

Abstract
To develop biodegradable polymers with favorable physicochemical and biological properties, we have synthesized a series of poly(terephthalate-co-phosphate)s using a two-step polycondensation. The diol 1,4-bis(2-hydroxyethyl) terephthalate was first reacted with ethylphosphorodichloridate (EOP), and then chain-extended with terephthaloyl chloride (TC). Incorporation of phosphate into the poly(ethylene terephthalate) backbone rendered the co-polymers soluble in chloroform and biodegradable, lowered the Tg, decreased the crystallinity and increased the hydrophilicity. With an EOP/TC molar feed ratio of 80 : 20, the polymer exhibited good film-forming property, yielding at 86.6 ± 1.6% elongation with an elastic modulus of 13.76 ± 2.66 MPa. This polymer showed a favorable toxicity profile in vitro and good tissue biocompatibility in the muscular tissue of mice. In vitro the polymer lost 21% of mass in 21 days, but only 20% for up to 4 months in vivo. It showed no deterioration of properties after sterilization by γ -irradiation at 2.5 Mrad on solid CO2. Release of FITC-BSA from the microspheres was diffusion-controlled and exceeded 80% completion in two days. Release of the hydrophobic cyclosporine-A from microspheres was however much more sustained and near zero-ordered, discharging 60% in 70 days. A limited structure–property relationship has been established for this co-polymer series. The co-polymers became more hydrolytically labile as the phosphate component (EOP) was increased and the side chains were switched from the ethoxy to the methoxy structure. Converting the methoxy group to a sodium salt further increased the degradation rate significantly. The chain rigidity as reflected in the Tg values of the co-polymers decreased according to the following diol structure in the backbone: ethylene glycol > 2-methylpropylene diol > 2,2-dimethylpropylene diol. The wide range of physicochemical properties obtainable from this co-polymer series should help the design of degradable biomaterials for specific biomedical applications.
Keywords: Poly(terephthalate-co-phosphate), biodegradability, polycondensation, chain extension
Introduction
Advances in drug and gene delivery and tissue engineering continue to fuel the development of new biodegradable polymers. Polyesters [1], poly(amino acid)s [2, 3], polyamides [4], polyurethanes [5], poly(orthoester)s [6], poly(anhydride)s [7], poly(carbonate)s [8, 9], poly(iminocarbonate)s [10] and poly(phosphazene)s [11, 12] have been the prominent players. In drug delivery and tissue engineering scaffolding applications, poly(lactide-co-glycolide) co-polymers still dominate the field [13–16]. However, with an increasingly broadening utility for biodegradable drug-carriers and scaffolds, no one single material can be expected to satisfy all requirements of different applications. For instance, a coating material for a drug-eluting stent might require the polymer to be film-forming and elastomeric; or a fibrous tissue-engineering scaffold would demand a polymer with fiber-forming and strong mechanical properties. In this study, we evaluated the potential of a new series of poly(ester–phosphoester) co-polymers.
Polyphosphoesters represent a class of biodegradable polymers having a phosphoester linkage in the backbone, which would include poly(phosphate)s, poly(phosphonate)s and poly(phosphite)s [17–19]. Much of the research into the basic science of these polymers has been pioneered by Penczek and his colleagues in the 1970s [20–22]. The interesting characteristics of these polymers include: (1) versatility and tunable properties, the physicochemical and degradation properties can be adjusted by changing the backbone and the side-chain structures; (2) favorable physicochemical properties, the flexible phosphoester bond in the backbone provide the plasticizing effect to lower the glass transition temperature and confer good solvent solubility and processibility of the polymers; (3) biocompatibility, the ultimate hydrolytic breakdown products of a polyphosphoester are phosphates, alcohols and diols. A biocompatible polyphosphoester can be prepared from innocuous alcohol and diol building blocks.
We have previously tested the concept of improving the solubility and degradation behavior of poly(d,l-lactide) (PDLA) by forming a co-polymer between PDLA and polyphosphate [18, 23, 24]. The poly(lactide-co-ethylphosphate) shows a faster and more linear degradation profile than PDLA, and is soluble in non-halogenated solvents such as ethyl acetate [23]. As a drug carrier for paclitaxel (PACLIMER®), it showed promise in preclinical studies by improving the pharmacokinetics and toxicity of paclitaxel when the drug-loaded microspheres were injected intraperitoneally into dogs. The efficacy of this delivery system was also demonstrated in the local-regional therapy of lung cancer tumor nodules in mice [25]. Using similar approach and with an interest of tissue engineering scaffolding applications, we aimed to develop a co-polymer between polyphosphate and a polyester with strong mechanical properties. For that we targeted the extensively studied poly(ethylene terephthalate) (PET).
Poly(ethylene terephthalate) (PET) is one of the most commonly used biomedical polyesters, such as in vascular graft applications [26, 27]. Its appeal partly lies in its superb mechanical properties, which are derived from the liquid crystalline characteristics of the ethylene terephthalate structure. Hypothesizing that a biodegradable polymer with good mechanical properties can be built on this structure, we introduced phosphate units into PET. The general structure of poly(terephthalate-co-phosphate)s is shown in Fig. 1. In this paper, we primarily focus on poly(terephthalate-co-phosphate)s synthesized from BHET (Fig. 1, R = —CH2CH2—). The synthesis, characterization and structure–properties relationships have been investigated in this study. The in vitro cellular toxicity and in vivo tissue compatibility have been studied in details. Illustrative applications of poly(terephthalate-co-phosphate) for drug delivery in the form of microspheres and film are also included.
Figure 1.
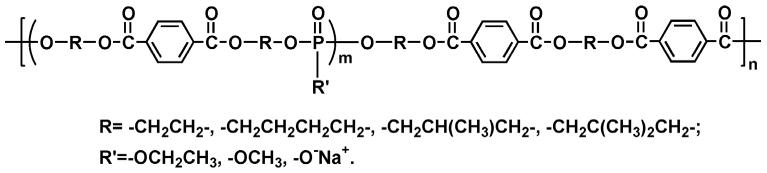
Structures of designed poly(terephthalate-co-phosphate)s, P(BHET-EOP/TC).
Materials and Methods
Terephthaloyl chloride, ethyl phosphorodichloridate, cobalt(II) acetate tetrahydrate, calcium acetate hydrate and all other reagents (if not specified) and solvents were purchased from Sigma-Aldrich (St. Louis, MO, USA). Terephthaloyl chloride was recrystallized from hexane before use, and ethyl phosphorodichloridate was redistilled. Dimethyl terephthalate and 4-dimethylaminopyridine (DMAP) were obtained from TCI America (Portland, OR, USA) and used without further purification. Cyclosporine A was obtained from Sigma.
Synthesis of 1,4-bis(2-hydroxyethyl) terephthalate (BHET)
Dimethyl terephthalate (277 g, 1.4 mol) and ethylene glycol (445 g, 7.2 mol) were weighed into a 1-l round bottom flask connected to a vacuum line. A catalytic amount of cobalt(II) acetate tetrahydrate (180 mg, 0.5 mol) and calcium acetate hydrate (90 mg, 0.4 mol) was added to prevent transesterification. The reaction mixture was heated at 160°C in an oil bath under mild vacuum and terminated after 18 h. While still molten, the mixture was precipitated into cold water. The precipitate was collected, dried under vacuum, redissolved into warm methanol and filtered to remove the sludge (oligomers). A precipitate formed when the solution was cooled to −20°C. After recrystallization from ethyl acetate and methanol, BHET was obtained as a white powder, mp. 108–110°C; Lit. 109–110°C [28], yield 68%.
Synthesis of poly(terephthalate-co-phosphate) P(BHET-EOP, 80 : 20)
Under argon stream, 10 g 1,4-bis(hydroxyethyl) terephthalate (BHET), 9.61 g 4-dimethylaminopyridine (DMAP) and 70 ml methylene chloride were transferred to a 250-ml flask equipped with an addition funnel. After BHET and DMAP were dissolved, the solution was cooled down to −40°C. A solution of 5.13 g ethyl phosphorodichloridate (EOP) in 20 ml methylene chloride was added dropwise through the funnel. Following the addition of EOP, the mixture was stirred at room temperature for 4 h and then a solution of 1.60 g terephthaloyl chloride (TC) in 20 ml methylene chloride was added drop-wise to the flask. The temperature was gradually brought up to 50°C, and allowed to react overnight. The solvent was removed by evaporation, and the residue was re-dissolved in 200 ml chloroform, washed with NaCl saturated solution three times and dried over Na2SO4. A white precipitate was obtained upon quenching the solution into cold methanol. The purification process was repeated twice, and then dried under vacuum at 60°C. The poly(terephthalate-co-phosphate) P(BHET-EOP, 80 : 20) was obtained as a tough, off-white solid, with a yield of 82%. Elemental analysis: Found % (Theoretical value, %): C: 49.61 (51.82); H: 4.95 (4.81); P: 6.24 (7.03). Mw = 7920, Mn = 6120 (GPC, polystyrene as standards).
