

Abstract
In the present study we tested the hypothesis that prenatal nicotine exposure increases heart susceptibility to ischemia/reperfusion (I/R) injury in adult offspring. Nicotine was administered to pregnant rats via subcutaneous osmotic minipumps throughout gestation. Nicotine treatment resulted in a rapid and transient decrease in food-intake and a moderate decrease in maternal body weight gain. Hearts were isolated from adult male and female offspring and subjected to I/R in a Langendorff preparation. Nicotine significantly attenuated left ventricle (LV) developed pressure, heart rate, and coronary flow rate in female but not male hearts at baseline. Additionally, nicotine significantly increased LV infarct size and attenuated postischemic recovery of LV function in both male and female offspring with more pronounced effects in females. In female but not male hearts, nicotine significantly decreased the postischemic coronary flow rate. However, coronary nitric oxide release was decreased in male but not female hearts. Caspase-3, -8, and -9 levels were not significantly changed in either female or male hearts. However, nicotine caused a significant decrease in protein levels of protein kinase (PK) Cε in both male and female hearts and a decrease in PKCδ levels in female hearts only. Control studies of maternal food restriction showed that a moderate decrease in maternal body weight gain had no effect on female hearts but significantly improved postischemic recovery of LV function in male hearts. The results suggest that prenatal nicotine exposure causes in utero programming of the PKC isozyme gene expression pattern in the developing heart and increases heart susceptibility to I/R injury in adult offspring.
Epidemiological studies have demonstrated that in utero exposure to maternal cigarette smoking is a significant risk factor for sudden infant death syndrome and is associated with elevated blood pressure and cardiovascular disease in offspring later in life (Beratis et al., 1996; Blake et al., 2000). As one of the major components in cigarette smoking, nicotine is likely to contribute to the development of cardiovascular disorders. Nicotine readily crosses the placenta and maternal cigarette smoking produces higher nicotine concentrations in fetal circulation than that experienced by the mother (Lambers and Clark, 1996). Because nicotine is an agonist of nicotinic acetylcholine receptors, exposure to nicotine early in life may cause permanent changes in nicotinic receptors and consequent cell function (Slotkin, 1998). We have recently demonstrated that fetal and neonatal nicotine exposure alters vascular function in adult offspring in a gender-specific manner, which may lead to an increased risk of cardiovascular dysfunction in adult life (Xiao et al., 2007).
The effects of prenatal nicotine exposure on fetal heart development and their long-term pathophysiological consequences in the adult heart have not been determined. Human epidemiological studies suggested a link between adverse intrauterine environments and an increased risk of ischemic heart disease in adulthood (Barker et al., 1993). Recent animal studies demonstrated that fetal exposure to hypoxia (Xiao et al., 2000; Bae et al., 2003; Li et al., 2003, 2004; Xu et al., 2006), glucocorticoids (Dodic et al., 2001), and cocaine (Bae et al., 2005; Bae and Zhang, 2005a) caused an epigenetic programming in the heart and resulted in increased heart susceptibility to ischemia and reperfusion (I/R) injury in adult offspring. It has been shown that maternal cigarette smoking acutely increases fetal heart rate, which is probably due to an increase in sympathetic activity (Mancia et al., 1997). The chronic effects of fetal nicotine exposure on the heart are somewhat different and to a large extent result from alterations in heart development. Prenatal nicotine exposure has been shown to alter the types of nicotinic receptors that facilitate excitatory inputs to cardiac vagal neurons, which may be responsible for the bradycardia observed in offspring (Huang et al., 2004). It has also been demonstrated that heart function does not change in rat pups exposed to nicotine prenatally in normal conditions, but fetal nicotine exposure produces intolerance to neonatal hypoxia, resulting in a rapid and profound fall in heart rate (Slotkin et al., 1997).
In the present study we tested in a rat model the hypothesis that prenatal nicotine exposure increases heart susceptibility to I/R injury in adult offspring. Postischemic recovery of left ventricle (LV) function and myocardial infarct size were determined in hearts isolated from adult offspring in a Langendorff preparation. To determine the potential gender effects of prenatal nicotine exposure on the heart, both male and female offspring were included in the studies. Given that apoptosis plays an important role in I/R injury (Kang and Izumo, 2000) and the finding that nicotine exposure leads to a gender-specific increase in apoptotic markers in the brain (Machaalani et al., 2005), we measured the levels of caspase-3, -8, and -9 in the hearts. In addition, we investigated possible mechanisms underlying nicotine-induced cardiac dysfunction and determined the protein levels of PKCε and PKCδ in the hearts, which have been shown to be important in the regulation of cardiac I/R injury (Murriel and Mochly-Rosen, 2003). Because maternal nicotine administration reduced food intake and caused a moderate decrease in maternal body weight gain during pregnancy, we also studied the effects of decreased maternal body weight gain with food restriction on heart vulnerability to I/R injury in adult offspring.
Materials and Methods
Experimental Animals
Time-dated pregnant Sprague-Dawley rats were purchased from Charles River Laboratories (Portage, MI). Nicotine was administrated through osmotic minipumps implanted s.c. as described previously (Xiao et al., 2007). In brief, on the 4th day of pregnancy, rats were anesthetized with ketamine and xylazine, and an incision was made on the back to insert osmotic minipumps (type 2ML4; Alza, Palo Alto, CA). The incision was closed with four sutures. Half of the pregnant rats were implanted with the minipumps containing nicotine at a concentration of 102 mg/ml, and the other half were implanted with the minipumps containing only saline, which served as the vehicle control. The flow rate of the minipumps was 60 μl/day, which delivered a dose of 2.1 mg of nicotine free-base per day. In rats of an average of 350 g b.wt., this corresponds to a dose rate of 6 mg/kg/day, which closely resembles doses occurring in moderate to heavy human smokers (Lichtensteiger et al., 1988; Slotkin, 1998). According to the manufacturer’s specifications, the delivery period for the pumps is 28 days, and thus delivery continued after birth until postnatal day 10. As previously reported (Xiao et al., 2007), nicotine treatment did not affect the litter size and the length of gestation, and all of the pregnancies reached full term. Maternal food intake and body weight gain were monitored daily. On day 21 of pregnancy, some rats were euthanized, and fetal hearts were isolated. Each litter was counted as an individual sample. Other rats were allowed to give birth naturally. Pups born to the dams were kept with their mothers until weaning. At weaning, male and female pups were separated and transferred to cages where they were housed in groups of two. Male and female offspring were sacrificed at 3 months of age, and hearts were isolated for Western blot, histological, and I/R functional studies. In separate studies, pregnant rats were divided into two groups and were fed with food equivalent to that determined in saline control and nicotine-treated animals, respectively. Hearts were isolated from adult male and female offspring and studied for I/R injury. Offspring in the present study were from four treatment groups of pregnant rats: saline control (four litters), nicotine treatment (four litters), control (six litters), and food restriction (five litters). All procedures and protocols used in the present study were approved by the Institutional Animal Care and Use Committee of Loma Linda University and followed the guidelines by the National Institutes of Health Guide for the Care and Use of Laboratory Animals (National Research Council, 1996).
Histological Analysis
Hearts were fixed in 4% formalin and embedded in paraffin. Both horizontal and vertical section cuts were conducted from the midline of hearts, and serial sections (5 μm) were stained with hematoxylin and eosin staining. The wall thicknesses of left and right ventricles and the ventricle septum were measured.
