

Abstract
Described are assemblies consisting of polymeric capsules, “polycaps,” formed from two calix[4]arene tetraureas covalently connected at their lower rims. In these structures self-assembly leads to reversibly formed capsule sites along a chain, reminiscent of beads on a string. Their dynamic behavior is characterized by 1H NMR spectroscopy through encapsulation of guest species, reversible polymerization, and the formation of sharply defined hybrid capsules.
Keywords: calixarene, self-assembly, molecular recognition, hydrogen bonding
Molecule-within-molecule complexes represent an unusual arrangement of matter (1) with promising applications in mechanistic and physical organic chemistry (2, 3). More complicated synthetic systems that begin to resemble complexes found in biology are molecules that assemble through weak intermolecular forces. Specifically, cup-shaped molecules with hydrogen bonding sites on their larger rims have interesting capacities for molecular assembly. When the hydrogen bonding sites are self-complementary, the molecules dimerize to form capsules. These assemblies reversibly encapsulate smaller guest species of suitable size and shape, but show dynamic qualities (4): the capsules form and dissipate at rates that vary from microseconds to hours—long enough for chemical reactions to occur within them (5). Several such systems derived from resorcinarenes and calixarenes have been extensively characterized (6–8).
In the calix[4]arenes (Fig. 1) the cup-like shape provides half of each capsule, while the ureas on its larger (upper) rim provide the hydrogen bonds that permit the molecules to assemble. The alkyl (R1) groups on the phenolic oxygens of the lower rim maintain (on average) the cup-like conformation (9), and the R2 groups on the distal urea nitrogen (Nβ) provide for reasonable solubility in organic media. Their ready synthetic accessibility has raised the possibility (8, 10) that covalent connection at their lower rims could lead to polymeric capsules, or polycaps, on assembly (Fig. 2).
Figure 1.

Two calixarenes assemble to provide a host capsule for small molecule guests (G). The cyclic array of ureas provide the hydrogen bonds that hold the capsule together and the peripheral R1 and R2 groups provide solubility in organic solvents.
Figure 2.

Schematic depiction of polymer capsule (polycap) formation. Two calixarenes are covalently attached at their lower rims. Encapsulation of guests (G) causes the formation of a linear polymer.
MATERIALS AND METHODS
General.
All chemicals were used without further purification unless otherwise specified. p-Heptylphenyl isocyanate was prepared from the corresponding acid based on a literature procedure (11). 5,11,17,23-Tetraoctylurea-25,26,27,28-tetrapropoxycalix[4]arene (1, R1 = n-propyl, R2 = n-octyl) was prepared from the known tetraamine (12) by treatment with octyl isocyanate in a manner similar to 6 and 7. The 5,11,17,23-tetra-tert-butyl-25,26,27-tripropoxy-28-hydroxycalix[4]arene 2 was prepared according to the literature (13). Proton (1H) NMR spectra were recorded on Bruker (Billerica, MA) DRX-600 (600 MHz) or AM-300 (300 MHz) spectrometers. Carbon (13C) spectra were recorded on a Bruker DRX-600 (151 MHz) spectrometer. IR spectra were recorded on a Perkin–Elmer Paragon 1000PC FT-IR spectrometer. The fast atom bombardment (FAB) positive ion mass spectra were obtained on a VG ZAB-VSE double-focusing high resolution mass spectrometer equipped with a cesium ion gun. Matrix-assisted laser desorption ionization mass spectrometry experiments were performed on a PerSeptive Biosystems (Cambridge, MA) Voyager-Elite mass spectrometer with delayed extraction. Melting points (mp) were obtained on a Thomas Hoover (Philadelphia) capillary melting point apparatus and are uncorrected.
5,11,17,23-Tetra-tert-butyl-25,26,27-tripropoxy-28-(ethoxycarbonyl)methoxycalix[4]arene (3).
The alcohol 2 was suspended in anhydrous N,N-dimethylformamide (DMF; 50 ml) under N2 prior to the addition of 0.23 g (5.8 mmol) of NaH (60% dispersion in mineral oil). The mixture was heated between 55 and 60°C for 1 h to effect complete deprotonation. To the pale yellow, cloudy mixture was added ethyl bromoacetate (480 μl, 4.32 mmol), and stirring was continued at the above temperature for 14 h. The cooled mixture was added to 300 ml of water and extracted with CHCl3 (2 × 150 ml). The organic layer was separated, washed with water (2 × 200 ml) and dried over MgSO4. Following filtration and concentration, the resulting oily solid was triturated with methanol affording a white powder. The solid (2.27 g, 2.64 mmol, 92%) was filtered and washed with methanol: mp 189–191°C. 1H NMR (CDCl3) δ 6.90 (s, 2H), 6.89 (s, 2H), 6.66 (s, 4H), 4.82 (s, 2H), 4.65 (d, 2H, J = 12.7 Hz), 4.40 (d, 2H, J = 12.4 Hz), 4.21 (q, 2H, J = 7.1 Hz), 3.85–3.71 (m, 6H), 3.17 (d, 2H, J = 13.0 Hz), 3.12 (d, 2H, J = 12.6 Hz), 2.10 (m, 2H), 1.97 (m, 4H), 1.29 (t, 3H, J = 7.2 Hz), 1.17 (s, 9H), 1.16 (s, 9H), 1.08–0.96 (m, 9 H), 0.99 (s, 18H). 13C NMR (CDCl3) 170.87, 154.08, 153.53, 153.48, 144.89, 144.59, 144.25, 134.64, 134.12, 133.28, 133.09, 125.50, 125.14, 124.85, 124.79, 76.83, 70.91, 60.21, 33.79, 33.62, 31.46, 31.41, 31.25, 30.91, 23.25, 23.19, 14.10, 10.35, 10.00. IR (thin film) 2961, 2874, 1765, 1480, 1389, 1361, 1300, 1201, 1125, 1070, 910, 870, 734 cm−1. High-resolution mass spectrometry (HRMS) (FAB; M + Cs+) calculated for C57H80O6Cs 993.5009, found 993.5035.