A series of other P(BHET-EOP/TC) co-polymers with different molar ratios of phosphates in the backbone were prepared according to the procedure described above. These poly(terephthalate-co-phosphate)s were defined by the different feed ratios of EOP to TC, x/(100 − x) (see Table 1).
Table 1.
Composition of poly(terephthalate-co-phosphate)s calculated from 1H-NMR spectra
| Poly(terephthalate-co-phosphate) | EOP/TC ratio (Charged) | BHET-EOP/TC ratio* (Found) |
|---|---|---|
| P(BHET-EOP/TC, 75 : 25) | 75 : 25 | 75.6 : 24.4 |
| P(BHET-EOP/TC, 80 : 20) | 80 : 20 | 81 : 19 |
| P(BHET-EOP/TC, 82.5 : 17.5) | 82.5 : 17.5 | 82.5 : 17.5 |
| P(BHET-EOP/TC, 85 : 15) | 85 : 15 | 84 : 16 |
| P(BHET-EOP/TC, 87.5 : 12.5) | 87.5 : 12.5 | 87 : 13 |
| P(BHET-EOP/TC, 90 : 10) | 90 : 10 | 89 : 11 |
| P(BHET-EOP/TC, 95 : 5) | 95 : 5 | 96 : 4 |
For the convenience of calculation and peak assignment, the structure was rearranged as (also see Fig. 3 for details):
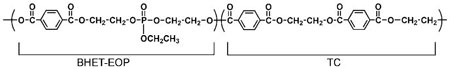
Synthesis of P(BHET-EOP/MOP/TC, 60 : 20 : 20)
To a cold (−40°C) solution of 20 g (78.66 mmol) BHET and 19.22 g (157.36 mmol) DMAP in 150 ml methylene chloride, was added drop-wise a solution of 7.69 g (47.20 mmol) ethyl phosphorodichloridate (EOP) and 2.34 g (15.73 mmol) methyl phosphorodichloridate (MOP) in 25 ml methylene chloride. The temperature of the mixture was brought up to refluxing gradually and maintained overnight. Another solution of 3.20 g terephthaloyl chloride (TC) in 20 ml methylene chloride was added through a funnel, and the mixture was refluxed for another 18 h. The mixture was washed three times with 200 ml of 0.1 M HCl saturated with NaCl, once with saturated NaCl solution and dried over anhydrous Na2SO4. The polymer was quenched out with ether and hexane, and obtained as white powder. Yield 79%, Mw = 9630, Mn = 4580 (GPC, polystyrene as standards).
Synthesis of P(BHET-EOP/MOP/TC, 40 : 40 : 20) was performed according to the same procedure. Yield 72%, Mw = 8020, Mn = 4580 (GPC, polystyrene as standards).
Synthesis of poly(terephthalate-co-phosphate) with negative charge
To a solution of 5 g P(BHET-EOP/MOP/TC, 60 : 20 : 20) in 50 ml chloroform, was added a solution of 0.858 g NaI in 25 ml acetone. The solution was refluxed under dried nitrogen or argon atmosphere overnight. The product was gradually precipitated. The pellet was washed and extracted extensively with acetone in a Soxhlet extractor. White powder was obtained after vacuum drying for 24 h.
Characterization
Chemical analysis
Gel-permeation chromatography (GPC) was performed on a Hewlett Packard 1090M liquid chromatograph. Chloroform was used as the mobile phase to elute the sample at a concentration of 2 mg/ml through a series of three PL gel polystyrene columns (102 Å, 103 Å and 105 Å pore size) (Hewlett Packard). Analysis was performed at 1 ml/min, 40°C and 270 nm (UV detection). The system was calibrated with monodispersed polystyrene standards (Polymer Laboratories). Fourier transfer infrared spectroscopy (FT-IR) was performed on a Perkin-Elmer 1600 Series machine. Samples were prepared by film casting on KBr pellets. 1H-NMR and 31P-NMR spectra were recorded on a Bruker AMX300 in CDCl3 or DMSO-d6, using H3PO4 as an external standard. Elemental analysis was performed by Galbraith Laboratories (Knoxville, TN, USA). The intrinsic viscosity in chloroform was measured in Ubbelohde viscometer (ASTM size OC) at 40°C.
Thermal properties
Differential scanning calorimetry was performed on a Seiko DSC220 at a heating rate of 10°C/min in sealed pans. Analysis was performed on a Seiko SDM 5500 Rheostation. The glass transition temperatures (Tg) were measured from the inflection points.
Contact angle analysis
Polymer thin film was deposited on coverslips (diameter of 10 mm, Corning) by spin-coating using 5% polymer solution in chloroform. Water-in-air contact angle was measured using a goniometer.
Tensile test
Polymer films were fabricated by casting polymers solution (15–20% in chloroform) in a Teflon mold. The film was air dried first, followed by extensive vacuum drying at 60°C for three days. The films were then cut into dumb-bell-shaped specimens and the tensile properties were analyzed on an Instron 5544 material testing system according to ASTM (American Society for Testing and Materials) method D638M.
Cytotoxicity of poly(terephthalate-co-phosphate)
Cytotoxicity of co-polymer by direct contact
The cytotoxicity of P(BHET-EOP, 80 : 20) was assessed by culturing HEK 293 (human embryonic kidney) cells on the P(BHET-EOP, 80 : 20) coated glass cover slips. Cell morphology and growth rate were determined after 72 h. As a control, HEK cells were also cultured directly on TCPS.
Cytotoxicity of co-polymer microspheres on GT3TKB tumor cells
P(BHET-EOP/TC, 80 : 20) microspheres were added to 96-well tissue-culture plates at different concentration, and then human gastric carcinoma cells (GT3TKB, RIKEN cell bank, Tsukuba, Japan) at 104 cells/well density were seeded. The cells were then incubated for 48 h at 37°C. Cell proliferation rate was analyzed by MTT assay. The results were plotted as % relative growth vs concentration of polymer microspheres in the tissue culture well.
Cytotoxicity of co-polymer degradation products
About 100–150 mg of P(BHET-EOP, 80 : 20) were degraded in 20 ml 1 M NaOH at 37°C for 1–2 days to ensure complete degradation. The solution was then neutralized with 1 M HCl. Poly(l-lactide) (PLLA, Mw = 14 000, Sigma) and poly(l-lysine) (PLL, Mw = 88 000, Sigma) were treated similarly and used as controls. About 200 μl of various concentrations of the degraded polymer products were placed in 96-well tissue culture plates, which were seeded with human gastric carcinoma cells (GT3TKB) at a density of 1 × 104 cells/well one day prior to the experiment. The degraded polymer products were incubated with the GT3TKB cells for 48 h. The results of the assay were plotted as % relative growth vs concentration of degraded polymer in the culture well.
Ames test and ISO agarose overlay tests
These two tests were performed at NamSA Laboratories (Northwood, OH, USA) according to the International Organization for Standardization ISO 10993-5 standard.
Degradation of poly(terephthalate-co-phosphate)s in PBS
P(BHET-EOP/TC, 80 : 20) and P(BHET-EOP/TC, 85 : 15) films were made by solution casting (15–20% in chloroform) and dried under vacuum for 3 days. Discs with 1 mm in thickness and 6 mm in diameter were cut from these sheets. Three discs of each co-polymer were placed in 4 ml of phosphate-buffered saline (PBS, 0.1 M; pH 7.4) at 37°C. The discs were taken out of the PBS at different points in time, washed with distilled water, and freeze-dried overnight. The samples were analyzed for change in molecular weight and mass loss over time.
In vivo tissue compatibility and degradation
P(BHET-EOP/TC, 80 : 20) discs, 8 mm in diameter, approx. 1 mm in thickness and 60–80 mg in weight, were prepared by solvent casting as described above. Poly(lactide-co-glycolide) (PLGA, RG755, Birmingham Polymers, Birmingham, AL, USA) was used as a control. PLGA discs, 8 mm in diameter, approx. 1.5 mm in thickness and 100 mg in weight were prepared by compression molding at room temperature using a manual Carver press. All discs were individually weighed, packaged in aluminum foil pouch and sterilized by γ -irradiation at 2.5 Mrad on dry ice. The initial molecular weights of the polymers were measured by GPC. Specific pathogen free (SPF) Sprague–Dawley rats (200–250 g) were randomly grouped and weighed individually before surgery. The sterilized discs were implanted into the muscles at the right hind limb of the rats under general anesthesia. The fascia over the implantation site was closed with a couple of stitches using 4-O Nylon suture to mark the location. The subcutaneous layer and skin were closed with absorbable suture (4-O Dexon). The rats were monitored for a few h post-operation and daily. At predetermined time points (days 3, 7, 14 and 1 month, 2, 4 and 6 months), three rats from each group were killed by CO2 inhalation. Blood was collected by cardiac puncture. Tissue samples from the implantation site were harvested, fixed in buffered formalin (10%), sectioned and stained with H&E for histological examination. The tissue reactions were evaluated using the criteria including inflammation and damage to the surrounding tissue. The extent of inflammation response at the implantation site was assessed by an independent pathologist. At each time point, polymer implants were retrieved from the rat muscle, rinsed with cold DI water several times to clean any attached tissue. Samples were lyophilized to constant weight and weight loss of the polymer disc comparing to its original weight was calculated.