Western Blot Analysis
Protein levels of procaspase-3, -8, and -9, PKCε, and PKCδ in the left ventricle were determined with Western blot analysis. Briefly, tissues were homogenized in a lysis buffer containing 150 mM NaCl, 50 mM Tris·HCl, 10 mM EDTA, 0.1% Tween 20, 0.1% β-mercaptoethanol, 0.1 mM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, and 5 μg/ml aprotinin, pH 7.4. Homogenates were then centrifuged at 4°C for 10 min at 10,000g, and supernatants were collected. Proteins were measured in the supernatant using a protein assay kit from Bio-Rad (Hercules, CA). Samples with equal proteins were loaded onto 10% polyacrylamide gel with 0.1% sodium dodecyl sulfate and were separated by electrophoresis at 100 V for 2 h. Proteins were then transferred onto nitrocellulose membranes. Nonspecific binding sites were blocked with overnight incubation at 4°C in a Tris-buffered saline solution containing 5% dry milk. The membranes were incubated with primary antibodies against caspase-3, -8, and-9, PKCε, or PKCδ (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), respectively. After washing, membranes were incubated with secondary horseradish peroxidase-conjugated antibodies. Proteins were visualized with enhanced chemiluminescence reagents, and blots were exposed to Hyperfilm. Results were quantified with the Kodak electrophoresis documentation and analysis system and Kodak ID image analysis software.
Perfused Hearts Subjected to I/R
Hearts were isolated from adult male and female offspring and retrogradely perfused via the aorta in a Langendorff preparation under constant pressure (70 mm Hg) with gassed (95% O2 and 5% CO2) Krebs-Henseleit buffer at 37°C as described previously (Bae and Zhang, 2005b). A pressure transducer connected to a saline-filled balloon inserted into the LV was used to assess ventricular function by measuring LV pressure (millimeters of mercury) and its first derivative (dP/dt). LV end-diastolic pressure (LVEDP) was set at approximately 5 mm Hg. After baseline recording, hearts were subjected to 25 min of global ischemia by stopping the perfusion followed by 60 min of reperfusion. LV functional parameters, LV developed pressure (LVDP), heart rate (HR), dP/dtmax, dP/dtmin, and LVEDP were continuously recorded with an online computer. Pressure rate product (PRP) was calculated as LVDP × HR. Pulmonary artery effluent was collected as an index of coronary flow.
Measurement of Myocardial Infarct Size
Myocardial infarct size was measured as described previously (Bae and Zhang, 2005b). Briefly, at the end of reperfusion, left ventricles were collected, cut into four slices, incubated with 1% triphenyltetrazolium chloride solution for 15 min at 37°C, and immersed in formalin for 60 min. Each slice was then photographed separately, and areas of myocardial infarction in each slice were analyzed by computerized planimetry (Image-Pro plus; Media Cybernetics, Inc., Silver Spring, MD), corrected for the tissue weight, summed for each heart, and expressed as a percentage of total LV weight.
Measurement of Nitric Oxide
NO was measured by the chemiluminescence method as described previously (Bae and Zhang, 2005a). Samples of coronary effluent (100 μl) were injected into a gas purge vessel containing 5 ml of vanadium(III) HCl to react for 1 min and to reduce nitrite/nitrate in the sample back to NO. To achieve high reducing efficiency, the reduction was performed at 90°C. NO in the sample was then “stripped” into the head-space by helium bubbling (12 ml/min) for 1 min. NO in the head-space was drawn into the NO analyzer (model 280; Sievers Instruments; Boulder, CO) and mixed with O3 in the front of a cooled Hamamatsu red-sensitive photomultiplier tube. Signals from the detector were analyzed with the use of an on-line computer as area under the peak. The measurement reflected the combined amount of nitrite, nitrate, and NO and was normalized to coronary flow rate and tissue weight, expressed as nanomoles per minute per gram.
Statistical Analysis
Data were expressed as means ± S.E.M. Statistical significance (P < 0.05) was determined by two-way ANOVA or t test, where appropriate.
Results
Effects of Prenatal Nicotine Exposure and Maternal Food Restriction on Body and Heart Weight
As shown in Fig. 1, maternal nicotine administration starting at day 4 of pregnancy caused a rapid and transient reduction in food intake and a moderate decrease in maternal body weight gain during the period of treatment. Control studies of maternal food restriction mimicking that observed in nicotine-treated animals resulted in a similar decrease in maternal body weight gain (Fig. 1). Nicotine treatment caused a significant decrease in body weight of near-term (21 day) fetuses (3.79 ± 0.05 versus 4.12 ± 0.04 g, n = 4–5 litters, P < 0.05). A two-factor ANOVA (age and treatment) showed significance differences (P < 0.0001) in age, nicotine treatment, and the treatment × age interaction, respectively, in rats at post-natal ages up to 30 days (Fig. 2, top). In contrast, although there was a significant difference (P < 0.0001) in age in rats at postnatal ages up to 30 days, neither maternal food restriction nor the treatment × age interaction showed significant differences (P > 0.05) (Fig. 2, bottom). In adult offspring, there were no significant differences in body weight (male, 498.7 ± 12.9 versus 468.6 ± 30.0 g, P > 0.05; female, 304.0 ± 14.4 versus 298.7 ± 8.7 g, P > 0.05) or heart weight (male, 1.49 ± 0.06 versus 1.43 ± 0.02 g, P > 0.05; female, 0.99 ± 0.04 versus 0.91 ± 0.03 g, P > 0.05) between saline control and prenatal nicotine-treated males or females. In addition, prenatal nicotine exposure had no significant effects on the thickness of left and right ventricular walls and the ventricular septum (data not shown).
Fig. 1.

Effect of nicotine administration on maternal food intake and body weight gain. Nicotine was administrated to pregnant rats through osmotic pumps, as described under Materials and Methods. Control rats received saline. Maternal food intake and body weight gain were measured daily. In separate studies, pregnant rats were divided into two groups and were fed with food equivalent to that determined in saline control and nicotine-treated animals, respectively. Data are means ± S.E.M. n = 4 to 6.
Fig. 2.
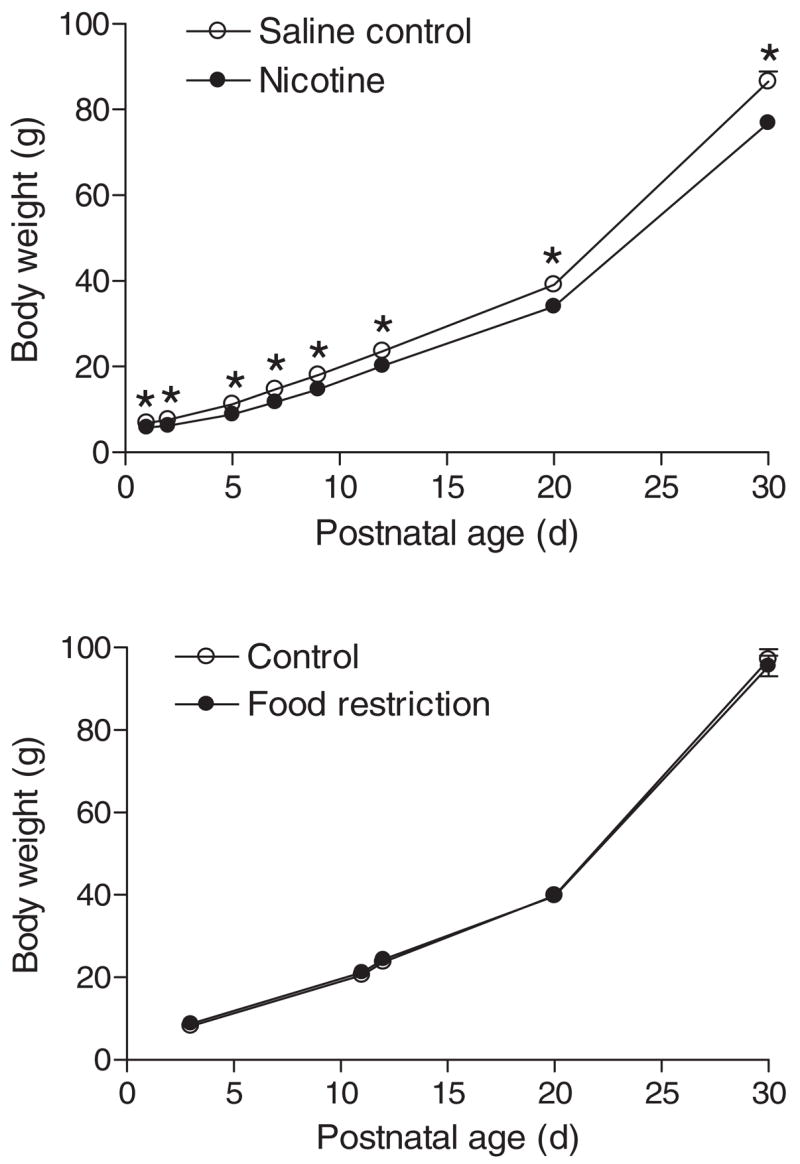
Effect of maternal nicotine administration and food restriction on neonatal body weight. Nicotine was administrated to pregnant rats through osmotic pumps, as described under Materials and Methods. Control rats received saline. In separate studies, pregnant rats were divided into two groups and were fed with equivalent food to that determined in saline control and nicotine-treated animals, respectively. Body weight was measured in offspring from 1 to 30 days of age. Data are means ± S.E.M. Data were analyzed by two-way ANOVA with age as one factor and nicotine treatment as the other.*, P < 0.05 versus nicotine treatment. n = 4 to 6 litters.
