). The dimer is formed by head-to-tail interactions between the N- and C-terminal domains. The TIM barrel is somewhat distorted, with the seventh ‘strand’ not properly formed and the preceding helix being of minimal length (labelled α9 in Figs. 3 ▶ b and 3 ▶ c). Lys41, the active-site residue which forms the Schiff base with the PLP cofactor, is part of a short left-handed α-helix (α2) which immediately follows the first strand of the TIM barrel. The C-terminal domain mainly comprises a rather asymmetric β-sandwich (top of Fig. 3 ▶ b). Strands β1 and β10, which immediately flank the TIM barrel in the primary sequence, form the core of a mixed β-sheet (β14–β13–β16–β10–β1–β19–β20). Strand β10 leads straight into strand β11, part of an antiparallel β-sheet (β15–β12–β11) which forms the other side of the sandwich, stacking against strands β14–β13–β16–β10. A β-hairpin (β17–β18) completes the C-terminal domain.
3.3. The PLP cofactor-binding site
The active site is formed between the loops at one end of the TIM-barrel domain in one subunit of the dimer and residues in the C-terminal domain of the other, mainly residues Lys41, Tyr45, Arg138, His168, Arg224, Tyr270′ and Tyr359′ (where primes are used to distinguish between subunits; Fig. 4 ▶ a). These residues are conserved between B. anthracis and B. stearothermophilus (Fig. 3 ▶ c). In particular, the interaction of Lys41, Arg138 and Arg224 with PLP is precisely conserved in both alanine racemases. The arginine residues each donate a hydrogen bond to the cofactor: Arg138 to the phenolic O atom and Arg224 to the pyridinyl N atom. Lys41 forms an aldimine linkage with the PLP, eliminating water to form the Schiff base (Fig. 4 ▶ a). The main-chain N atoms of Ser209, Gly226 and Ile227 (the last two not labelled in Fig. 4 ▶) stabilize the PLP phosphate, with the help of Ser209 Oγ, Tyr45 Oη and Tyr359 Oη. A stacking interaction between the imidazole ring of His168 and the PLP pyridine may provide additional stabilization.
Figure 4.

The active site of B. anthracis alanine racemase. (a) The structure of the active site (atom-coloured ball-and-stick representation; carbon in grey) highlighting the PLP adduct (carbon in lime green) and showing 2F obs − F calc electron density contoured at 1.0σ (grey surface with black lines). Residues surrounding the active site are labelled, with the exceptions of Arg224–Ile227 and Tyr359, which form the back and front faces of the site as shown, respectively. The electron density shows the formation of the Schiff base between Lys41 and the PLP cofactor. (b) An equivalent view of the structure in the presence of inhibitor. The ball-and-stick models with lime green C atoms show the refined positions for a racemic mixture of PLP–l-Ala-P and PLP–d-Ala-P.
3.4. The substrate-binding site
The inhibitor-bound structure of B. anthracis alanine racemase was obtained by soaking apo crystals with the inhibitor l-Ala-P (see §2). There are no significant structural changes induced by inhibitor binding; the r.m.s. Cα position deviation between the apo and inhibitor-bound structures is 0.2 Å. The active sites of the apo and inhibitor-bound structures are also very similar: the largest conformational change occurs at Lys41, which no longer forms an aldimine linkage with PLP, and the PLP itself rotates by ∼15° in the plane of the pyridoxal ring to form a new aldimine link to the inhibitor (Fig. 4 ▶ b). The phosphate group of l-Ala-P is fixed by interactions with the amide N atom of Met317′, the released Nζ atom of Lys41, the guanidinium N atoms of Arg138 and the Oη atom of Tyr270′, which interact with the phosphate O atoms. Three water molecules also form bridging interactions between this phosphate group and the phenolic O atom of PLP. In common with all other bacterial alanine racemases, residues Tyr270′ and Lys41 are the best candidates to act as the catalytic bases positioned to abstract protons from l-alanine and d-alanine, respectively (Fig. 1 ▶).