Preparation of microspheres encapsulating fluorescein isothiocyanate-labeled bovine serum albumin (FITC-BSA)
FITC-BSA solution (10 mg/ml, 100 μl) was added to a solution of 100 mg P(BHET-EOP, 80 : 20) in 1 ml methylene chloride, and emulsified with a vortexer for 15 s on ice. The resulting emulsion was immediately poured into 5 ml 1% polyvinyl alcohol (PVA)–5% NaCl solution. The vortexing was continued for 1 min. The resulting emulsion was poured into 20 ml of an aqueous solution of 0.3% PVA and 5% NaCl, which was being stirred vigorously. Isopropanol solution (2%, 25 ml) was added, and the mixture was kept stirring for 1 h to ensure complete extraction. The microspheres were collected by centrifugation at 3000 × g, washed three times with water and lyophilized. Empty microspheres were prepared in the same way except that water was used as the inner aqueous phase.
Encapsulation efficiency was determined by comparing the quantity of FITC-BSA entrapped with the amount added to the emulsion. The amount entrapped was determined by measuring the quantity of FITC-BSA (assayed at 492 nm on a Shimadzu UV 160 spectrophotometer, Shimadzu North America, Columbia, MD, USA) remaining in the supernatant following the collection of the microspheres and subtracting it from the initial FITC-BSA amount.
To determine the loading level, 5 mg microspheres were digested in 1 ml 0.5 M NaOH for 6–8 h and assayed for FITC-BSA content at 492 nm in a UV spectrophotometer. The loading level was determined by assaying for FITC-BSA after hydrolyzing the microspheres with 0.5 M NaOH for 8 h. The loading level was calculated from the amount of drug incorporated and the initial weight of microspheres.
In vitro release of FITC-BSA from microspheres
5 mg P(BHET-EOP, 80 : 20) microspheres containing FITC-BSA was suspended into 1 ml PBS (pH 7.4) and placed into a 37°C shaker. At various time points, the suspension was spun at 3000 × g for 10 min and 500 μl of the supernatant was replaced by fresh PBS. The release of FITC-BSA from the microspheres was followed by measuring the fluorescence intensity at 519 nm on a Shimadzu UV 160 spectrophotometer.
Preparation of microspheres containing cyclosporine A
In order to determine the loading level and release kinetics of cyclosporine A, tritium-labeled cyclosporine A (Amersham-Pharmacia) was included in the microspheres. A solution of 200 mg P(BHET) in 1 ml methylene chloride was added to 20 mg cyclosporine A with 10 μCi radioactivity. The mixture was vortexed for 1 min, then added to 5 ml 1% PVA–5% NaCl solution while vortexing for 3 min. This mixture was added to 8 ml 0.3% PVA–5% NaCl solution while vortexing for 3 min. The microspheres were washed 5 times by centrifugation and resuspension in deionized water. The suspension was stirred in a magnetic stirrer overnight. The microspheres were suspended in 2% mannitol and lyophilized for at least 48 h.
Approximately 30 mg of polymer microspheres was weighed and placed in 10 ml 0.1 M PBS (pH 7.4) and placed in a shaking incubator at 37°C. At various time points, microspheres were centrifuged and the supernatant was replaced by fresh PBS. 200 μl of the supernatant was added to 2 ml of scintillation fluid and counted in a scintillation counter under a tritium specific program measuring counts per min. At the end of the study, the microspheres were dissolved in DMSO. 200 μl of this solution was added to 2 ml of scintillation fluid and counted similarly. The standard curve constructed with known amount of tritiated cyclosporine A was used to calculate the concentration of the released cyclosporine A.
Preparation of P(BHET-EOP/TC, 80 : 20) film containing paclitaxel
Solvent casting method was used to prepare the co-polymer film with or without 10% nominal paclitaxel loading. The film was air-dried followed by vacuum dried at 60°C for two days to remove any residue solvent that might influence the drug release rate (<0.05 to 5 ppm, depending upon the thickness of the film). The film was then cut into small discs with 6 mm in diameter and incubated in 1.2 ml of PBS (pH 7.4) layered with 0.3 ml of octanol. Discs with paclitaxel loaded were used as the negative control, and paclitaxel floated to the octanol layer was used as the positive control. At various time points, the octanol layer was collected and diluted 10 times with methanol, and analyzed on a HPLC system (Agilent Technologies) equipped with Zorbax Rx C18 column (4.6 mm × 15 cm) using a mixture mobile phase (water/methanol/acetonitrile, 37.5 : 50 : 12.5 (v/v/v)) at a flow rate of 1.5 ml/min. The release rate was calculated from the amount of paclitaxel released and the total amount of drug loaded.
Results and Discussion
Synthesis and structural characterization of poly(terephthalate-co-phosphate)s
Our initial attempt was to synthesize a polyphosphoester (P(BHET-EOP)) by polycondensing 1,4-bis(hydroxyethyl) terephthalate (BHET) with ethyl phosphorodichloridate (EOP) at stoichiometric ratios (Fig. 2). The resulting P(BHET-EOP) was a very viscous semi-solid at room temperature. The Tg of P(BHET-EOP) was 19°C, with a water-in-air contact angle of 8°. To raise the Tg and improve mechanical properties of the polymer, the scheme shown in Fig. 3 was developed to reduce the phosphoester content in the polymer. This two step polymerization scheme led to the synthesis of a series of poly(terephthalate-co-phosphate)s with different contents of phosphoester component (EOP). In this scheme, BHET was first reacted with EOP to yield a hydroxyl-terminated prepolymer, which was further polymerized with terephthaloyl chloride (TC). The BHET/EOP/TC ratio was controlled to be 100 : x : (100 − x). The ratio in the notation of the P(BHET-EOP/TC) series (Fig. 1) refers to the molar ratio of EOP to TC (x/(100 − x)).
Figure 2.
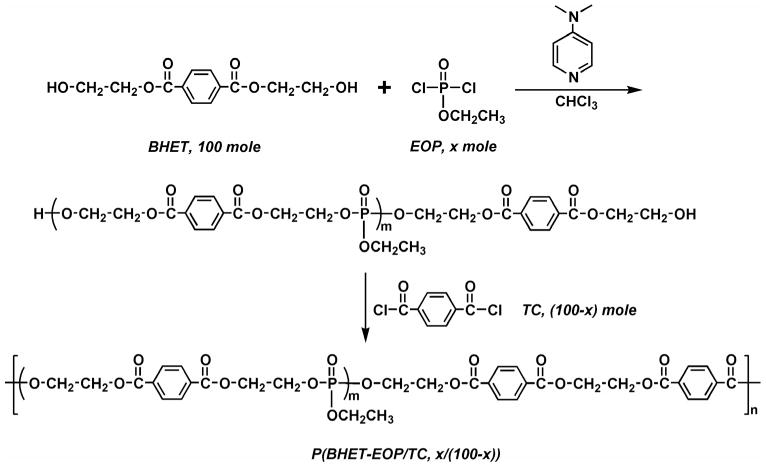
Synthetic scheme of P(BHET-EOP).
Figure 3.
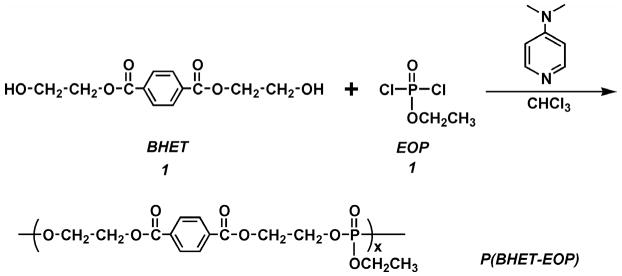
Synthetic scheme of poly(terephthalate-co-phosphate)s, P(BHET-EOP/TC).
The addition sequence and the ratio of reactive chlorides affected the molecular weight and solubility of the polymers. Adding the terephthaloyl chloride first or adding the two reactive dichlorides at the same time yielded a polymer that was only slightly soluble in DMF and DMSO, but not in chloroform or methylene chloride. The polymers had high crystallinity with a Tm of 184°C. We hypothesize that the higher reactivity of the TC monomer compared with EOP towards the hydroxyl group might have produced longer oligo-terephthalate blocks in the backbone to account for this high Tm. The two-step polycondensation shown in Fig. 3 prevented the formation of long segments of oligo-terephthalate, rendering the co-polymers with good solubility in common organic solvents (chloroform, methylene chloride and NMP) and relatively higher molecular weights. The solubility of the co-polymers decreased as the phosphate content decreased. When the EOP to TC feeding ratio was 1, the resulting co-polymer P(BHET-EOP/TC, 50 : 50) became insoluble in chloroform. Table 2 summarizes the solubility property vs EOP/TC feeding ratio.
Table 2.