Effect of Prenatal Nicotine Exposure on Postischemic Recovery of LV Function
Table 1 shows the preischemic values of LV function and coronary flow rate in isolated hearts from adult male and female offspring in a Langendorff preparation. Prenatal nicotine exposure showed no significant effects on baseline LV function and coronary flow in male hearts, but significantly attenuated LVDP, HR, and coronary flow rate at baseline levels in female hearts (Table 1). As shown in Figs. 3 and 4, global ischemia for 25 min caused a significant impairment in LV function in both male and female hearts. Consistent with the previous findings (Bae and Zhang, 2005b), the postischemic recoveries of LVDP, dP/dtmax, dP/dtmin, and the pressure-rate product were significantly better in female hearts than those in male hearts in saline control animals (Figs. 3 and 4). Prenatal nicotine exposure resulted in significant decreases in postischemic recovery of LV function in both male and female hearts with the detrimental effects in female hearts being more pronounced (Figs. 3 and 4). In prenatal nicotine-treated animals, there were no significant differences in the postischemic recovery of LV function in male and female hearts (Figs. 3 and 4). The infarct size of LV at the end of 60-min reperfusion after 25-min ischemia is shown in Fig. 5. Ischemia and reperfusion caused LV myocardial infarction in both male and female hearts. Prenatal nicotine treatment significantly increased infarct size in both male and female hearts (Fig. 5). In prenatal nicotine-treated animals, the postischemic infarct size of LV was significantly greater in female than in male hearts (Fig. 5).
TABLE 1.
Preischemic left ventricle functional parameters of 3-month-old rat offspring
| LVDP | HR | dP/dtmax | dP/dtmin | Coronary Flow | |
|---|---|---|---|---|---|
| mm Hg | beats/min | mm Hg/s | ml/min/g | ||
| Control M (n = 11) | 91.7 ± 1.6 | 266 ± 5 | 2586 ± 168 | 1689 ± 76 | 9.1 ± 0.5 |
| Nicotine M (n = 11) | 88.5 ± 2.4 | 267 ± 8 | 2297 ± 129 | 1522 ± 97 | 9.1 ± 0.7 |
| Control F (n = 9) | 99.8 ± 0.9 | 273 ± 3 | 3209 ± 85 | 1808 ± 77 | 10.4 ± 0.8 |
| Nicotine F (n = 11) | 90.3 ± 3.2* | 253 ± 8* | 2999 ± 213 | 1681 ± 88 | 7.1 ± 0.7* |
M, male; F, female.
P < 0.05 versus control.
Fig. 3.

Effect of maternal nicotine administration on postischemic recovery of LV function in adult male offspring. Nicotine was administrated to pregnant rats through osmotic pumps, as described under Materials and Methods. Control rats received saline. Hearts were obtained from 3-month-old male offspring and were subjected to 25 min of ischemia and 60 min of reperfusion in a Langendorff preparation. Data are means ± S.E.M. Data were analyzed by two-way ANOVA with ischemia-reperfusion as one factor and nicotine treatment as the other.*, P < 0.05, versus saline control for the entire curve. n = 11.
Fig. 4.

Effect of maternal nicotine administration on postischemic recovery of LV function in adult female offspring. Nicotine was administrated to pregnant rats through osmotic pumps, as described under Materials and Methods. Control rats received saline. Hearts were obtained from 3-month-old female offspring and were subjected to 25 min of ischemia and 60 min of reperfusion in a Langendorff preparation. Data are means ± S.E.M. Data were analyzed by two-way ANOVA with ischemia-reperfusion as one factor and nicotine treatment as the other.*, P < 0.05, versus saline control for the entire curve. n = 9 to 11.
Fig. 5.
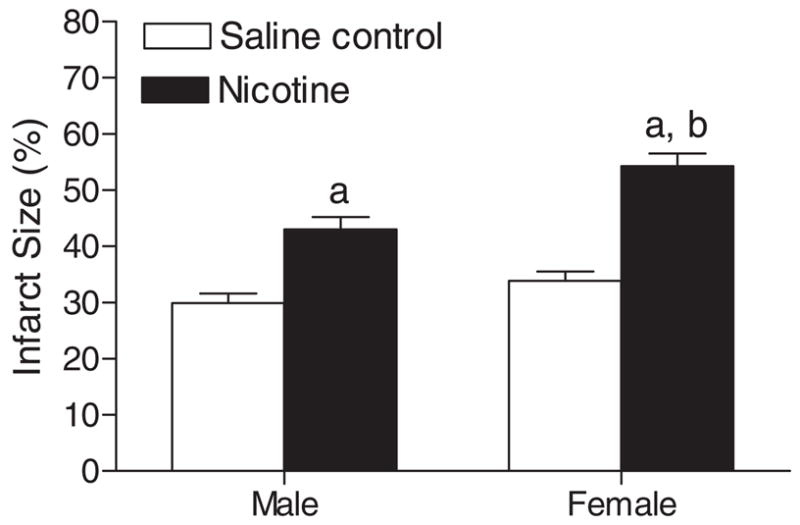
Effect of maternal nicotine administration on I/R-induced myocardial infarction in adult offspring. Nicotine was administrated to pregnant rats through osmotic pumps, as described under Materials and Methods. Control rats received saline. Hearts were obtained from 3-month-old male and female offspring and were subjected to 25 min of ischemia and 60 min of reperfusion in a Langendorff preparation. Left ventricles were collected at the end of reperfusion, and myocardial infarct size was determined with 1% triphenyltetrazolium chloride staining and expressed as a percentage of the total left ventricular weight. Data are means ± S.E.M. a, P < 0.05 versus saline control; b, P < 0.05 versus male. n = 9 to 11.
Effect of Prenatal Nicotine Exposure on Coronary Flow and NO Production
Pulmonary artery effluent was collected as an index of coronary flow, and NO in coronary effluent was measured by the chemiluminescence method as described under Materials and Methods. In female hearts, prenatal nicotine exposure significantly decreased coronary flow rate (milliliters per minute per gram of heart wet weight) at the baseline and during postischemic recovery (Fig. 6). In contrast, nicotine had no effects on coronary flow rate in male hearts (Fig. 6). Unlike the changes observed in coronary flow rate, prenatal nicotine exposure significantly decreased coronary NO levels at the baseline and during postischemic recovery in male hearts but not in female hearts (Fig. 7).
Fig. 6.