The electron density for the inhibitor-bound structure, while generally good, is not continuous around the C1 atom of l-Ala-P (equivalent to the alanine Cα atom in the natural substrate; Fig. 4 ▶ b). Furthermore, the density appears to fit the remaining atoms of the PLP–l-Ala-P moiety quite ‘loosely’. In an attempt to understand this, the final round of refinement was repeated in parallel with (i) no PLP–l-Ala-P or PLP–d-Ala-P moiety, (ii) with only PLP–l-Ala-P, (iii) with only PLP–d-Ala-P and (iv) with overlapping PLP–l-Ala-P and PLP–d-Ala-P moieties each with 50% occupancy. For each refinement, the resulting 2F obs − F calc and F obs −F calc difference maps were examined. While all maps clearly show that soaking with l-Ala-P has broken the Lys41–PLP linkage, leading to a repositioning of Lys41, we cannot be certain of the exact chemical identity of the PLP-containing moiety in the crystal. The most likely interpretation (shown in Fig. 4 ▶ b) is that the PLP–l-Ala-P moiety is itself a tightly binding substrate of alanine racemase and has been partially converted to PLP–d-Ala-P, with the electron density representing this mixture. The two components of this mixture adopt slightly different orientations within the active site (explaining the ‘looseness’ of the electron density), while the C1 atom which is repositioned upon racemization is almost invisible in the electron density. The stable binding of the PLP–l-Ala-P and PLP–d-Ala-P moieties within the enzyme active site would then explain the inhibition by l-Ala-P.
3.5. Comparison with related structures
Our structural interpretation largely supports the work of Stamper et al. (1998 ▶) in their study of the complex between the B. stearothermophilus homologue (57% sequence identity) and l-Ala-P. Overall, the structures obtained for the B. stearothermophilus and B. anthracis proteins are very similar (r.m.s. deviations of between 0.92 and 1.01 Å for >90% of the Cα-atom positions in pairs of structures) and the relative dispositions of the N- and C-terminal domains are essentially identical. However, Stamper and coworkers observed no racemization of the l-Ala-P, a conclusion that is supported for this enzyme by both NMR and kinetic data. One possible explanation for this difference is that the l-Ala-P was racemized prior to or as part of our soaking experiment; another is that Stamper and coworkers obtained crystals by cocrystallization whereas our crystals were obtained by soaking. However, we consider it more likely that small but genuine differences around the active site, as discussed below, allow racemization with the B. anthracis enzyme. A second structural difference is that Stamper and coworkers reported a 20° twist of the plane of the PLP pyridine ring on formation of the l-Ala-P adduct, whereas our electron density indicates no significant twist; however, we note that the geometry of the pyridine ring in their original uninhibited structure (PDB code 1sft) is significantly distorted from its ideal planar conformation.
A subsequent study by Watanabe et al. (2002 ▶) elucidated the structures of complexes between B. stearothermophilus alanine racemase and two inactive reduced forms of the natural PLP–l-Ala and PLP–d-Ala intermediates (PDB codes 1l6f and 1l6g, respectively), which are closely related to the PLP–l-Ala-P and PLP–d-Ala-P adducts. This study proposed a revised mechanism for alanine racemase activity that avoids the formation of a quinonoid intermediate (as shown in Fig. 1 ▶), which they argued would be destabilized by Arg219 (residue 224 in B. anthracis numbering). Given the chemical difference, the refined conformations of PLP–l-Ala-P and PLP–d-Ala-P in our inhibited structure agree well with the structures of Watanabe and coworkers.
Overall, the active-site conformations of the structures discussed above are very similar [the r.m.s. deviations between Cα-atom positions of active-site residues, as defined in Fig. 3 ▶(c), are <0.3 Å], but there are localized perturbations that arise from sequence differences between the two species. Most significantly, residue Thr316 (cf. Cys311 in the B. stearothermophilus enzyme) makes direct contact with the alanine phosphate moiety (Fig. 4 ▶ b). However, the increased bulk of this side chain also displaces a key conserved water molecule, which may result in added flexibility in the binding mode of l-Ala-P and perhaps explains the observed racemization. Further differences arise from Ile225 (cf. Tyr220) and a compensatory change at Ile26 (cf. Leu24), from Pro89 (cf. Ala87), which causes a change in main-chain conformation between residues 86 and 90, and from Asn131 (cf. Lys129), which repositions Tyr166. Designing variants on the l-Ala-P inhibitor which target these differences may allow inhibitors to be developed with greater selectivity for B. anthracis.
Supplementary Material
PDB reference: alanine racemase, without l-Ala-P, 2vd8, r2vd8sf
PDB reference: with l-Ala-P, 2vd9, r2vd9sf
Acknowledgments
The OPPF is funded by the UK Medical Research Council (MRC) and Biotechnology and Biological Sciences Research Council (BBSRC) and this work was carried out as part of the Structural Proteomics in Europe (SPINE) consortium (European Commission Grant No. QLG2-CT-2002-00988). KA is supported by an MRC/Hutchison Whampoa Dorothy Hodgkin Postgraduate Award, while DIS and RME are also supported by the MRC. We thank the staff of the UK MAD beamline, BM14, at the ESRF for support.
References
- Alzari, P. M. et al. (2006). Acta Cryst. D62, 1103–1113.