Solubility of poly(terephthalate-co-phosphate)s
| Co-polymer | CHCl3 | CH2Cl2 | NMP | DMF | DMSO |
|---|---|---|---|---|---|
| P(BHET-EOP) | +++ | +++ | ++ | ++ | ++ |
| P(BHET-EOP/TC, 95 : 5) | +++ | +++ | ++ | ++ | ++ |
| P(BHET-EOP/TC, 90 : 10) | +++ | +++ | ++ | ++ | ++ |
| P(BHET-EOP/TC, 85 : 15) | ++ | ++ | ++ | ++ | ++ |
| P(BHET-EOP/TC, 80 : 20) | ++ | ++ | ++ | ++ | ++ |
| P(BHET-EOP/TC, 50 : 50) | — | — | +/Δ | +/Δ | +/Δ |
NMP, N-methylpyrrolidone; DMF, dimethylformamide; DMSO, dimethylsulfoxide; +++, readily soluble; ++, soluble; +/Δ, soluble on heating.
The structures of these co-polymers were confirmed by 1H-NMR, 31P-NMR and FT-IR spectra. A set of spectra for P(BHET-EOP : TC, 80/20) is shown in Fig. 4. The 1H-NMR chemical shift assignments were listed in Fig. 4a. Using ratio of integrations of peaks δ 4.5 ppm (or δ 4.4 ppm) and δ 4.7 ppm, we could calculate the phosphate to terephthalate contents. The compositions of poly(terephthalate-co-phosphate)s as calculated by this method were fairly consistent with the feeding ratios of the monomers (Table 1). The 31P-NMR spectrum of P(BHET-EOP/TC) showed one single peak at δ 2.49 ppm. This indicated that there were very few terminal phosphate groups. The FT-IR spectrum of P(BHET-EOP/TC, 80 : 20) revealed the characteristic absorption peaks for carbonyl groups (1721 cm−1, stretching), P=O groups (1274 cm−1, stretching) and P—O—C groups (1035 cm–1, P—O stretching) (data not shown). All other co-polymers showed similar spectrum characteristics.
Figure 4.
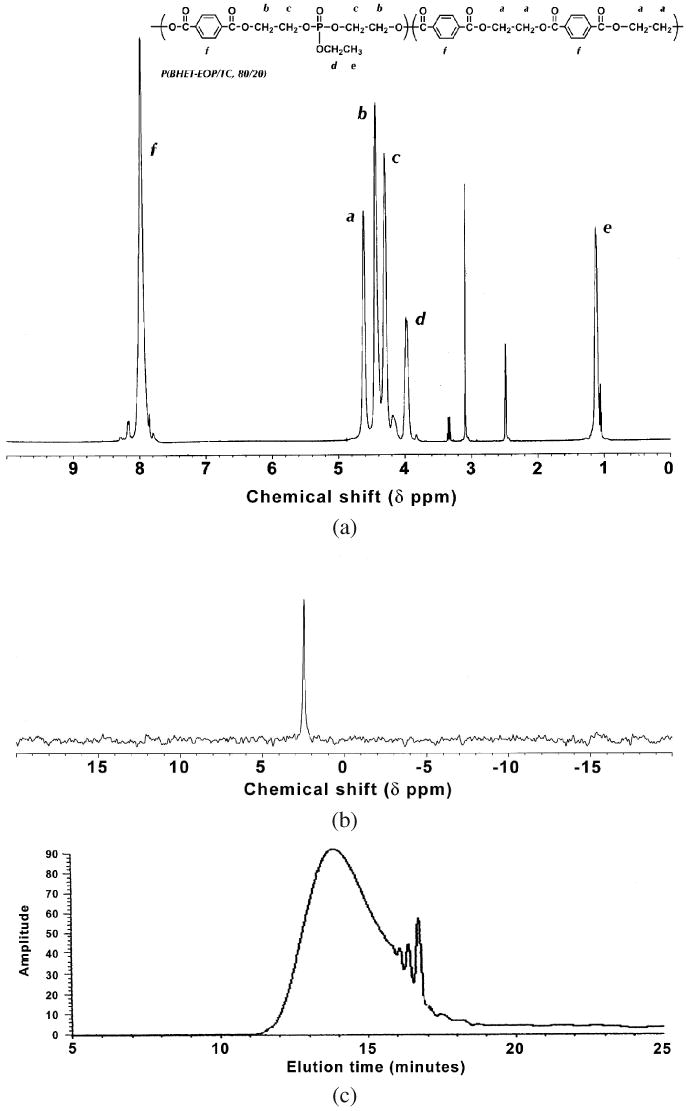
(a) 1H-NMR spectrum for P(BHET-EOP/TC, 80 : 20). (b) 31P-NMR spectrum for P(BHET-EOP/TC, 80 : 20). (c) GPC chromatogram for P(BHET-EOP/TC, 80 : 20).
GPC analysis of the co-polymer showed a polydispersed distribution typical of polycondensation products (Fig. 4c). The average molecular weight of poly(terephthalate-co-phosphate)s increased with the TC content, which was also confirmed by the intrinsic viscosity data (Table 3). This confirms that the reaction of the extended diol with terephthaloyl chloride is more energetic than that with ethyl phosphorodichloridate. Interestingly, we found that a 2–5% excess of EOP yielded a higher molecular weight polymer. This might be due to the high susceptibility of EOP degradation to moisture. In addition, increasing the batch size significantly increases the molecular weight of the resulting polymer (Table 3). For example, in the synthesis of P(BHET-EOP/TC, 80 : 20), increasing the amount of BHET from 10 g to 100 g increased the weight average molecular weight by 7-fold to 62 585. This is most likely because at larger batch size, it is much easier to maintain the stoichiometry of BHET : (EOP + TC), which is crucial to obtaining a high-molecular-weight polymer through a polycondensation scheme.
Table 3.
Intrinsic viscosity and molecular weights of poly(terephthalate-co-phosphate)s determined by GPC analysis using polystyrene as standards
| Poly(terephthalate-co-phosphate) | Batch size | Mw | Mn | [η] (dl/g) |
|---|---|---|---|---|
| P(BHET-EOP/TC, 80 : 20) | S | 7920 | 6120 | 0.180 |
| M | 15 520 | 6322 | N.D. | |
| L | 62 585 | 32 940 | N.D. | |
| P(BHET-EOP/TC, 85 : 15) | S | 5180 | 2270 | 0.146 |
| P(BHET-EOP/TC, 90 : 10) | S | 4200 | 1590 | 0.148 |
| M | 14 050 | 4790 | N.D. | |
| P(BHET-EOP/TC, 95 : 5) | S | 1480 | 600 | 0.089 |
Batch size was defined according to the amount of BHET used as starting material. S: 10–20 g of BHET, M: 50 g of BHET and L: 100 g of BHET. Mw and Mn were calculated by GPC using polystyrene as standard. Intrinsic viscosity was measured at 40°C in chloroform using an Ubbelohde viscometer.
A unique feature of this series of co-polymers, as compared to other commonly used biodegradable polymers such as poly(lactide-co-glycolide)s, is the availability of functional side groups. This is an advantage of the pentavalency of the phosphorus atom in the backbone, allowing the chemical linkage of drug molecules or other agents to the polymer [29]. For instance, drugs with hydroxyl groups may be coupled to the phosphorus via an ester bond, which is hydrolyzable. Cell-adhesion molecules can be conjugated to the surface of the poly(terephthalate-co-phosphate) scaffolds, improving the cell adhesion for tissue-engineering applications [30]. These poly(phosphoester)s are natural candidates for a completely biodegradable pendant delivery system. In this study, we investigated the feasibility of introducing phosphate ion to the polymer chains.
Phosphate ion incorporating polymer was prepared by synthesizing co-polymers with methoxy side chains and then cleaving the methoxy side chain with NaI [30]. Replacing all the EOP with methyl phosphorodichloridate (MOP) yielded a co-polymer P(BHET-MOP/TC, 80 : 20) with relatively low molecular weight, maybe due to the increased instability of MOP compared with EOP (reacting to moisture). This change also dramatically increased the hydrophilicity of the polymer (data not shown). We here reported two co-polymers prepared with MOP equivalent to 40% (mol) and 20% (mol) of BHET used in the polymerization, respectively (Fig. 5). The molecular weights of these two co-polymers were slightly lower than P(BHET-EOP/TC, 80 : 20). Furthermore, converting the methoxy groups into sodium salt renders the polymer less soluble in chloroform and methylene chloride [30].
Figure 5.