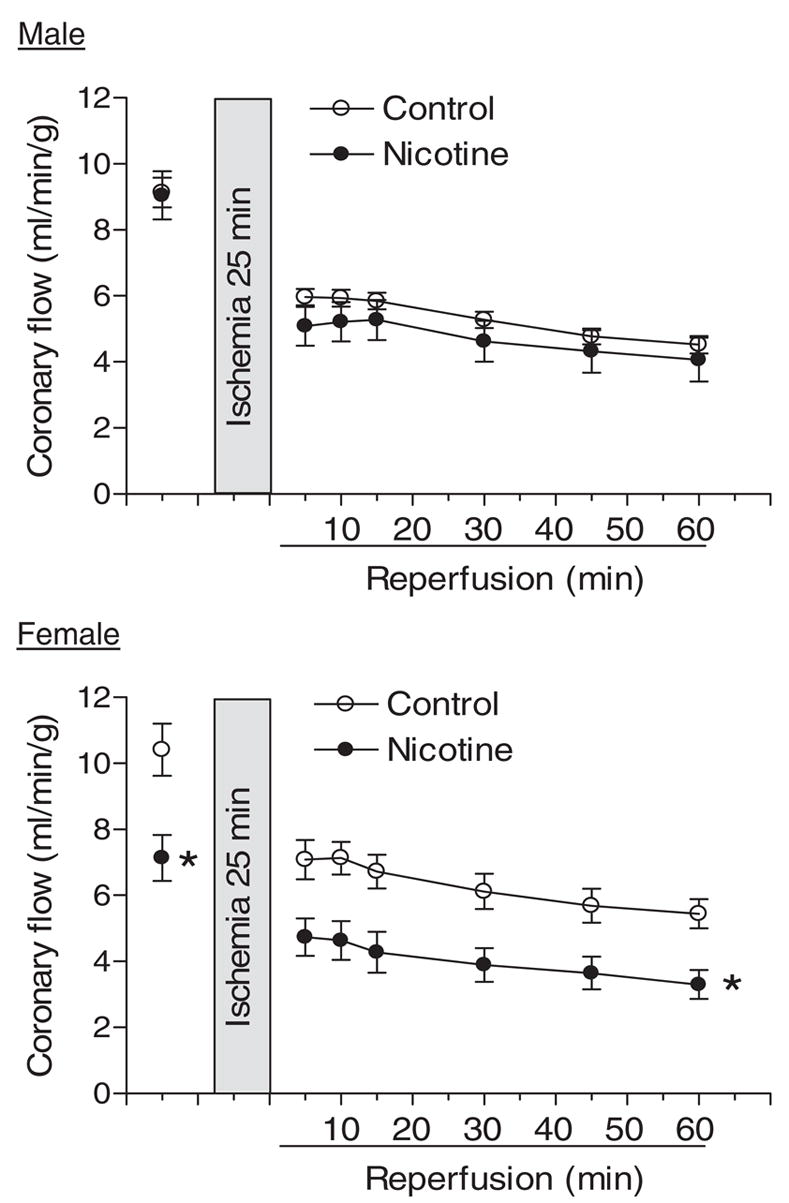
Effect of maternal nicotine administration on coronary flow rate in adult offspring. Nicotine was administrated to pregnant rats through osmotic pumps, as described under Materials and Methods. Control rats received saline. Hearts were obtained from 3-month-old male and female offspring and were subjected to 25 min of ischemia and 60 min of reperfusion in a Langendorff preparation. Pulmonary artery effluent was collected as an index of coronary flow (milliliters per minute per gram of heart wet weight). Data are means ± S.E.M. Data were analyzed by two-way ANOVA with ischemia-reperfusion as one factor and nicotine treatment as the other. *, P < 0.05 versus saline control for the entire curve. n = 9 to 11.
Fig. 7.
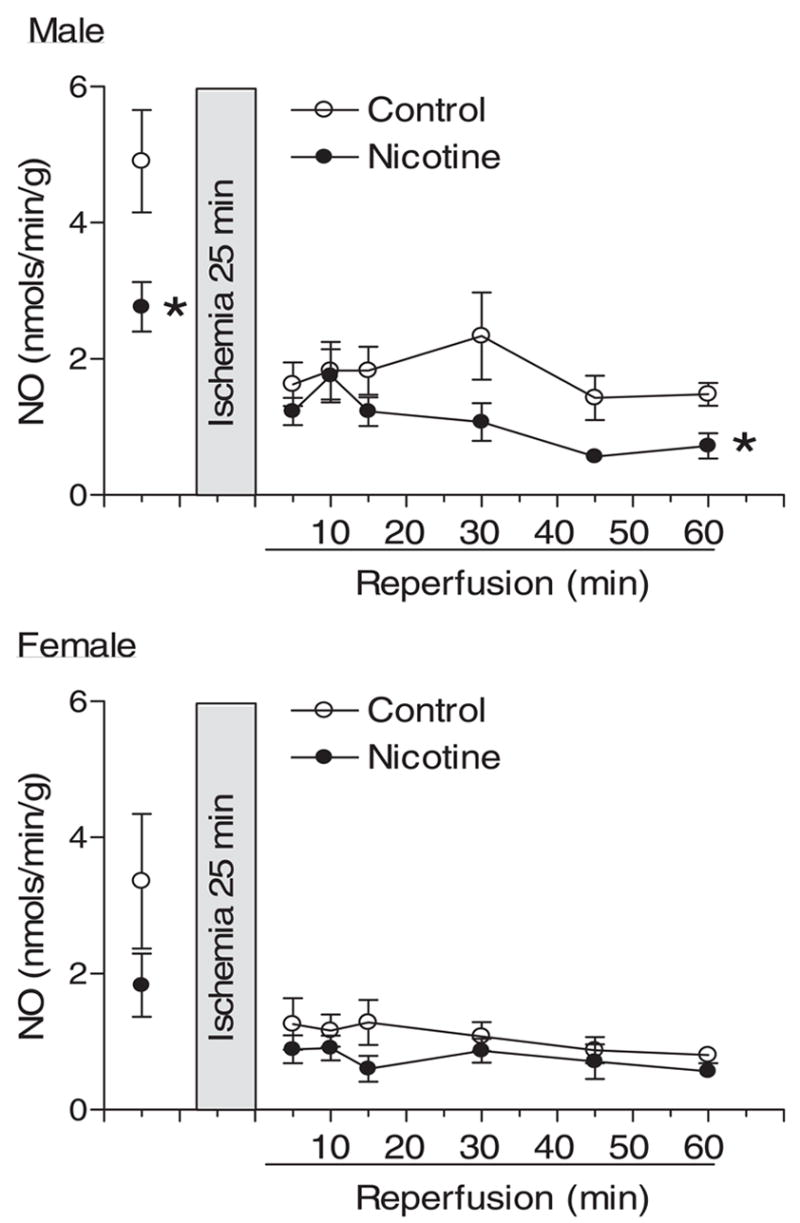
Effect of maternal nicotine administration on coronary NO release in adult offspring. Nicotine was administrated to pregnant rats through osmotic pumps, as described under Materials and Methods. Control rats received saline. Hearts were obtained from 3-month-old male and female offspring and were subjected to 25 min of ischemia and 60 min of reperfusion in a Langendorff preparation. Pulmonary artery effluent was collected and NO (nanomoles per minute per gram of heart wet weight) was measured using the chemiluminescence method, as described under Materials and Methods. Data are means ± S.E.M. Data were analyzed by two-way ANOVA with ischemia-reperfusion as one factor and nicotine treatment as the other. *P < 0.05, versus saline control for the entire curve. n = 9 to 11.
Effect of Prenatal Nicotine Exposure on Cardiac Caspase and PKC Isozyme Expression
The protein levels of caspase-3, -8, and -9 and PKCε and PKCδ expression in the heart were determined by Western blot analyses. As shown in Fig. 8, differential expression of caspase-3, -8, and -9 in male and female hearts was observed. Caspase-3 and -8 levels were significantly higher in male hearts than those in female hearts. In male hearts, levels of caspase-3 were significantly higher than those of caspase-8 and -9. Unlike in male hearts, in female hearts levels of caspase-9 were significantly higher than those of caspase-3 and -8. Prenatal nicotine exposure had no significant effects on caspase-3, -8, or -9 levels in either male or female hearts (Fig. 8).
Fig. 8.