5,11,17,23-Tetranitro-25,26,27-tripropoxy-28-(ethoxycarbonyl)methoxycalix[4]arene (4).
Calixarene 3 (2.26 g, 2.62 mmol) was dissolved in CH2Cl2 (55 ml) and glacial acetic acid (27 ml). To this colorless solution was added fuming HNO3 (16.5 ml), resulting in a color change to an opaque purple/black. After 1.5 h the clear yellow/orange solution was added to 300 ml of water and further diluted with CH2Cl2 (100 ml). The organic layer was separated and the aqueous layer was washed with CH2Cl2 (50 ml). The combined organic extracts were washed with water (300 ml), dried over MgSO4, and filtered. Evaporation of the filtrate to dryness gave a yellow oil which upon trituration with MeOH yielded 1.57 g (1.92 mmol, 73%) of the desired product as a faint yellow, sticky powder: mp 140°C (dec). 1H NMR (CDCl3) δ 7.84 (s, 4H), 7.34 (m, 4H), 4.76 (s, 2H), 4.76 (d, 2H, J = 14.2 Hz), 4.53 (d, 2H, J = 13.9 Hz), 4.24 (q, 2H, J = 7.1 Hz), 4.01 (t, 2H, J = 7.7 Hz), 3.90 (m, 4H), 3.46 (d, 2H, J = 14.9 Hz), 3.41 (d, 2H, J = 14.6 Hz), 1.95–1.87 (m, 6H), 1.31 (t, 3H, J = 7.1 Hz), 1.05 (t, 6H, J = 7.4 Hz), 0.98 (t, 3H, J = 7.5 Hz). 13C NMR (CDCl3) 168.76, 162.40, 161.33, 161.10, 143.28, 142.94, 136.10, 135.94, 134.91, 134.82, 124.55, 124.41, 123.67, 123.63, 77.90, 70.59, 61.20, 31.25, 30.88, 23.09, 23.06, 13.99, 10.09, 9.68. IR (thin film) 2968, 2937, 2881, 1755, 1586, 1523, 1452, 1347, 1213, 1193, 1097, 1060, 988, 909, 746 cm−1. HRMS (FAB; M + Cs+) calculated for C41H44N4O14Cs 949.1908, found 949.1934.
5,11,17,23-Tetraamino-25,26,27-tripropoxy-28-(ethoxycarbonyl)methoxycalix[4]arene (5).
Tetranitro compound 4 (1.5 g, 1.8 mmol) and Raney nickel (cat) were suspended in EtOH (100 ml) under H2 (atm). The mixture was heated to 55°C for 4 h prior to filtration through a Celite pad. Concentration of the filtrate to dryness yielded the reduction product as a brown solid which was used without further purification (1.17 g, 1.68 mmol, 91%): mp 160°C (dec). 1H NMR (CDCl3) δ 6.24 (s, 2H), 6.22 (s, 2H), 5.90 (s, 4H), 4.62 (s, 2H), 4.51 (d, 2H, J = 13.5 Hz), 4.31 (d, 2H, J = 13.1 Hz), 4.17 (q, 2H, J = 7.2 Hz), 3.78–3.61 (m, 6H), 2.97 (d, 4H, J = 14.4 Hz), 2.92 (d, 4H, J = 14.4 Hz), 2.69 (s, 8H), 191–1.78 (m, 6H), 1.27 (t, 3H, J = 7.2 Hz), 0.98 (t, 6H, J = 7.4 Hz), 0.92 (t, 3H, J = 7.5 Hz). 13C NMR (CDCl3) 170.71, 150.52, 149.70, 149.45, 140.80, 140.30, 140.24, 136.46, 136.16, 134.88, 134.78, 115.95, 115.85, 115.74, 76.73, 70.38, 60.03, 31.26, 30.88, 22.98, 22.92, 14.01, 10.34, 9.93. IR (thin film) 3345, 3207, 2961, 2874, 1759, 1607, 1468, 1384, 1279, 1216, 1187, 1069, 1007, 853, 732 cm−1. HRMS (FAB; M + Cs+) calculated for C41H52N4O6Cs 829.2941, found 829.2917.