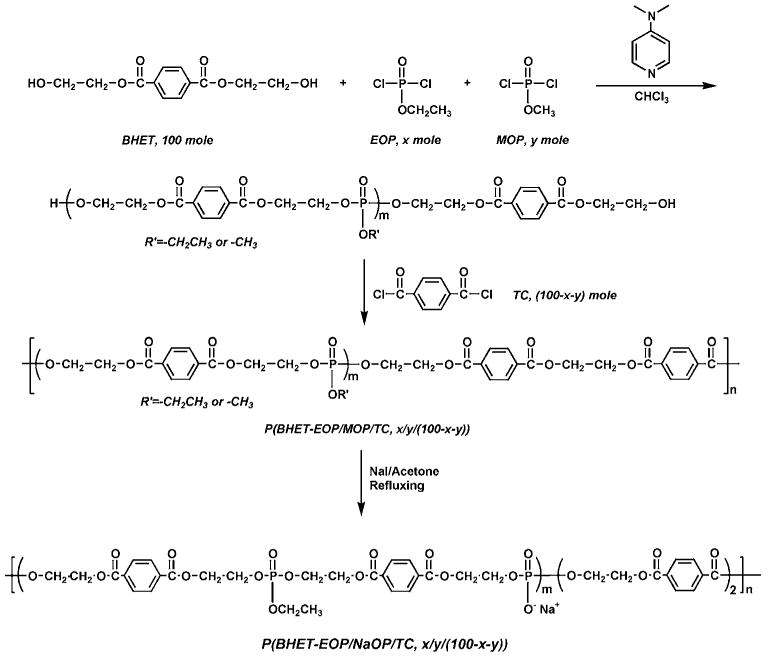
Synthetic scheme of P(BHET-EOP/MOP/TC) and P(BHET-EOP/NaOP/TC).
Physicochemical properties of poly(terephthalate-co-phosphate)s
Dacron (PET) is a highly crystallized polymer (Tm = 265°C, Tg = 69°C) [31]. Introduction of ethyl phosphate bond into the backbone significantly changes the thermal properties of the polymer. In this P(BHET-EOP/TC) co-polymer series, when the EOP/TC ratio was higher than 50 : 50, the PPEs were amorphous. No melting point (Tm) was observed in differential scanning calorimetric (DSC) analysis. The co-polymers began to soften at around 150°C. When the EOP/TC charging ratio was reduced to 50 : 50, DSC graph showed a melting peak at 201°C, indicating the presence of crystalline phase.
As expected, the glass transition temperature (Tg) increased as the EOP/TC feeding ratio decreased. Nevertheless, the Tg increment was minor until the EOP/TC ratio decreased to below 75 : 25. There was a dramatic increase of Tg from P(BHET-EOP/TC, 80 : 20) (Tg = 25°C) to P(BHET-EOP/TC, 50 : 50) (Tg = 64°C). This significant increase is hypothesized to be related to the crystalline phase in P(BHET-EOP/TC, 50 : 50). Figure 6a and 6b shows the DSC curves for these two co-polymers. The Tg values of a series of other P(BHET-EOP/TC) co-polymers of differing EOP/TC feed ratios were determined, and the results are shown in Fig. 6c and in Table 4.
Figure 6.
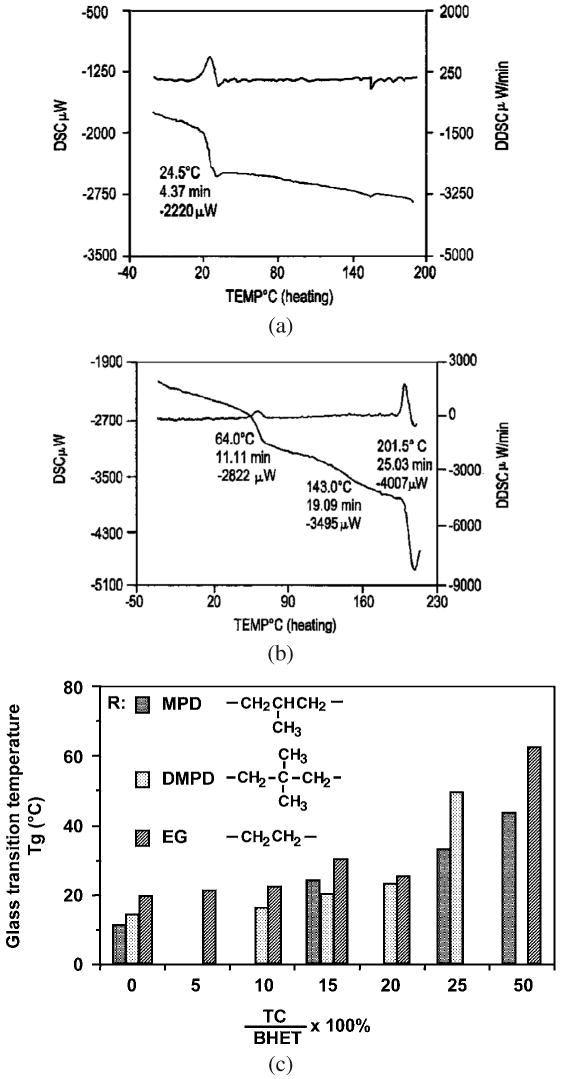
(a) DSC graph for P(BHET-EOP/TC, 80 : 20). (b) DSC graph for P(BHET-EOP/TC, 50 : 50). (c) Glass transition temperature as a function of terephthalate (or phosphate) content in poly(terephthalate-co-phosphate) and the structure of the diol residue in the co-polymer backbone.
Table 4.
Glass transition temperatures and water-in-air contact angles of selected poly(terephthalate-co-phosphate)s
| Poly(terephthalate-co-phosphate) | Tg (°C) | Water-in-air contact angle (deg) |
|---|---|---|
| P(BHET-EOP/TC, 50 : 50) | 64 | N.D. |
| P(BHET-EOP/TC, 80 : 20) | 25 | 65 |
| P(BHET-EOP/TC, 85 : 15) | 28 | 54 |
| P(BHET-EOP/TC, 90 : 10) | 21 | N.D. |
| P(BHET-EOP/TC, 95 : 5) | 21 | 10 |
| P(BHET-EOP/TC, 100 : 0) | 19 | 8 |
| P(BHET-EOP/MOP/TC, 60 : 60 : 20) | 32 | 14 |
| P(BHET-EOP/MOP/TC, 40 : 40 : 20) | 39 | 9 |
| P(BHET-NaOP/TC, 80 : 20) | 58 (Tm = 180) | N.D. |
| P(BHET-EOP/NaOP/TC, 60 : 20 : 20) | 52 | N.D. |
| P(BHET-EOP/NaOP/TC, 40 : 40 : 20) | 63 | N.D. |
Introducing sterically hindered diol to the polymer backbone, by substituting ethylene glycol with 2-methylpropylene diol (MPD) or 2,2-dimethylpropylene diol (DMPD) residues, reduced its glass transition temperature (Fig. 6c). The Tg values of the co-polymers with different aliphatic diol residues decreased in the order of diol residue sequence: EG > DMPD > MPD. This result indicated that the co-polymer chain rigidity reduced in the same order.
The relative hydrophobicity of the P(BHET-EOP/TC) series was assessed by measuring the water-in-air contact angle. The contact angle decreased dramatically from 65° for P(BHET-EOP/TC, 80 : 20) to 8° for P(BHET-EOP). While the charging molar ratio of EOP/TC was maintained at 80 : 20, substituting the ethoxy side chain with methoxy side chain (MOP) also markedly increased the hydrophilicity (Table 4). At the ratio of EOP/MOP/TC of 40 : 40 : 20, the contact angle decreased to a value close to that of P(BHET-EOP).
Furthermore, introducing negative charges to phosphate sidechains by converting the methoxy groups into sodium salt rendered the polymer less soluble in chloroform and methylene chloride. The negatively charged P(BHET-ENaOP/TC)s became hydrophilic by gross observation. As expected, the methoxy group substituted P(BHET-EOP/TC) and the negatively charged poly(terephthalate-co-phosphate)s had much higher Tg values than their parent polymers (Table 4). P(BHET-NaOP/TC, 80 : 20) has a clear sharp melting peak at 180°C as determined by DSC.
Poly(terephthalate-co-phosphate)s in general had good film-forming properties, particularly at EOP/TC feeding ratios of 80 : 20 and 85 : 15. These two co-polymers formed elastomeric films with different thickness by solvent casting. Poly(terephthalate-co-phosphate)s showed typical tensile properties for amorphous or low crystalline thermoplastics. At an EOP/TC ratio higher than 90 : 10, the film became weak and difficult to handle. P(BHET-EOP/TC, 80 : 20) film yielded at 86.6 ± 1.6% elongation (Fig. 7), with a modulus of elasticity of 13.76±2.66 MPa. Substituting EOP with MOP partially (40 : 40 or 60 : 20, see Table 5) did not affect the film forming property significantly, except that the film became more brittle and less ductile. P(BHET-EMOP/TC)s had nearly 100% higher moduli of elasticity than P(BHET-EOP/TC, 80 : 20). In addition, fibers could be drawn from co-polymer melts at 160–180°C from all three co-polymers listed in Table 5 (with a 20% molar ratio of TC).
Figure 7.