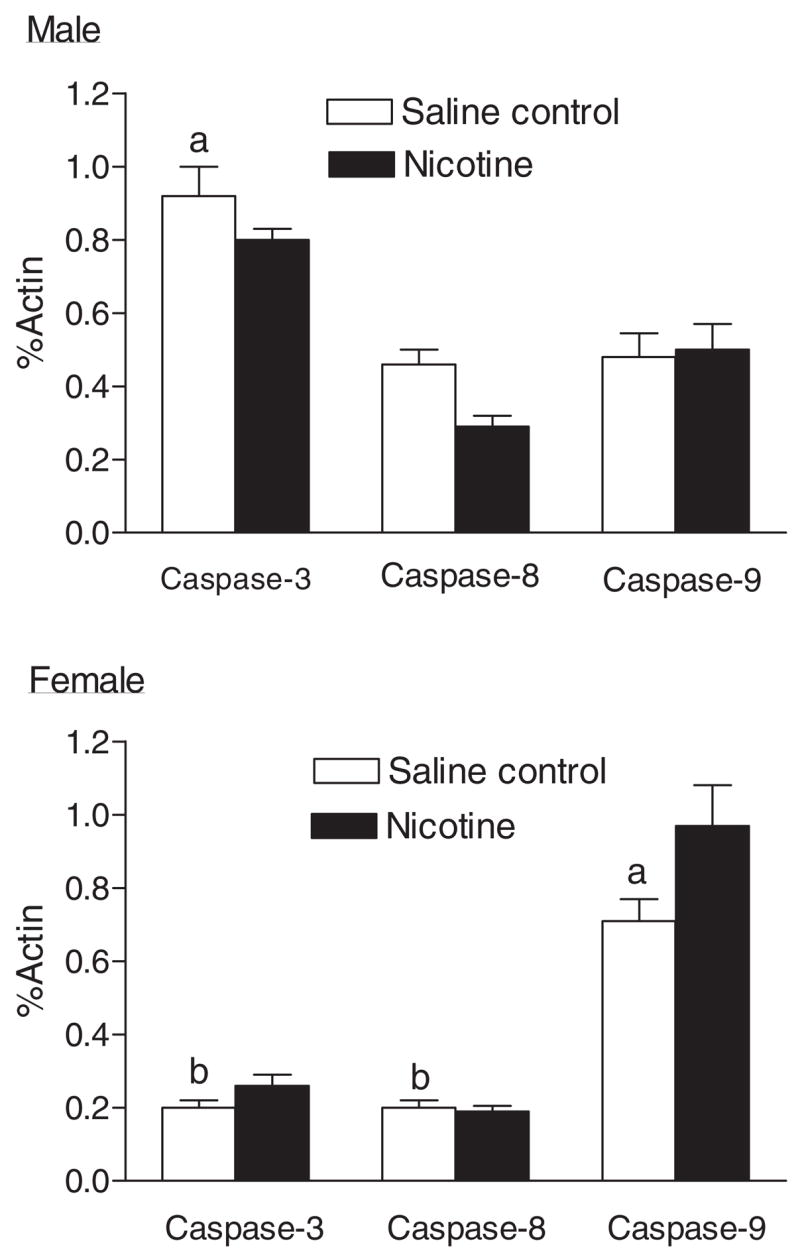
Effect of maternal nicotine administration on caspase protein levels in the hearts of adult offspring. Nicotine was administrated to pregnant rats through osmotic pumps, as described under Materials and Methods. Control rats received saline. Hearts were obtained from 3-month-old male and female offspring, and caspase-3, -8, and -9 protein levels were determined using Western blot analyses. Data are means ± S.E.M. a, P < 0.05 versus other caspases in the same sex; b, P < 0.05 versus the same caspase in male hearts. n = 4.
The gender dimorphism of PKC isozyme expression levels was also identified in the heart (Fig. 9). Whereas there was no significant difference in PKCε protein levels in male and female hearts, the levels of PKCδ were significantly higher in female hearts than those in male hearts. In female hearts, there was no significant difference in PKCε and PKCδ protein levels. However, in male hearts there were significant higher levels of PKCε than of PKCδ. Prenatal nicotine exposure caused a significant decrease in the expression of PKCε in both male and female hearts and a significant decrease in PKCδ expression only in female hearts (Fig. 9).
Fig. 9.
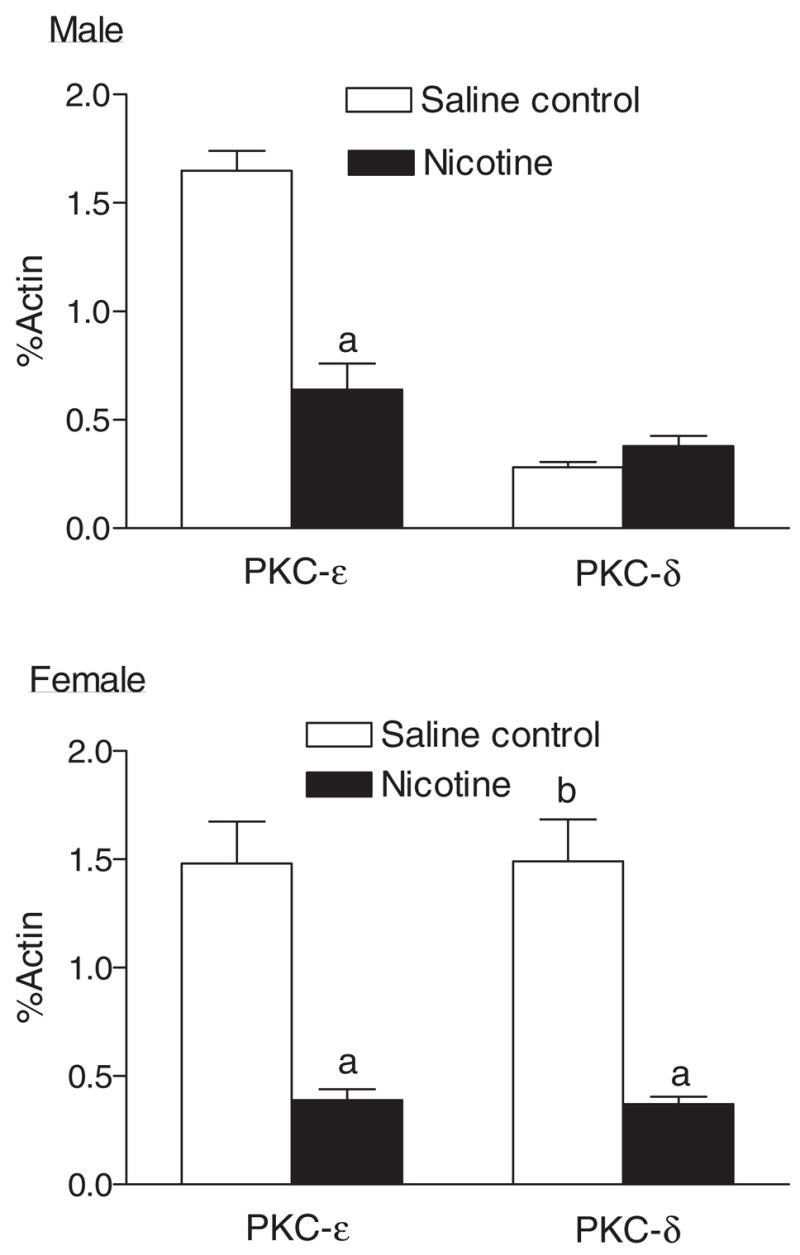
Effect of maternal nicotine administration on PKCε and PKCδ protein levels in the hearts of adult offspring. Nicotine was administrated to pregnant rats through osmotic pumps, as described under Materials and Methods. Control rats received saline. Hearts were obtained from 3-month-old male and female offspring and protein levels of PKCε and PKCδ were determined using Western blot analyses. Data are means ± S.E.M. a, P < 0.05 versus saline control; b, P < 0.05 versus male. n = 4.