5,11,17,23-Tetra-p-tolylurea-25,26,27-tripropoxy-28-(ethoxycarbonyl)methoxycalix[4]arene (6).
Tetraamino compound 5 (0.929 g, 1.33 mmol) was dissolved in dry CH2Cl2 (75 ml) under N2. To the homogeneous solution was added p-tolyl isocyanate (840 μl, 6.66 mmol) and the reaction was stirred at room temperature for 4 h. The solvent was then removed in vacuo giving a tan solid. Trituration with MeOH furnished a tan powder that was subsequently filtered and washed well with MeOH. The urea (1.49 g, 1.21 mmol, 91%) was used without further purification: mp >300°C (dec). 1H NMR [dimethyl sulfoxide (DMSO)-d6] δ 8.31 (s, 1H), 8.30 (s, 2H), 8.29 (s, 1H), 8.12 (s, 2H), 8.04 (s, 2H), 7.27 (d, 4H, J = 8.1 Hz), 7.17 (d, 4H, J = 8.3 Hz), 7.04 (d, 4H, J = 8.2 Hz), 6.99 (d, 4 H, J = 8.4 Hz), 6.98 (s, 2H), 6.97 (s, 2H), 6.64 (s, 2H), 6.63 (s, 2H), 4.74 (s, 2H), 4.55 (d, 2H, J = 13.0 Hz), 4.33 (d, 2H, J = 12.7 Hz), 4.15 (q, 2H, J = 7.1 Hz), 3.81–3.66 (m, 6H), 3.12 (d, 2H, J = 13.2 Hz), 3.10 (d, 2H, J = 12.8 Hz), 2.22 (s, 6H), 2.20 (s, 6H), 1.96 (m, 2H), 1.87 (m, 4H), 1.24 (t, 3H, J = 7.1 Hz), 1.00 (t, 6H, J = 7.4 Hz), 0.95 (t, 3H, J = 7.4 Hz). 13C NMR (DMSO-d6) 170.05, 152.71, 152.60, 151.50, 150.90, 150.84, 137.41, 135.18, 134.97, 134.15, 133.91, 133.84, 133.76, 133.54, 130.47, 130.46, 130.32, 129.22, 129.15, 118.48, 118.38, 118.21, 118.08, 76.73, 76.48, 70.63, 59.96, 40.58, 40.05, 31.03, 30.53, 22.63, 20.23, 13.97, 10.31, 9.91. IR (thin film) 3341, 2962, 2916, 2872, 1758, 1664, 1601, 1550, 1511, 1474, 1416, 1314, 1213, 1069, 1001, 816, 733 cm−1. HRMS (FAB; M + Cs+) calculated for C73H80N8O10Cs 1361.5052, found 1361.5075.
5,11,17,23-Tetra-p-tolylurea-25,26,27-tripropoxy-28-carboxymethoxycalix[4]arene (8).
The ester 6 (1.48 g, 1.21 mmol) was dissolved in a mixture of tetrahydrofuran (THF; 60 ml) and water (12 ml). To this suspension was added LiOH⋅H2O (1.0 g, 24 mmol) and the mixture was stirred at room temperature for 14 h. After this period, the resulting clear, copper-colored solution was poured into 300 ml of water and treated with 1 M HCl until strongly acidic. The tan precipitate was filtered and washed with water yielding 1.42 g (1.18 mmol, 98%) of the crude acid (further purified by trituration with hot MeOH): mp >300°C (dec). 1H NMR (DMF-d7) δ 12.39 (s, 1H), 8.53 (s, 3H), 8.50 (s, 1H), 8.33 (s, 2H), 8.26 (s, 2H), 7.41 (d, 4H, J = 8.4), 7.30 (d, 4H, J = 8.4), 7.17 (s, 4H), 7.10 (d, 4H, J = 8.3), 7.06 (d, 4H, J = 8.4), 6.79 (s, 2H), 6.78 (s, 2H), 4.76 (s, 2H), 4.63 (d, 2H, J = 13.2 Hz), 4.48 (d, 2H, J = 12.9 Hz), 3.95–3.81 (m, 6H), 3.22 (d, 2H, J = 13.1 Hz), 3.19 (d, 2H, J = 12.8 Hz), 2.26 (s, 6H), 2.24 (s, 6H), 2.01–1.91 (m, 6H), 1.04 (t, 6H, J = 7.5 Hz), 0.98 (t, 3H, J = 7.5 Hz). 13C NMR (DMSO-d6) 171.52, 152.76, 152.69, 152.56, 151.29, 150.79, 150.59, 137.49, 135.03, 134.83, 134.35, 134.22, 134.08, 134.01, 133.75, 130.46, 130.44, 130.36, 129.25, 129.20, 118.43, 118.23, 118.22, 118.14, 118.04, 76.92, 76.70, 70.92, 31.03, 30.57, 22.58, 20.27, 10.27, 10.03. IR (thin film) 3351, 2960, 2925, 2872, 1759, 1665, 1603, 1550, 1514, 1477, 1414, 1315, 1213, 998, 816 cm−1. HRMS (FAB; M + Cs+) calculated for C71H76N8O10Cs 1333.4739, found 1333.4790.