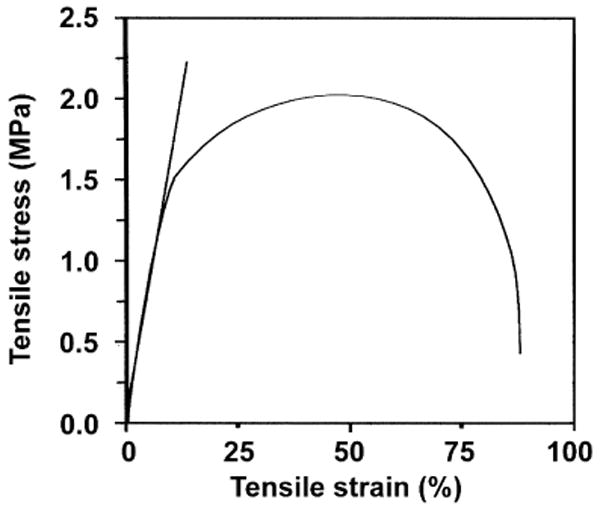
Tensile stress–strain curve for P(BHET-EOP/TC, 80 : 20) thin film prepared by the solvent casting method.
Table 5.
Tensile properties of selected poly(terephthalate-co-phosphate)s
| Poly(terephthalate-co-phosphate) | Tensile stress at yield (MPa) | Elongation at break (%) | Modulus of elasticity (MPa) |
|---|---|---|---|
| P(BHET-EOP/TC, 80 : 20) | 1.47 ± 0.31 | 86.6 ± 1.6 | 13.76 ± 2.66 |
| P(BHET-EOP/MOP/TC, 60 : 20 : 20) | 2.55 ± 0.47 | 74.6 ± 8.7 | 26.77 ± 2.19 |
| P(BHET-EOP/MOP/TC, 40 : 40 : 20) | 2.79 ± 1.01 | 48.9 ± 1.6 | 27.59 ± 9.36 |
Due to the above described favorable properties of P(BHET-EOP/TC, 80 : 20) and P(BHET-EOP/TC, 85 : 15), we focused on this polymer in the evaluation of cytotoxicity, degradation, tissue compatibility and drug-release kinetics.
Cytotoxicity of poly(terephthalate-co-phosphate)s
The cytotoxicity of P(BHET-EOP/TC, 80 : 20), as a representative, was assessed by three cell culture assays: (1) cytotoxicity of the polymer itself by directly culturing cells on the co-polymer thin film; (2) cytotoxicity of the polymer microspheres to cells because of potential applications in drug delivery; (3) cytotoxicity of the degradable products of the co-polymer.
The first assay was performed by culturing human embryonic kidney cells (HEK293 cell line, ATCC) on co-polymer-coated coverslips. After 3 days of culture, HEK293 cells exhibited a normal morphology that was comparable to the TCPS control group; they also proliferated at a rate comparable to the control (Fig. 8a).
Figure 8.
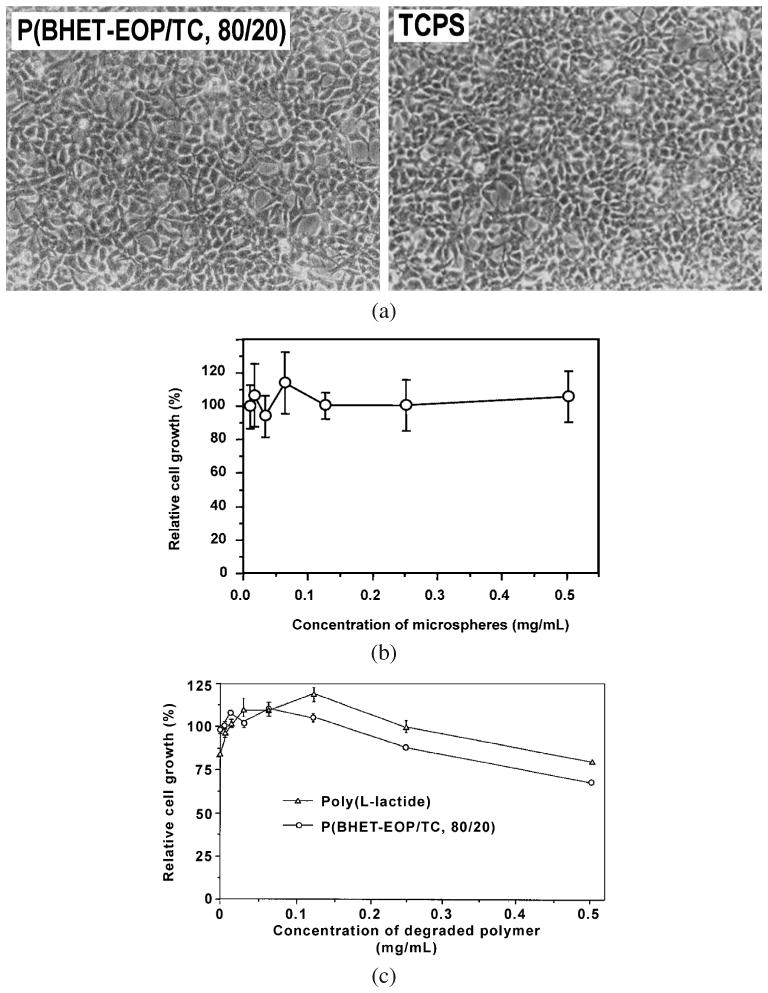
(a) Phase-contrast images of HEK293 cells cultured on P(BHET-EOP/TC, 80 : 20) coated cover slip in comparison with that cultured on tissue-culture polystyrene surface. (b) Effect of P(BHET-EOP/TC, 80 : 20) microspheres (2–10 μm) on the proliferation of human GT3TKB gastric tumor cells in reference to cells cultured on tissue culture plate. (c) Effect of the degradation products of P(BHET-EOP/TC, 80 : 20) and PLLA on the proliferation of human GT3TKB gastric tumor cells in reference to cells cultured on tissue culture plate.
The second assay on cytotoxicity of co-polymer microspheres was most relevant to particulate-mediated drug delivery, where the polymer microspheres might or might not be taken up by the cells. When cultured with polymer microspheres (<10 μm), the proliferation assay on human gastric carcinoma cells (GT3TKB) showed no inhibition of cell growth for concentration of P(BHET-EOP/TC, 80 : 20) microspheres up to at least 1.0 mg/ml (Fig. 8b).
The degradation products of P(BHET-EOP/TC, 80 : 20) and poly(l-lactide) did not show any inhibitory effect on cellular proliferation at doses lower than 125 μg/ml (Fig. 8c). At higher doses (0.25–0.5 mg/ml) both P(BHET-EOP/TC, 80 : 20) and PDLA did show some inhibition. In separate Ames test and ISO agarose overlay test, neither mutagenicity nor cytotoxicity was observed for P(BHET-EOP/TC, 80 : 20), P(BHET-EOP/TC, 85 : 15) and the BHET monomer (data not shown). These results indicated that there no toxic leachable or degradation products produced from polymers and BHET, or the leached products could not diffuse through the agarose layer.
Degradation and stability of poly(terephthalate-co-phosphate)s
Figure 9a shows the cumulative mass loss of P(BHET-EOP/TC, 80 : 20) and P(BHET-EOP/TC, 85 : 15) discs at 37°C in 0.1 M PBS. P(BHET-EOP/TC, 80 : 20) degraded at a slower rate than P(BHET-EOP/TC, 85 : 15), reflecting the influence of the phosphate in accelerating the degradation. The former showed a gradual increase of mass loss over time, losing 21% total mass after 18 days of incubation. The latter had a 22% mass loss after 24 h of incubation, followed by a gradual increase to about 43% on day 18. SEM analysis of the cross-sections of the samples on day 18 indicated that P(BHET-EOP/TC, 80 : 20) swelled much less than P(BHET-EOP/TC, 85 : 15). P(BHET-EOP/TC, 85 : 15) discs developed a thick swollen degradation layer, compared with P(BHET-EOP/TC, 80 : 20) discs (Fig. 9b). GPC analysis of the recovered samples on day 18 showed that the molecular weights of P(BHET-EOP/TC, 80 : 20) and P(BHET-EOP/TC, 85 : 15) discs had declined by 21% and 43% of their original values, respectively. Figure 9c shows a GPC chromatograph of P(BHET-EOP/TC, 80 : 20) sample after incubation for 2 weeks at 37°C in 0.1 M PBS. The GPC trace of the degraded sample resembled that of the original sample, but shifted towards the lower molecular weight direction. This result suggested that the degradation of P(BHET-EOP/TC, 80 : 20) occurred as a relatively uniform random chain scission. Given the fact that the phosphoester bond in the backbone is more susceptible to hydrolysis than the terephthalate ester bond, this finding is consistent with the previous conclusion that this co-polymer is largely amorphous and the distribution of the phosphate groups in the backbone is relatively uniform and random throughout the polymer chain.
Figure 9.