Effect of Maternal Food Restriction on Postischemic Recovery of LV Function
Control studies of maternal food restriction and moderated decreases in maternal body weight gain (Fig. 1) had no significant effects on body weight and heart weight in both male and female adult offspring. In addition, the baseline values for LV function and coronary flow rate as determined in a Langendorff preparation were not significantly different. Consistent with the finding in saline control animals, the postischemic recovery of LV function was significantly better in female than in male hearts in food control animals (Fig. 10). In contrast to the finding with prenatal nicotine treatment, maternal food restriction and moderated decreases in maternal body weight gain resulted in a significant increase in the postischemic recovery of LV function in hearts of adult male offspring (Fig. 10). However, the postischemic recovery of LV function in female hearts was not significantly affected (Fig. 10). In offspring of maternal food restriction rats, there was no significant difference in the postischemic recovery of LV function in male and female hearts (Fig. 10).
Fig. 10.

Effect of maternal food restriction on postischemic recovery of LV function in adult offspring. Pregnant rats were divided into two groups and were fed with equivalent food to that determined in saline control and nicotine-treated animals, respectively. Hearts were obtained from 3-month-old male and female offspring and were subjected to 25 min of ischemia and 60 min of reperfusion in a Langendorff preparation. Data are means ± S.E.M. Data were analyzed by two-way ANOVA with ischemia-reperfusion as one factor and maternal food restriction as the other. *, P < 0.05 versus control for the entire curve. n = 5.
Discussion
The present study demonstrated that prenatal nicotine exposure significantly increased LV myocardial infarct size and decreased postischemic recovery of LV function after 25 min of ischemia in adult offspring. This result is in agreement with previous studies showing that prenatal hypoxia and cocaine treatments increased cardiac vulnerability to I/R injury in adult offspring (Li et al., 2003; Bae et al., 2005; Xu et al., 2006) and is consistent with epidemiological studies showing an association of adverse intrauterine environment and an increased risk of ischemic heart disease in later adult life (Barker et al., 1993). It has been shown that second-hand smoke exposure in utero for 3 weeks increases infarct size in young pups that were subjected to 17 min of left coronary artery occlusion and 2 h of reperfusion (Zhu et al., 1997). Similar studies have demonstrated that prenatal nicotine exposure produced intolerance to neonatal hypoxia by interfering with the maintenance of cardiac function (Slotkin et al., 1997).
In contrast with previous findings that prenatal cocaine and hypoxia treatments predominantly affected the male heart in adult offspring (Li et al., 2003; Bae et al., 2005; Xu et al., 2006), prenatal nicotine exposure resulted in more pronounced effects in female hearts. The finding that postischemic recovery of female hearts was significantly better than that of male hearts in saline control animals is in agreement with previous studies (Bae et al., 2005; Bae and Zhang, 2005b). Unlike prenatal cocaine treatment that increased heart vulnerability to I/R injury only in male adult offspring (Bae et al., 2005), nicotine resulted in a significant decrease in postischemic recovery of LV function in both male and female hearts with the detrimental effects in female hearts being more pronounced and abolished the difference in postischemic recovery of LV function between male and female hearts. These findings suggest different gender mechanisms of in utero cardiac programming caused by fetal nicotine and cocaine exposure. Additionally, our previous study demonstrated that prenatal nicotine treatment significantly increased norepinephrine-induced contractions of aortas in male but not female adult offspring (Xiao et al., 2007), suggesting an organ and/or tissue specificity of gender-dependent programming induced by prenatal nicotine exposure.
The present finding of increased sensitivity of female hearts in manifestation of heart vulnerability to I/R injury in adult offspring after prenatal nicotine exposure is novel and intriguing. The gender dichotomy in fetal programming of adult disease has been demonstrated in several animal models. Although the results are conflicting, it has generally been shown that female offspring are less sensitive in manifestation of hypertension caused by adverse prenatal stimuli (do Carmo Pinho Franco et al., 2003). It is likely that multiple mechanisms are involved in the gender-dependent effects of prenatal nicotine exposure on heart vulnerability to I/R injury. The finding that coronary flow rate was significantly decreased in female but not male hearts at the baseline and during postischemic recovery in nicotine-treated animals suggests a mechanism for the increased sensitivity of female hearts. The moderate decrease in baseline coronary flow rate may not be critical for cardiac function at rest but is likely to contribute to the increased heart susceptibility to I/R injury in the female hearts. Given that NO plays an important role in the regulation of coronary flow, we measured NO release in coronary effluent. We found that prenatal nicotine treatment significantly decreased coronary NO release in male but not in female hearts. This finding is consistent with our previous study demonstrating a gender-specific decrease in endothelial nitric-oxide synthase activity in the aorta of male offspring after prenatal nicotine treatment (Xiao et al., 2007) and suggests that the decreased coronary flow rate observed in female hearts is mediated by increased coronary vascular tone or changes in coronary vasculature rather than by a decrease in NO release.
In addition to altered coronary flow, intrinsic changes in cardiomyocytes contribute significantly to cardiac programming resulting from an adverse intrauterine environment (Li et al., 2003; Bae and Zhang, 2005a; Bae et al., 2005; Xu et al., 2006). It has been clearly demonstrated that apoptosis plays an important role in I/R injury (Kang and Izumo, 2000; Condorelli et al., 2001). Our previous studies demonstrated that increased myocyte apoptosis played a key role in the increased heart vulnerability to I/R injury in adult offspring after prenatal insults (Bae et al., 2003, 2005; Li et al., 2003; Bae and Zhang, 2005a). In the present study, we demonstrated a differential expression pattern of caspase-3, -8, and -9 in male and female hearts in saline control animals. The finding that caspase-3 and -8 levels were significantly higher in male than in female hearts suggests a possible mechanism for the increased susceptibility of male hearts compared with female hearts to I/R injury in control animals. It has been shown that heart-targeted overexpression of caspase-3 in mice increases I/R-induced myocyte infarct size and depresses cardiac function (Condorelli et al., 2001). Nevertheless, prenatal nicotine treatment had no significant effects on caspase-3, -8, or -9 levels in either male or female hearts, suggesting that they may not be involved in nicotine-induced programming of heart vulnerability to I/R injury in adult offspring.
Our previous studies demonstrated that prenatal insults caused an epigenetic modification of the PKCε gene in the fetal heart resulting in a down-regulation of PKCε expression in the heart of adult offspring (Li et al., 2004; Bae et al., 2005; Zhang et al., 2007). A similar finding has been obtained in the present study showing that prenatal nicotine exposure significantly decreased cardiac PKCε protein expression in adult offspring. These findings suggest a common mechanism of PKCε in cardiac programming in response to intrauterine adverse stimuli. PKCε plays a pivotal role of cardio-protection during cardiac I/R injury through inhibition of apoptosis (Sugden and Clerk, 2001; Murriel and Mochly-Rosen, 2003; Gray et al., 2004; Gregory et al., 2004). Recent studies in a PKCε knockout mouse model have demonstrated that PKCε expression is not required for normal cardiac function under physiological conditions, but PKCε activation is necessary and sufficient for acute cardioprotection during cardiac I/R (Gray et al., 2004). Our previous study demonstrated that prenatal cocaine exposure caused a decrease in PKCε protein levels only in adult male hearts, which corresponded to the functional study showing that only male hearts were affected in postischemic recovery of LV function (Bae et al., 2005). In the present study, we found that PKCε protein levels were decreased in both male and female hearts after prenatal nicotine treatment and that postischemic recovery of LV function was significantly decreased in both male and female hearts. These findings suggest a gender dichotomy of the PKCε gene expression pattern in the heart and reinforce a key role for PKCε in programming of heart vulnerability to I/R injury in adult offspring.
Unlike PKCε, the role of PKCδ in I/R injury is less clear and is somewhat controversial. Inhibition of PKCδ during reperfusion has been shown to decrease reperfusion-induced injury (Murriel and Mochly-Rosen, 2003). Other studies demonstrated the cardioprotective effects of δ Kawamura et al., 1998; Zhao et al., 1998; Bouwman et al., 2006). It has been demonstrated that estrogen deficiency decreases ischemic tolerance in the aged rat heart through decreases in both PKCδ and PKCε levels (Hunter et al., 2007). The present finding that female hearts had higher levels of PKCδ and had greater resistance to I/R injury than male hearts suggests a possible mechanism of PKCδ in the gender dichotomy of heart susceptibility to I/R injury. The significantly decreased PKCδ protein levels in female but not male hearts is likely to contribute to the increased heart susceptibility to I/R injury in female than in male adult offspring after prenatal nicotine exposure. The lack of effect of nicotine on PKCδ in male hearts may be due in part to the low basal level of PKCδ in the male heart.