1,4-Bis{5,11,17,23-tetra-p-tolylurea-25,26,27-tripropoxy-28-[(aminocarbonyl)methoxy]calix[4]arene}xylene (11).
The acid 8 (0.195 g, 0.163 mmol) was dissolved in anhydrous DMF (3 ml) under N2. To this solution was added 1-yl-oxy-tris-pyrrolidino-phosphonium hexafluorophosphate [(Novabiochem, San Diego); 0.104 g, 0.200 mmol], triethylamine (27.8 μl, 0.200 mmol), and xylylenediamine (0.0104 g, 0.0766 mmol). The reaction solution was stirred at room temperature for 1 h, heated at 55°C for 7 h, and subsequently stirred overnight at room temperature. Most of the DMF was then removed in vacuo, and the resulting yellow oil was diluted to 20 ml with CHCl3. This solution was washed with 1 M HCl (2 × 30 ml), 1 M NaOH (2 × 30 ml), and brine (30 ml). The combined organic extracts were dried over MgSO4, filtered, and concentrated to dryness. The residue was redissolved in a minimum amount of THF and purified in two parts by preparative TLC (15:1 CH2Cl2/MeOH, Rf = 0.4). Trituration with MeOH gave the chromatographed product as an off-white powder (0.142 g, 0.0567 mmol, 74%): mp >300°C (dec). 1H NMR (DMSO-d6) δ 8.70 (t, 2H, J = 5.9 Hz), 8.30 (s, 4H), 8.29 (s, 2H), 8.25 (s, 2H), 8.16 (s, 4H), 8.05 (s, 4H), 7.28 (s, 4H), 7.25 (d, 4H, J = 8.4 Hz), 7.24 (d, 4H, J = 8.5 Hz), 7.16 (d, 8H, J = 8.4 Hz), 7.02 (d, 8H, J ≈ 8 Hz), 6.98 (d, 8H, J = 8.5 Hz), 6.93 (s, 4H), 6.90 (s, 4H), 6.67 (s, 8H), 4.57 (s, 4H), 4.51 (m, 4H), 4.41 (d, 4H, J = 12.7 Hz), 4.29 (d, 4H, J = 12.5 Hz), 3.76 (t, 4H, J = 7.3 Hz), 3.71 (t, 8H, J = 7.4 Hz), 3.13 (d, 4H, J = 13.6 Hz), 3.10 (d, 4H, J = 13.4 Hz), 2.21 (s, 6H), 2.20 (s, 6H), 2.19 (s, 12H), 1.82 (m, 4H), 1.72 (m, 8H), 0.89 (t, 6H, J = 7.4 Hz), 0.82 (t, 12H, J = 7.4 Hz). IR (thin film) 3346, 2960, 2923, 2872, 1665, 1603, 1549, 1514, 1476, 1415, 1314, 1210, 1000, 815 cm−1. Low-resolution mass spectrometry (FAB; M + Cs+) calculated for C150H160N18O18Cs 2634, found 2634.
5,11,17,23-Tetra-p-heptylphenylurea-25,26,27-tripropoxy-28-(ethoxycarbonyl)methoxycalix[4]arene (7).
Tetraamino compound 5 (0.161 g, 0.230 mmol) was dissolved in dry CH2Cl2 (5 ml) and added to a solution of p-heptylphenylisocyanate (0.250 g, 1.15 mmol) in dry CH2Cl2. After stirring at room temperature for 4.5 h the solvent was removed in vacuo and the resulting oily solid was triturated with MeOH. The resulting off-white powder was filtered, washed well with MeOH, and used without further purification (0.297 g, 0.190 mmol, 83%): mp 200°C (dec). 1H NMR (DMSO-d6) δ 8.31 (s, 1H), 8.30 (s, 1H), 8.28 (s, 1H), 8.27 (s, 1H), 8.11 (s, 2H), 8.02 (s, 2H), 7.27 (d, 4H, J ≈ 8 Hz), 7.16 (d, 4H, J = 8.4 Hz), 7.03 (d, 4H, J = 8.2 Hz), 6.98 (d, 4 H, J = 8.3 Hz), 6.98 (s, 2H), 6.97 (s, 2H), 6.64 (s, 4H), 4.74 (s, 2H), 4.55 (d, 2H, J = 12.9 Hz), 4.33 (d, 2H, J = 13.1 Hz), 4.15 (q, 2H, J = 7.1 Hz), 3.81–3.67 (m, 6H), 3.12 (d, 2H, J = 11.6 Hz), 3.10 (d, 2H, J = 11.7 Hz), 2.50–2.44 (m, 8H), 1.96 (m, 2H), 1.87 (m, 4H), 1.51 (m, 8H), 1.26–1.22 (m, 35H), 1.00 (t, 6H, J = 7.4 Hz), 0.95 (t, 3H, J = 7.4 Hz), 0.86–0.82 (m, 12H). 13C NMR (THF-d8) 170.82, 153.85, 153.69, 153.65, 153.31, 152.75, 152.66, 139.24, 139.14, 136.90, 136.83, 136.64, 135.84, 135.69, 135.63, 135.49, 135.21, 129.41, 129.37, 119.60–119.57 (m), 119.30, 119.21, 77.89, 77.86, 71.87, 60.82, 36.20, 36.17, 32.96, 32.83, 32.46, 32.13, 30.29, 30.27, 24.23, 24.18, 23.64, 14.74, 14.52, 11.08, 10.73. IR (thin film) 3337, 2956, 3189, 3119, 2925, 2854, 1764, 1666, 1604, 1552, 1514, 1470, 1418, 1316, 1215, 1070, 1043, 1008, 848 cm−1. HRMS (FAB; M + Cs+) calculated for C97H128N8O10Cs 1697.8808, found 1697.8826.