- Au, K. et al. (2006). Acta Cryst. D62, 1267–1275. [DOI] [PubMed]
- Badet, B., Roise, D. & Walsh, C. T. (1984). Biochemistry, 23, 5188–5194. [DOI] [PubMed]
- Berrow, N. S., Alderton, D., Sainsbury, S., Nettleship, J., Assenberg, R., Rahman, N., Stuart, D. I. & Owens, R. J. (2007). Nucleic Acids Res.35, e45. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Copié, V., Faraci, W. S., Walsh, C. T. & Griffin, R. G. (1988). Biochemistry, 27, 4966–4970. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Erion, M. D. & Walsh, C. T. (1987). Biochemistry, 26, 3417–3425. [DOI] [PubMed]
- Esnouf, R. M. (1997). J. Mol. Graph. Model.15, 132–134. [DOI] [PubMed]
- Faraci, W. S. & Walsh, C. T. (1989). Biochemistry, 28, 431–437. [DOI] [PubMed]
- Gouet, P., Courcelle, E., Stuart, D. I. & Métoz, F. (1999). Bioinformatics, 15, 305–308. [DOI] [PubMed]
- Grishin, N. V., Phillips, M. A. & Goldsmith, E. J. (1995). Protein Sci.4, 1291–1304. [DOI] [PMC free article] [PubMed]
- Hoffmann, K., Schneider-Scherzer, E., Kleinkauf, H. & Zocher, R. (1994). J. Biol. Chem.269, 12710–127145. [PubMed]
- Huang, C. M., Elmets, C. A., Tang, D. C., Li, F. & Yusuf, N. (2004). Genomics Proteomics Bioinformatics, 2, 143–151. [DOI] [PMC free article] [PubMed]
- Huang, C. M., Foster, K. W., DeSilva, T. S. & Elmets, C. A. (2004). Proteomics, 4, 2653–2661. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol.372, 774–797. [DOI] [PubMed]
- Lovell, S. C., Davis, I. W., Arendall, W. B. III, de Bakker, P. I., Word, J. M., Prisant, M. G., Richardson, J. S. & Richardson, D. C. (2003). Proteins, 50, 437–450. [DOI] [PubMed]
- Merritt, E. A. & Bacon, D. J. (1997). Methods Enzymol.277, 505–524. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A., Lebedev, A., Wilson, K. S. & Dodson, E. J. (1999). Acta Cryst. D55, 247–255. [DOI] [PubMed]
- Nettleship, J. E., Brown, J., Groves, M. R. & Geerlof, A. (2008). Methods Mol. Biol.426, 299–318. [DOI] [PubMed]
- Neuhaus, F. C. (1967). Antimicrob. Agents Chemother.7, 304–313. [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Read, T. D. et al. (2003). Nature (London), 423, 81–86.
- Redmond, C., Baillie, L. W. J., Hibbs, S., Moir, A. J. G. & Moir, A. (2004). Microbiology, 150, 355–363. [DOI] [PubMed]
- Ren, J., Sainsbury, S., Berrow, N. S., Alderton, D., Nettleship, J. E., Stammers, D. K., Saunders, N. J. & Owens, R. J. (2005). BMC Struct. Biol.10, 13. [DOI] [PMC free article] [PubMed]
- Shaw, J. P., Petsko, G. A. & Ringe, D. (1997). Biochemistry, 36, 1329–1342. [DOI] [PubMed]
- Stamper, C. G. F., Morollo, A. A. & Ringe, D. (1998). Biochemistry, 37, 10438–10445. [DOI] [PubMed]
- Stuart, D. I., Levine, M., Muirhead, H. & Stammers, D. K. (1979). J. Mol. Biol.134, 109–142. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.
- Walter, T. S., Diprose, J., Brown, J., Pickford, M., Owens, R. J., Stuart, D. I. & Harlos, K. (2003). J. Appl. Cryst.36, 308–314.
- Walter, T. S. et al. (2005). Acta Cryst. D61, 651–657. [DOI] [PMC free article] [PubMed]
- Walter, T., Meier, C., Assenberg, R., Au, K., Ren, J., Verma, A., Nettleship, J., Owens, R., Stuart, D. & Grimes, J. (2006). Structure, 14, 1617–1622. [DOI] [PMC free article] [PubMed]
- Watanabe, A., Yoshimura, T., Mikami, B., Hayashi, H., Kagamiyama, H. & Esaki, N. (2002). J. Biol. Chem.277, 19166–19172. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: alanine racemase, without l-Ala-P, 2vd8, r2vd8sf
PDB reference: with l-Ala-P, 2vd9, r2vd9sf