The present finding that a transient maternal food restriction during early gestation and moderately decreased maternal body weight gain, mimicking that observed in nicotine-treated animals, significantly improved postischemic recovery of LV function in male but lacked the effect in female adult offspring is very intriguing and suggests an additional explanation for why males were less sensitive than females in heart susceptibility to I/R injury after prenatal nicotine treatment. It is possible that the reduced food intake and decreased maternal weight gain observed in nicotine-treated animals produced cardiac protection that counteracted nicotine-mediated detrimental effects in male adult offspring. Additionally, these studies reinforce a specific effect of nicotine in programming of the heart resulting in increased heart susceptibility to I/R injury in adult offspring. To our knowledge, this is the first report that transient maternal food restriction during early gestation induces a gender-specific cardioprotective effect in adult male offspring. Previous studies in several animal models showed that sustained maternal food restriction or a low-protein diet during the late gestational period or throughout pregnancy caused vascular and cardiac dysfunction in adult offspring (do Carmo Pinho Franco et al., 2002; Brawley et al., 2003; Cheema et al., 2005; Williams et al., 2005) and impaired postischemic recovery of the heart (Xu et al., 2006; Elmes et al., 2007). Taken together, these studies suggest that specific gestational periods and duration of the treatment are critical in determining the outcomes of cardiac programming and the effects on heart function in adult offspring.
In conclusion, we have demonstrated that prenatal nicotine exposure causes cardiac programming and increases heart susceptibility to I/R injury in adult offspring. Together with the previous studies, the present finding reinforces the notion that multiple adverse factors during pregnancy can affect heart development in utero and impair heart function in adulthood. Given that maternal cigarette smoking with fetal nicotine exposure is one of the most widespread prenatal insults, the present finding has obvious clinical significance. As is often the case with novel findings, the present study may raise more questions than it answers. For instance, are the effects on heart development mediated by a direct effect of nicotine on the fetal heart or indirectly through its effect on the mother? In addition, what are the epigenetic mechanisms involved in the gender-dependent regulation of PKC isozyme gene expression pattern in the heart, which persists into adulthood? Does prenatal nicotine treatment alter the coronary vasculature in female hearts? What are the mechanisms for cardiac protection in adult offspring after transient maternal food restriction during early gestation? Undoubtedly, these questions warrant continuing investigations.
Acknowledgments
This work was supported in part by National Institutes of Health Grants HL82779 (L.Z.), HL83966 (L.Z.), and S06GM073842 (S.Y.), and by Tobacco-related Disease Research Program Award 14FT-0075 (D.X.).
ABBREVIATIONS
- I/R
ischemia/reperfusion
- LV
left ventricle
- PK
protein kinase
- LVEDP
left ventricle end diastolic pressure
- LVDP
left ventricle developed pressure
- HR
heart rate
- PRP
pressure rate product calculated as LVDP × HR
- NO
nitric oxide
- ANOVA
analysis of variance
Footnotes
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
References
- Bae S, Gilbert RD, Ducsay CA, Zhang L. Prenatal cocaine exposure increases heart susceptibility to ischaemia-reperfusion injury in adult male but not female rats. J Physiol. 2005;565:149–158. doi: 10.1113/jphysiol.2005.082701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S, Xiao Y, Li G, Casiano CA, Zhang L. Effect of maternal chronic hypoxic exposure during gestation on apoptosis in fetal rat heart. Am J Physiol. 2003;285:983–990. doi: 10.1152/ajpheart.00005.2003. [DOI] [PubMed] [Google Scholar]
- Bae S, Zhang L. Prenatal cocaine exposure increases apoptosis of neonatal rat heart and heart susceptibility to ischemia-reperfusion injury in 1-month-old rat. Br J Pharmacol. 2005a;144:900–907. doi: 10.1038/sj.bjp.0706129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S, Zhang L. Gender differences in cardioprotection against ischemia/reperfusion injury in adult rat hearts. J Pharmacol Exp Ther. 2005b;315:1125–1135. doi: 10.1124/jpet.105.090803. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Gluckman PD, Godfrey KM, Harding JE, Owens JA, Robinson JS. Fetal nutrition and cardiovascular disease in adult life. Lancet. 1993;341:938–941. doi: 10.1016/0140-6736(93)91224-a. [DOI] [PubMed] [Google Scholar]
- Beratis NG, Panagoulias D, Varvarigou A. Increased blood pressure in neonates and infants whose mothers smoked during pregnancy. J Pediatr. 1996;128:806–812. doi: 10.1016/s0022-3476(96)70333-5. [DOI] [PubMed] [Google Scholar]
- Blake KV, Gurrin LC, Evans SF, Beilin LJ, Landau LI, Stanley, Newnham JP. Maternal cigarette smoking during pregnancy, low birth weight and subsequent blood pressure in early childhood. Early Hum Dev. 2000;57:137–147. doi: 10.1016/s0378-3782(99)00064-x. [DOI] [PubMed] [Google Scholar]
- Bouwman RA, Salic K, Padding FG, Eringa EC, van eek-Harmsen BJ, Matsuda T, Baba A, Musters RJ, de Lange JJ, Boer C. Cardioprotection via activation of protein kinase C-ε depends on modulation of the reverse mode of the Na+/Ca2+ exchanger. Circulation. 2006;114:I226–I232. doi: 10.1161/CIRCULATIONAHA.105.000570. [DOI] [PubMed] [Google Scholar]
- Brawley L, Itoh S, Torrens C, Barker A, Bertram C, Poston L, Hanson M. Dietary protein restriction in pregnancy induces hypertension and vascular defects in rat male offspring. Pediatr Res. 2003;54:1–7. doi: 10.1203/01.PDR.0000065731.00639.02. [DOI] [PubMed] [Google Scholar]
- Cheema KK, Dent MR, Saini HK, Aroutiounova N, Tappia PS. Prenatal exposure to maternal undernutrition induces adult cardiac dysfunction. Br J Nutr. 2005;93:471–477. doi: 10.1079/bjn20041392. [DOI] [PubMed] [Google Scholar]
- Condorelli G, Roncarati R, Ross J, Pisani A, Stassi G, Todaro M, Trocha S, Drusco A, Gu Y, Russo MA, et al. Heart-targeted overexpression of caspase3 in mice increases infarct size and depresses cardiac function. Proc Natl Acad Sci U S A. 2001;98:9977–9982. doi: 10.1073/pnas.161120198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- do Carmo Pinho Franco M, Nigro D, Fortes ZB, Tostes RC, Carvalho MH, Lucas SR, Gomes GN, Coimbra TM, Gil FZ. Intrauterine undernutrition—renal and vascular origin of hypertension. Cardiovasc Res. 2003;60:228–234. doi: 10.1016/s0008-6363(03)00541-8. [DOI] [PubMed] [Google Scholar]
- Dodic M, Samuel C, Moritz K, Wintour EM, Morgan J, Grigg L, Wong J. Impaired cardiac functional reserve and left ventricular hypertrophy in adult sheep after prenatal dexamethasone exposure. Circ Res. 2001;89:623–629. doi: 10.1161/hh1901.097086. [DOI] [PubMed] [Google Scholar]