5,11,17,23-Tetra-p-heptylphenylurea-25,26,27-tripropoxy-28-carboxymethoxycalix[4]arene (9).
The ester 7 (0.243 g, 0.155 mmol) was dissolved in a mixture of THF (10 ml) and water (2.5 ml). To this suspension was added LiOH⋅H2O (0.130 g, 3.10 mmol). After stirring at room temperature for 12 h the solution was treated with 1 M HCl until strongly acidic and subsequently extracted with CHCl3 (20 ml). The organic layer was separated, evaporated to dryness, and triturated with MeOH to give the desired acid as a white powder (0.214 g, 0.139 mmol, 90%): mp 200°C (dec). 1H NMR (DMSO-d6) δ 12.23 (s, 1H), 8.32 (s, 1H), 8.31 (s, 1H), 8.29 (s, 1H), 8.26 (s, 1H), 8.16 (s, 2H), 8.06 (s, 2H), 7.27 (d, 2H, J = 8.1 Hz), 7.26 (d, 2H, J = 8.3 Hz), 7.18 (d, 4H, J = 8.3 Hz), 7.04 (d, 2H, J = 8.5 Hz), 7.03 (d, 4 H, J = 8.5 Hz), 6.99 (d, 4H, J = 8.3 Hz), 6.98 (s, 4H), 6.73 (s, 2H), 6.72 (s, 2H), 4.61 (s, 2H), 4.51 (d, 2H, J = 12.8 Hz), 4.35 (d, 2H, J = 12.3 Hz), 3.83–3.69 (m, 6H), 3.13 (d, 2H, J = 12.4 Hz), 3.12 (d, 2H, J = 11.8 Hz), 2.50–2.45 (m, 6H), 1.93 (m, 2H), 1.89 (m, 4H), 1.51 (m, 8H), 1.26–1.23 (m, 32H), 0.98 (t, 6H, J = 7.4 Hz), 0.94 (t, 3H, J = 7.4 Hz), 0.85–0.82 (m, 12H). 13C NMR (DMSO-d6) 171.22, 152.72, 152.62, 151.30, 150.66, 150.48, 137.63, 135.60, 135.56, 135.46, 135.14, 134.91, 134.43, 134.11, 134.04, 133.93, 133.74, 128.52, 128.46, 118.46, 118.31, 118.26, 118.23, 118.14, 76.95, 76.67, 70.68, 34.46, 34.44, 31.27, 31.12, 30.99, 30.97, 30.55, 28.54, 28.50, 22.58, 22.54, 22.04, 13.84, 10.26, 9.97. IR (thin film) 3368, 3194, 3128, 2926, 2854, 1762, 1667, 1605, 1554, 1514, 1478, 1418, 1316, 1216, 1000, 849, 761 cm−1. HRMS (FAB; M + Cs+) calculated for C95H124N8O10Cs 1669.8495, found 1669.8470.
1,4-Bis{5,11,17,23-tetra-p-heptylphenylurea-25,26,27tripropoxy-28-[(aminocarbonyl)methoxy]calix[4]arene}xylene (12).