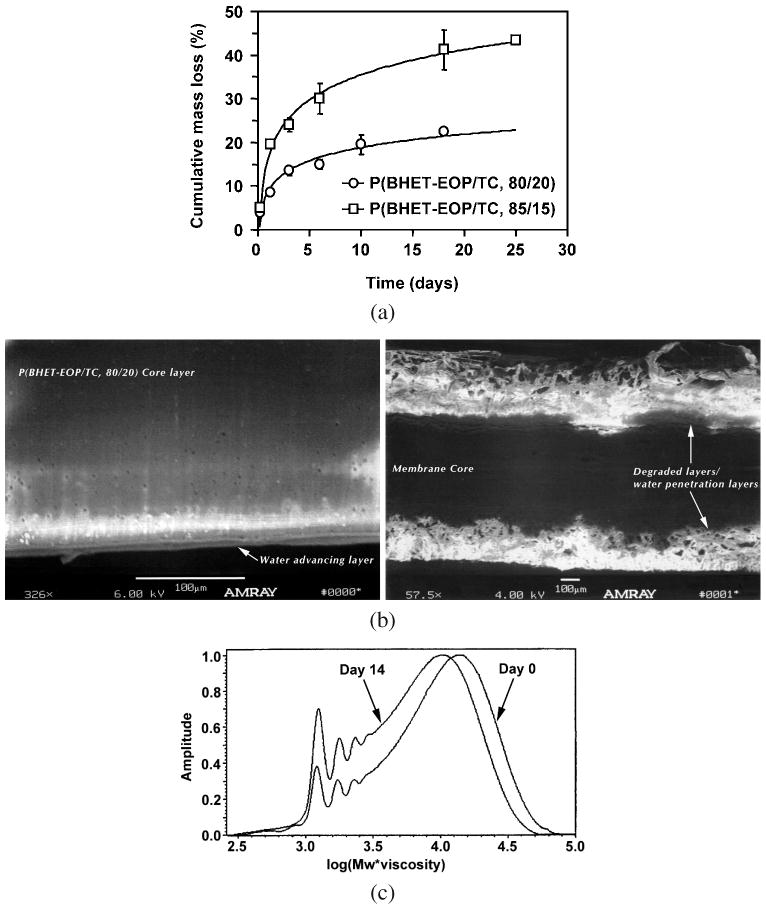
(a) In vitro degradation of P(BHET-EOP/TC) discs in 0.1 M PBS at 37°C. (b) SEM images of P(BHET-EOP/TC, 80 : 20) (left panel) disc and P(BHET-EOP/TC, 85 : 15) disc (right panel) after incubation in 0.1 M PBS at 37°C for 18 days. (c) GPC analysis of P(BHET-EOP/TC, 80 : 20) disc after incubation in 0.1 M PBS at 37°C for 14 days, in comparison with the original sample.
This degradation study highlighted the hydrolytic sensitivity of the co-polymer to the phosphate content in the backbone. Increasing the TC content in the polymer would significantly retard the degradation rate. It is worth noting that molecular weight is also an important parameter affecting the degradation rate of the co-polymer. Apparently, higher molecular weight PPEs with the same composition degraded at a slower rate than that with a lower Mw. For example, P(BHET-EOP/TC, 80 : 20) with a Mw of 14 300 showed only a 5% weight loss in 14 days of incubation under the same condition (data not shown).
As discussed above, in the P(BHET-EOP/TC, 80 : 20) series, substituting ethoxy with methoxy group yielded co-polymers with increased hydrophilicity, which in turn accelerated the in vitro degradation as demonstrated in Fig. 10. Furthermore, converting the methoxy group to a sodium salt dramatically increased the degradation rate.
Figure 10.
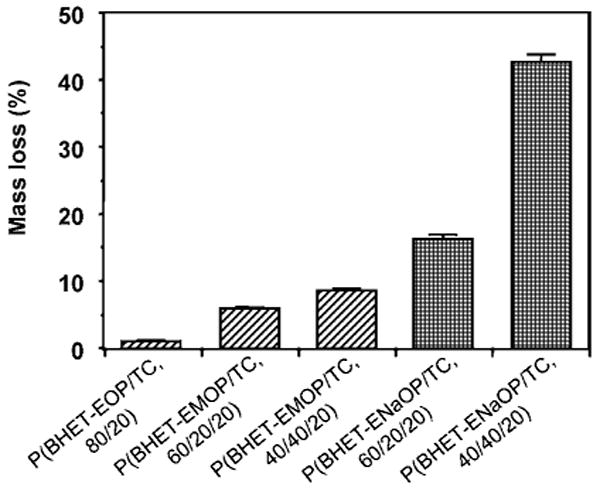
Mass loss of P(BHET-EOP/TC, 80 : 20) discs after incubation in 0.1 M PBS at 37°C. Molecular weights of the sample tested are: P(BHET-EOP/TC, 80 : 20), Mw = 12 500; P(BHET-EOP/MOP/TC, 60 : 20 : 20), Mw = 9350; P(BHET-EOP/MOP/TC, 40 : 40 : 20), Mw = 7830. No Mw data were available for P(BHET-EOP/NaOP/TC, 60 : 20 : 20) and P(BHET-EOP/NaOP/TC, 40 : 40 : 20).
These data demonstrated the feasibility of fine-tuning the degradation rate of the co-polymers by adjusting the EOP/TC ratio, changing the side-chain structure (ethyl group vs methyl group) and controlling the molecular weight of the co-polymers. The co-polymers became more hydrolytically labile as the phosphate component (EOP) was increased and the side chains were switched from the ethoxy to the methoxy structure.
Storage stability and sterilization stability
Designed to be biodegradable, poly(terephthalate-co-phosphate)s are naturally susceptible to hydrolysis, even in air (to moisture in the air). Nevertheless, compared with other degradable polymers, for example, aliphatic polyanhydrides, poly(terephthalate-co-phosphate)s were relatively stable when stored under dry condition, even without vacuum or inert gas packaging. When P(BHET-EOP/TC, 80 : 20) was stored in a desiccator at room temperature, the molecular weight drop was insignificant after one month (data not shown).
Sterilizability of a biomedical polymeric material often determines its clinical utility. When P(BHET-EOP/TC, 80 : 20) was γ -irradiated at 2.5 Mrad in an aluminum foil pouch on solid CO2, neither the chemical structure nor the molecular weight were altered, as monitored by GPC, intrinsic viscosity measurement and FT-IR (data not shown).
In vivo degradation and biocompatibility
The biodegradation and biocompatibility of poly(terephthalate-co-phosphate)s were evaluated in a SPF Sprague–Dawley rat model by implanting polymer samples in the right hind limb muscle. P(BHET-EOP/TC, 80 : 20) was investigated as a representative compared with PLGA (75 : 25, RG755). Tissue reaction surrounding the implantation site was characterized by the average monocyte and macrophage count per observation field under 400×. Four areas per side were counted and the arithmetic mean was calculated and plotted against implantation time in Fig. 11. The inflammation response in the surrounding tissue was rated according to the following scoring system: Score 0 for ‘no irritation’; Score 1–200 for ‘slight irritation’; Score 201–400 for ‘mild irritation’; Score 401–600 for ‘moderate irritation’ and score of 601 and more for severe irritation. As illustrated in Fig. 11, inflammation reactions to P(BHET-EOP/TC, 80 : 20) implants were similar to that of PLGA wafers, and were characterized as slightly irritating from day 3 to the 6-month time point. Hematology data, differential leukocyte counts, cellular morphology findings and clinical chemistry values were overall unremarkable and comparable between the two groups at each interval. No histomorphological change in all major organs at the 6-month time point was observed (data not shown).
Figure 11.
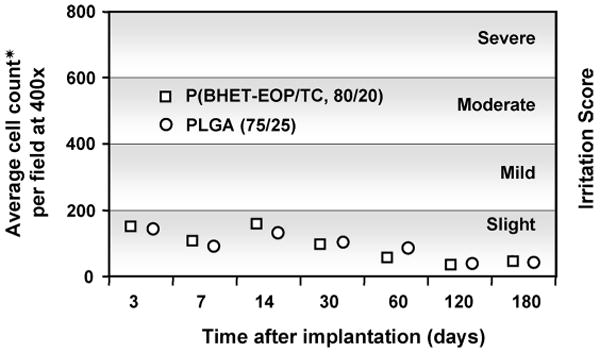
Tissue reaction scores (average monocyte and macrophage counts per field, magnification 400×) for poly(phosphoester) discs implanted between the muscle layers of the hind limb of male Sprague–Dawley rats were plotted as a function of implantation time. Four areas per side were counted and the arithmetic mean was shown in the figure. Tissue reaction was graded according to the following system: no irritation (0); slight irritation (1–200); mild irritation (201–400); moderate irritation (401–600); severe irritation (>601).
The analysis of polymer discs retrieved at different time points showed a typical biphasic degradation behavior of PLGA (Fig. 12). P(BHET-EOP/TC, 80 : 20), on the other hand, showed approx. 20% mass loss in 2 weeks, and remained unchanged for up to 4 months. This degradation behavior is speculated to be a reflection of the poor solubility of the di-terephthalate units in the physiological fluid. With higher phosphate content in the backbone, as in P(BHET-EOP/TC, 85 : 15), which not only increases the hydrophilicity but also reduces the di-terephthalate segments in the breakdown products, the in vitro degradation study showed a mass loss of over 40% in 25 days. As discussed above for the in vitro degradation rate of the P(BHET-EOP/TC)s, it is expected that their in vivo degradation rates would be accelerated by increasing the phosphate content in the backbone.