- Elmes MJ, Gardner DS, Langley-Evans SC. Fetal exposure to a maternal low-protein diet is associated with altered left ventricular pressure response to ischaemia-reperfusion injury. Br J Nutr. 2007;98:93–100. doi: 10.1017/S000711450769182X. [DOI] [PubMed] [Google Scholar]
- Franco MMC, Arruda RMM, Dantas AP, Kawamoto EM, Carvalho MHC, Tostes RCA, Nigro D. Intrauterine undernutrition: expression and activity of the endothelial nitric oxide synthase in male and female adult offspring. Cardiovasc Res. 2002;56:145–153. doi: 10.1016/s0008-6363(02)00508-4. [DOI] [PubMed] [Google Scholar]
- Gray MO, Zhou HZ, Schafhalter-Zoppoth I, Zhu P, Mochly-Rosen D, Messing RO. Preservation of base-line hemodynamic function and loss of inducible car-dioprotection in adult mice lacking protein kinase Cε. J Biol Chem. 2004;279:3596–3604. doi: 10.1074/jbc.M311459200. [DOI] [PubMed] [Google Scholar]
- Gregory KN, Hahn H, Haghighi K, Marrees Y, Odley A, Dorn GW, 2nd, Kranias EG. Increased particulate partitioning of PKCε reverses susceptibility of phospholamban knockout hearts to ischemic injury. J Mol Cell Cardiol. 2004;36:313–318. doi: 10.1016/j.yjmcc.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Huang ZG, Wang X, Evans C, Gold A, Bouairi E, Mendelowitz D. Prenatal nicotine exposure alters the types of nicotinic receptors that facilitate excitatory inputs to cardiac vagal neurons. J Neurophysiol. 2004;92:2548–2554. doi: 10.1152/jn.00500.2004. [DOI] [PubMed] [Google Scholar]
- Hunter JC, Kostyak JC, Novotny JL, Slimpson AM, Korzick DH. Estrogen deficiency decreases ischemic tolerance in the aged rat heart: roles of PKCδ, PKCε, Akt, and GSK3β. Am J Physiol. 2007;292:R800–R809. doi: 10.1152/ajpregu.00374.2006. [DOI] [PubMed] [Google Scholar]
- Kang PM, Izumo S. Apoptosis and heart failure: a critical review of the literature. Circ Res. 2000;86:1107–1113. doi: 10.1161/01.res.86.11.1107. [DOI] [PubMed] [Google Scholar]
- Kawamura S, Yoshida K, Miura T, Mizukami Y, Matsuzaki M. Ischemic preconditioning translocates PKC-δ and -ε, which mediate functional protection in isolated rat heart. Am J Physiol. 1998;275:H2266–H2271. doi: 10.1152/ajpheart.1998.275.6.H2266. [DOI] [PubMed] [Google Scholar]
- Lambers DS, Clark KE. The maternal and fetal physiologic effects of nicotine. Semin Perinatol. 1996;20:115–126. doi: 10.1016/s0146-0005(96)80079-6. [DOI] [PubMed] [Google Scholar]
- Li G, Bae S, Zhang L. Effect of prenatal hypoxia on heat stress-mediated cardioprotection in adult rat heart. Am J Physiol. 2004;286:H1712–H1719. doi: 10.1152/ajpheart.00898.2003. [DOI] [PubMed] [Google Scholar]
- Li G, Xiao Y, Estrella JL, Ducsay CA, Gilbert RD, Zhang L. Effect of fetal hypoxia on heart susceptibility to ischemia and reperfusion injury in the adult rat. J Soc Gynecol Investig. 2003;10:265–274. doi: 10.1016/s1071-5576(03)00074-1. [DOI] [PubMed] [Google Scholar]
- Lichtensteiger W, Ribary U, Schlumpf M, Odermatt B, Widmer HR. Prenatal adverse effects of nicotine on the developing brain. Prog Brain Res. 1988;73:137–157. doi: 10.1016/S0079-6123(08)60502-6. [DOI] [PubMed] [Google Scholar]
- Machaalani R, Waters KA, Tinworth KD. Effects of postnatal nicotine exposure on apoptotic markers in the developing piglet brain. Neuroscience. 2005;132:325–333. doi: 10.1016/j.neuroscience.2004.12.039. [DOI] [PubMed] [Google Scholar]
- Mancia G, Groppelli A, Di Rienzo M, Castiglioni P, Parati G. Smoking impairs baroreflex sensitivity in humans. Am J Physiol. 1997;273:H1555–H1560. doi: 10.1152/ajpheart.1997.273.3.H1555. [DOI] [PubMed] [Google Scholar]
- Martin JC, Martin DC, Chao S, Shores P. Interactive effects of chronic maternal ethanol and nicotine exposure upon offspring development and function. Neurobehav Toxicol Teratol. 1982;4:293–298. [PubMed] [Google Scholar]
- Murriel CL, Mochly-Rosen D. Opposing roles of δ and ε PKC in cardiac ischemia and reperfusion: targeting the apoptotic machinery. Arch Biochem Biophys. 2003;420:246–254. doi: 10.1016/j.abb.2003.08.038. [DOI] [PubMed] [Google Scholar]
- National Research Council. Guide for the Care and Use of Laboratory Animals. National Academy Press; Washington, DC: 1996. [Google Scholar]
- Slotkin TA. Fetal nicotine or cocaine exposure: which one is worse? J Pharmacol Exp Ther. 1998;285:931–945. [PubMed] [Google Scholar]
- Slotkin TA, Saleh JL, McCook EC, Seidler FJ. Impaired cardiac function during postnatal hypoxia in rats exposed to nicotine prenatally: implications for perinatal morbidity and mortality, and for sudden infant death syndrome. Teratology. 1997;55:177–184. doi: 10.1002/(SICI)1096-9926(199703)55:3<177::AID-TERA2>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Sugden PH, Clerk A. Akt like a woman: gender differences in susceptibility to cardiovascular disease. Circ Res. 2001;88:975–977. doi: 10.1161/hh1001.091864. [DOI] [PubMed] [Google Scholar]
- Williams SJ, Campbell ME, McMillen IC, Davidge ST. Differential effects of maternal hypoxia or nutrient restriction on carotid and femoral vascular function in neonatal rats. Am J Physiol. 2005;288:R360–R367. doi: 10.1152/ajpregu.00178.2004. [DOI] [PubMed] [Google Scholar]
- Xiao D, Ducsay CA, Zhang L. Chronic hypoxia and developmental regulation of cytochrome c expression in rats. J Soc Gynecol Investig. 2000;7:279–283. [PubMed] [Google Scholar]
- Xiao D, Huang X, Lawrence J, Yang S, Zhang L. Fetal and neonatal nicotine exposure differentially regulates vascular contractility in adult male and female offspring. J Pharmacol Exp Ther. 2007;320:654–661. doi: 10.1124/jpet.106.113332. [DOI] [PubMed] [Google Scholar]
- Xu Y, Williams SJ, O’Brien D, Davidge ST. Hypoxia or nutrient restriction during pregnancy in rats leads to progressive cardiac remodelling and impairs postischemic recovery in adult male offspring. FASEB J. 2006;20:1251–1253. doi: 10.1096/fj.05-4917fje. [DOI] [PubMed] [Google Scholar]
- Zhang H, Darwanto A, Linkhart TA, Sowers LC, Zhang L. Maternal cocaine administration causes an epigenetic modification of PKCε gene expression in fetal rat heart. Mol Pharmacol. 2007;71:1319–1328. doi: 10.1124/mol.106.032011. [DOI] [PubMed] [Google Scholar]
- Zhao J, Renner O, Wightman L, Sugden PH, Stewart L, Miller AD, Latchman DS, Marber MS. The expression of constitutively active isotypes of protein kinase C to investigate preconditioning. J Biol Chem. 1998;273:23072–23079. doi: 10.1074/jbc.273.36.23072. [DOI] [PubMed] [Google Scholar]
- Zhu BQ, Sun YP, Sudhir K, Sievers RE, Browne AE, Gao L, Hutchison SJ, Chou TM, Deedwania PC, Chatterjee K, et al. Effects of second-hand smoke and gender on infarct size of young rats exposed in utero and in the neonatal to adolescent period. J Am Coll Cardiol. 1997;30:1878–1885. doi: 10.1016/s0735-1097(97)00364-1. [DOI] [PubMed] [Google Scholar]