The acid 9 (0.174 g, 0.113 mmol) was dissolved in anhydrous DMF (3 ml) under N2. To this solution was added PyBOP (0.0706 g, 0.136 mmol), triethylamine (18.9 μl, 0.136 mmol), and xylylenediamine (0.0071 g, 0.052 mmol). The reaction solution was stirred at room temperature for 1 h and then heated at 40–45°C for 12 h. Most of the DMF was removed in vacuo, and the resulting yellow oil was diluted to 20 ml with CHCl3. This solution was washed with 1 M HCl (2 × 30 ml), 1 M NaOH (2 × 30 ml), and brine (30 ml). The combined organic extracts were dried over MgSO4, filtered, and concentrated to dryness. The residue was redissolved in THF and purified by preparative TLC (30:1 CH2Cl2/MeOH, Rf = 0.3). Trituration with MeOH gave the chromatographed product as an off-white powder (0.145 g, 0.0457 mmol, 88%): mp 220°C (dec). 1H NMR (DMSO-d6) δ 8.68 (t, 2H, J = 5.9 Hz), 8.29 (s, 6H), 8.24 (s, 2H), 8.17 (s, 4H), 8.04 (s, 4H), 7.27 (s, 4H), 7.25 (d, 4H, J = 8.3 Hz), 7.24 (d, 4H, J = 8.5 Hz), 7.17 (d, 8H, J = 8.4 Hz), 7.02 (d, 4H, J = 8.4 Hz), 7.01 (d, 4H, J = 8.5 Hz), 6.97 (d, 8H, J = 8.4 Hz), 6.95 (s, 4H), 6.93 (s, 4H), 6.67 (s, 8H), 4.58 (s, 4H), 4.51 (m, 4H), 4.41 (d, 4H, J = 13.2 Hz), 4.30 (d, 4H, J = 12.8 Hz), 3.76 (t, 4H, J = 7.3 Hz), 3.72 (t, 8H, J = 7.4 Hz), 3.13 (d, 4H, J ≈ 15.0 Hz), 3.10 (d, 4H, J = 14.0 Hz), 2.50–2.44 (m, 16H), 1.82 (m, 4H), 1.72 (m, 8H), 1.49 (m, 16H), 1.26–1.22 (m, 64H), 0.89 (t, 6H, J = 7.3 Hz), 0.85–0.82 (m, 36H). IR (thin film) 3359, 3189, 3119, 2957, 2926, 2854, 1668, 1606, 1555, 1514, 1476, 1418, 1317, 1214, 1151, 1002, 848, 762, 695 cm−1. Low-resolution mass spectrometry (matrix-assisted laser desorption ionization; M + Na+) calculated for C198H256N18O18Na 3198, found 3198.
RESULTS
The synthesis of new calixarenes suitable for polymerization is shown in Fig. 3, wherein the functional group manipulations follow well-trodden paths. The known tripropoxy calixarene (13) shown as 2, was alkylated with ethyl bromoacetate to give the ester 3. Ipso-nitration of 3 with nitric acid followed by reduction with Raney nickel in EtOH yielded the tetraamine 5. The ureas were prepared in high yield from the aniline with p-tolyl isocyanate for 6 and p-heptylphenyl isocyanate for 7.
Figure 3.

The synthesis of polycap subunits involves the conversion of a previously described calixarene 2 to the monofunctionalized ureas 6–9. Derivatives 6-9 dimerize into capsules in organic solvents and give both proximal and distal regioisomers.
These monofunctionalized systems do indeed form capsules, and show all of the expected spectroscopic characteristics (6, 7, 14, 15) but lack the high symmetry of their unfunctionalized counterparts. There are two distinct regioisomers, in which the odd ester substituents are either proximal or distal (as shown schematically in Fig. 3). The number of different Nβ-H sites in each of the dimeric forms is 8. These sites give rise to the furthest downfield signals in the 1H NMR spectrum and appear between 9 and 10 ppm. Fig. 4 shows this region of the 600 MHz 1H NMR spectrum of the dimeric form of 6 in CDCl3, in which some 10 of these signals can be discerned; for dimeric 7 (in deuterated p-xylene) 8 signals were resolved in this region. The complexity of these spectra did not bode well for the use of NMR for studying the polymeric form of the capsules; nonetheless 1H NMR was quite effective for this purpose. For example, addition of benzene (a good guest for the capsule) to the solution of dimeric 7 leads to a new set of signals in the 1H NMR spectrum that represent capsules containing benzene. The encapsulated benzene gives rise to the sharp singlet at 4.2 ppm, the integration of which indicates that only one benzene molecule is accommodated by the interior of the capsule (spectrum not shown). This behavior is as expected from earlier studies in solution, but encapsulation of benzene has even been observed in the solid state (16).
Figure 4.
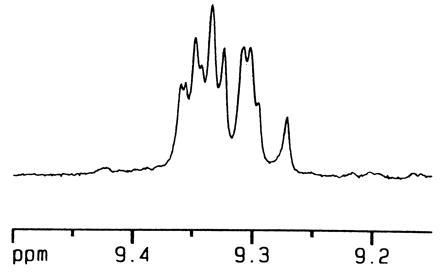
The downfield region of the 1H NMR spectrum of dimeric 6 in CDCl3, representing proton Nβ-H (see Fig. 3). A complex set of peaks arises from the two regioisomers of the assembly.
For the polycaps, two calixarenes units were covalently bound at the carboxyl functions on their lower rims. The connection was made in such a way that the hydrogen bonding surfaces of the upper rims are held apart—i.e., are directed in a divergent manner. This was accomplished by using a para-disubstituted benzene 10 (Fig. 3) as a spacer, a relatively rigid structure that orients the urea-bearing rims, on average, in opposite directions. Saponification of the single ester of 6 or 7 to the corresponding acids 8 and 9, followed by coupling to diamine 10 using PyBOP (17) gave the fundamental units for the polycap, the compounds 11 and 12 (Fig. 5).
Figure 5.

The basic unit of a polycap involves covalent connection of two subunits through a spacer unit. The para-disubstituted benzene orients the urea functions of the two ends in opposite directions.
Neither of these materials was soluble in p-xylene (a favorable solvent for encapsulation studies) or other common aromatics, but dissolved at reasonable concentrations in CDCl3. Fig. 6a shows a partial 1H NMR spectrum of 11 that results. It is abundantly clear that assembly has taken place. The signals for the hydrogen bonded Nβ-H of ureas appear in the expected downfield region, and while the signals are broadened throughout the spectrum, the assignments can easily be made. This is fortunate because the number of different capsules is enormous: each capsule can be formed as a proximal or distal regioisomer, the circular array of ureas can be arranged in a clockwise or counterclockwise sense, and every site is in a slightly different magnetic environment along the polymer chain. For encapsulation, p-difluorobenzene was used to compete with CDCl3 solvent for the interior of the polycaps of 11. The series of spectra in Fig. 6 b–d show the effects of added increments of p-difluorobenzene; it is clear that a new type of capsule is being generated at the expense of the original (solvent-filled) capsules. The overall polymeric nature of the assembly stays intact. When 12 was suspended in deuterated p-xylene, and benzene as a guest was added, the dissolution behavior was curious: the solid suspension gave way over the course of a few hours to a viscous, denser phase that eventually became homogeneous with the solution. A relevant section of the 1H NMR spectrum is shown in Fig. 7; the intense peak at 4.3 ppm is the resonance of the benzene inside the polycaps of 11.
Figure 6.

Titration of 11 at 7 mM in CDCl3 with added increments of p-difluorobenzene (vol/vol % in solution) gives rise to a new species of polycaps. The peak at δ 8.67 represents the amide-NH of the spacer for the original species, whereas the one at δ 8.74 arises from the polycaps with encapsulated p-difluorobenzene.
Figure 7.

The 1H NMR spectrum of 12 in a mixture of p-xylene-d10 and benzene (13:1, vol/vol) shows a sharp peak near δ 4.3 representing encapsulated benzene (the smaller sharp peak is due to a solvent impurity).
In addition to guest encapsulation by the polycaps, polymerization and depolymerization experiments were used to characterize the system: polycap formation induced by guest species, and polycap destruction through the formation of dumbbell-shaped heterodimers. In the first set of experiments a technique described by Whitesides and coworkers (18) was adapted for the polycaps. In this method a solvent such as DMSO, which competes for the hydrogen bond donors of the urea, is added to the CDCl3 solvent until the “melting point” of the assembly is reached. At this solvent composition equal amounts of monomer and assembly are present in solution: at higher DMSO amounts the monomeric form dominates, and at lower amounts the assembly dominates. The system is most vulnerable to the effects of added guests at the melting point, as shifts in equilibrium concentrations can be induced by guest encapsulation. Fig. 8 shows the partial 1H NMR spectrum using 2.7% deuterated DMSO which represents the approximate melting point of the polycaps from 11. Addition of p-difluorobenzene results in the changes shown in the series of spectra. It is clear that the guest nucleates the formation of more polycaps and does so at the expense of the monomeric species; polymerization of the assembly is driven by the encapsulation of the guest.
Figure 8.

Nucleation effects observed upon addition of p-difluorobenzene (vol/vol % in solution) to 11 in a 2.7% DMSO-d6 solution (in CDCl3). Spectrum a represents roughly equal amounts of monomeric 11 (δ 8.83) and polymeric 11 containing CDCl3 (δ 8.67). The new signal at δ 8.74 represents polycaps containing p-difluorobenzene.
Finally, depolymerization and capping experiments were performed using two different calixarene subunits. Earlier work by Böhmer and coworkers (7) had shown that two different calixarene capsules can form heterodimers—i.e., disproportionation occurs from the homodimeric states driven by mere entropy. Applied to the polycaps, an excess of a simple dimeric capsule could depolymerize the linear array as the formation of heterodimers takes place. The heterodimers are at ever-decreasing string sizes, leading ultimately to the dumbbell-like 2:1 complex shown in Fig. 9. This indeed proved to be the case. Fig. 10 shows the effects of titration of the polymeric form of 11 with a simple dimeric capsule (compound 1, R1 = n-propyl, R2 = n-octyl) on the downfield region of the 1H NMR spectrum. The spectrum begins to simplify and at 4.5 equivalents (Fig. 10d) of the capsule the result is what can be expected for the dumbbell 2:1 complex mixed with the symmetrical dimer. The final 1H NMR spectrum is quite sharp, although it represents the two diastereomers of the dumbbell which differ by the sense of clockwise vs. counterclockwise array of the ureas on either end. Apparently, this level of information is not communicated from one end of the dumbbell to the other.
Figure 9.

“Dumbbell-shaped” units are formed through disproportionation of polycaps with simple homodimeric calixarene dimers.
Figure 10.

Titration of 11 in CDCl3 with a symmetrical tetraurea calix[4]arene cap (equivs. cap: equivs. 11) gives rise to a discrete species, the “dumbbell.” Four sharp singlets are observed at low field for the Nβ-H protons of 11, and the amide proton of the spacer appears as a triplet at δ 8.83 (J = 6.0 Hz).
In summary, experiments have shown that hydrogen bonded polymeric systems can be well characterized in solution (19–21) by 1H NMR methods and some degree of control can be exercised on the assembly process. We are currently exploring conventional polymer characterization—polymer length, molecular weight distributions, viscosity, etc.—to determine its applicability to these dynamic and reversible systems. It is likely that rigid spacers that orient the cup-shaped subunits could lead to assemblies that include predictable arrays such as macrocycles. In addition, it is worth noting that the use of a triamine or tetraamine spacer can lead to a cross-linked polymer network. Additionally, the polycaps’ ability to function as receptors for small complementary molecules should permit new developments as sensors (22). We will report on these cases in due course.
Acknowledgments
We thank the National Institutes of Health and the Skaggs Research Foundation for support of this work.
ABBREVIATIONS
- DMSO
dimethyl sulfoxide
- DMF
N,N-dimethylformamide
- FAB
fast atom bombardment
- HRMS
high-resolution mass spectrometry
- PyBOP
1-yl-oxy-tris-pyrrolidino-phosphonium hexafluorophosphate
- THF
tetrahydrofuran
References
- 1.Cram D J, Cram J M. Container Molecules and Their Guests. Cambridge, MA: RSC; 1994. [Google Scholar]
- 2.Cram D J, Tanner M E, Thomas R. Angew Chem Int Ed Engl. 1991;30:1024–1027. [Google Scholar]
- 3.Timmerman P, Verboom W, van Veggel F C J M, van Duynhoven J P M, Reinhoudt D N. Angew Chem Int Ed Engl. 1994;33:2345–2348. [Google Scholar]
- 4.Rebek, J., Jr. (1996) Chem. Soc. Rev. 255–264.
- 5.Kang J, Rebek J., Jr Nature (London) 1997;385:50–52. doi: 10.1038/385050a0. [DOI] [PubMed] [Google Scholar]
- 6.Shimizu K D, Rebek J., Jr Proc Natl Acad Sci USA. 1995;92:12403–12407. doi: 10.1073/pnas.92.26.12403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mogck O, Böhmer V, Vogt W. Tetrahedron. 1996;52:8489–8496. [Google Scholar]
- 8.Chapman R G, Sherman J C. J Am Chem Soc. 1995;117:9081–9082. [Google Scholar]
- 9.Böhmer V. Angew Chem Int Ed Engl. 1995;34:713–745. [Google Scholar]
- 10.Rebek J., Jr Acta Chem Scand. 1996;50:707–716. doi: 10.3891/acta.chem.scand.50-0707. [DOI] [PubMed] [Google Scholar]
- 11.Capson T L, Poulter C D. Tetrahedron Lett. 1984;25:3515–3518. [Google Scholar]
- 12.van Wageningen, A. M. A. (1996) Thesis (Univ. of Twente, Enschede, The Netherlands).
- 13.Iwamoto K, Araki K, Shinkai S. J Org Chem. 1991;56:4955–4962. [Google Scholar]
- 14.Hamann B C, Shimizu K D, Rebek J., Jr Angew Chem Int Ed Engl. 1996;35:1326–1329. [Google Scholar]
- 15.Castellano R K, Rudkevich D M, Rebek J., Jr J Am Chem Soc. 1996;118:10002–10003. [Google Scholar]
- 16.Mogck, O., Paulus, E. F., Böhmer, V., Thondorf, I. & Vogt, W. (1996) J. Chem. Soc. Chem. Commun. 2533–2534.
- 17.Coste J, Le-Nguyen D, Castro B. Tetrahedron Lett. 1990;31:205–208. [Google Scholar]
- 18.Mammen M, Simanek E E, Whitesides G M. J Am Chem Soc. 1996;118:12614–12623. [Google Scholar]
- 19.Gallant M, Viet M T P, Wuest J D. J Org Chem. 1991;56:2284–2286. [Google Scholar]
- 20.Granja J R, Ghadiri M R. J Am Chem Soc. 1994;116:10785–10786. [Google Scholar]
- 21.Yang J, Fan E, Geib S J, Hamilton A D. J Am Chem Soc. 1993;115:5314–5315. [Google Scholar]
- 22.Schierbaum K-D, Weiss T, van Velzen E U T, Engbersen J F J, Reinhoudt D N, Göpel W. Science. 1994;265:1413–1415. doi: 10.1126/science.265.5177.1413. [DOI] [PubMed] [Google Scholar]