Figure 12.
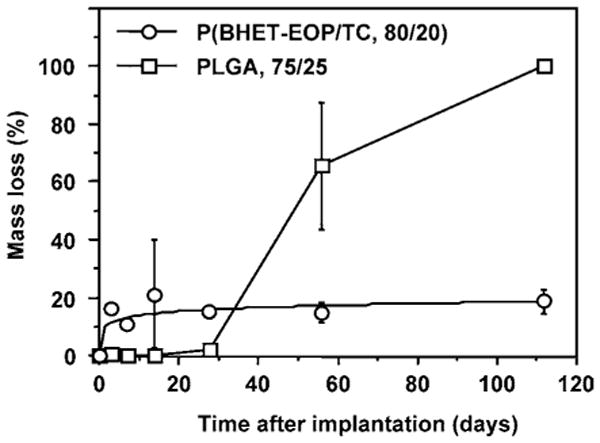
Mass loss of P(BHET-EOP/TC, 80 : 20) and PLGA (75 : 25) discs following implantation in the hind limb of male Sprague–Dawley rats.
FITC-labeled BSA-containing microspheres: preparation and release kinetics
P(BHET-EOP/TC, 80 : 20) microspheres were prepared via a double-emulsion solvent-extraction method using FITC-BSA as a model protein drug. The preparation procedure was adapted from Cleek et al. [32] and Uchida et al. [33] with some modifications. The preparation parameters were optimized to increase the encapsulation efficiency, improve the microsphere morphology and minimize the burst release of protein drug. Three different batches of microspheres with different loading levels (1.5, 14.1 and 22.8 wt%) were prepared. Under the optimal conditions as described in Materials and Methods, the size of the microspheres prepared with P(BHET-EOP/TC, 80 : 20) ranged between 2 and 20 μm with a smooth surface morphology (Fig. 13a). At lower loading level (1–5%), the protein (FITC-BSA) encapsulation efficiency was about 90%. At higher loading level (10–20%), the encapsulation efficiency decreased slightly to 70–80%.
Figure 13.
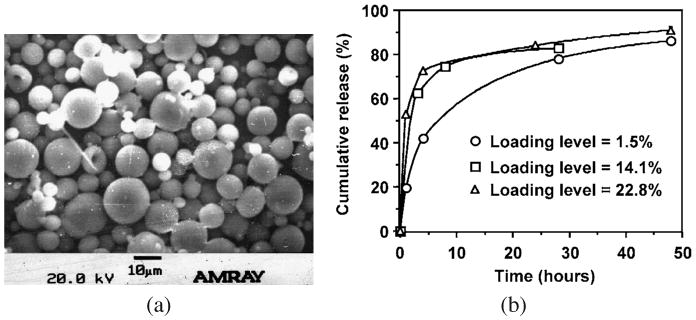
(a) SEM image of P(BHET-EOP/TC, 80 : 20) microspheres with 1.5% loading of FITC-BSA. (b) Effect of loading level on the release kinetics of FITC-BSA from P(BHET-EOP/TC, 80 : 20) microspheres. Mw of P(BHET-EOP/TC, 80 : 20) used in this study was 8200 (polystyrene as standards).
Figure 13b shows the release profiles of FITC-BSA from the three batches of P(BHET-EOP/TC, 80 : 20) microspheres with different loading levels. The in vitro release of FITC-BSA was relatively fast for all three batches of microspheres. Microspheres with loading levels of 14.1% or 22.8% released about 80% of the encapsulated protein within the first day, with a burst of 60–70% in the first 4 h. This was followed by a much lower release rate during the next 24 h. There remained about 5% of FITC-BSA in the microspheres, even after 10 days of release at 37°C in PBS (data not shown in the figure). Microspheres with a loading level of 1.5% showed a more gradual release kinetics, with minimal burst.
Cyclosporine-A-containing microspheres: preparation and release kinetics
The release kinetics of cyclosporine A are shown in Fig. 14a. The release was sustained with no burst effect. About 60% of the encapsulated cyclosporine A was released by day 70. This led to a projected release time of approx. 120 days in PBS at an average release rate of 0.2 μg/day per mg microspheres.
Figure 14.
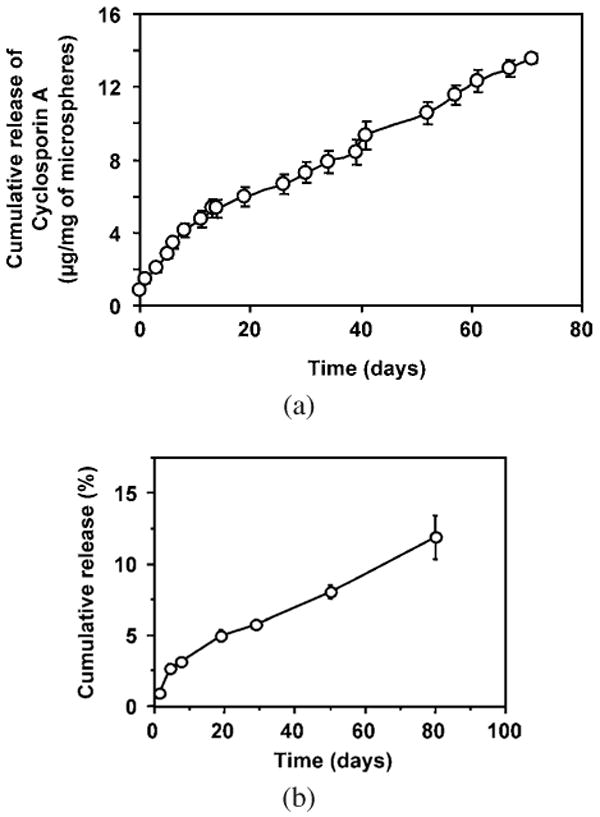
(a) Cumulative release of cyclosporine A (loading level of 2.3%) from P(BHET-EOP/TC, 80 : 20) microspheres in 0.1 M PBS at 37°C. (b) Cumulative release of paclitaxel (loading level of 10%) from P(BHET-EOP/TC, 80 : 20) microspheres in 0.1 M PBS at 37°C.
Cyclosporine-A-encapsulated P(BHET-EOP/TC, 80 : 20) microspheres were prepared for a potential application of treating chronic uveitis, which is a blinding disease that may require long-term treatment [34]. Cyclosporine A is a 1.2-kDa cyclic peptide that is poorly insoluble in water, facilitating the nearly linear release profile observed in the study. The drug has been shown in several human trials to be effective in treating chronic uveitis, especially that associated with Bechet's disease [34]. However, side effects from systemic administration, including nephrotoxic effects and hypertension, limit its use [35]. The systemic toxic effects of cyclosporine A could be minimized by delivering the drug locally. Direct intravitreal injection has been shown to prevent experimental autoimmune uveitis, but the intravitreal half-life of 6 h [36, 37] would require repeated injections of cyclosporine A. A non-biodegradable sustained release device for implantation to the vitreous has been developed [36, 38]. There are several limitations of this system primarily relating to the size (2.5 mm). The implantation procedure carries a significant risk of retinal detachment and requires replacement after the drug is depleted. The replacement surgery is further complicated by a high rate of tissue encapsulation of the device.
A microsphere formulation described here would offer an attractive alternative. Microspheres prepared by this method were small in size (<10 μm) with an aim to reduce the blockage of vision when injected in the vitreous. Using a single emulsion procedure, a drug loading level of 2.3% was obtained. The release rate of 0.2 μg/day per mg microspheres obtained in this study is comparable to that achieved by a non-biodegradable system implanted in the vitreous, which maintains a vitreous cyclosporine A level between 0.3 and 0.5 μg/ml [36, 38]. In addition, we have established that the presence of up to 5 mg of P(BHET-EOP/TC, 80 : 20) microspheres in the vitreous was biocompatible and did not affect retinal function (data not shown). These results demonstrated that this poly(terephthalate-co-phosphate) microsphere system would be a promising approach to delivery cyclosporine A intravitreously.
Paclitaxel-containing film: preparation and release kinetics
Paclitaxel (10 wt%) was embedded in P(BHET-EOP/TC, 80 : 20) film (approx. 0.7 mm) during solvent casting. The film was flexible with a smooth surface. DSC and X-ray analyses did not show any crystallinity of the drug in the film. This film could deliver paclitaxel for several months (Fig. 14b) at a constant rate of 0.15% per day. This corresponded to an average of 15 μg paclitaxel per day for a 100-mg film (approx. 0.7 mm). A total of 12% of paclitaxel was released for 80 days. During the same period, less than 30% of mass loss of the co-polymer was observed.
This paclitaxel release profile remarkably resembled the release profile of cyclosporine A from P(BHET-EOP/TC, 80 : 20) microspheres, despite the vastly different surface area and different drug loading levels. This indicates that the release mechanism of these two drugs is drug-dissolution controlled, suggesting that many drug release configurations would work for sustained release of paclitaxel.
Conclusions
In summary, a series of new biodegradable poly(terephthalate-co-phosphate)s with chemical structures analogous to PET, one of the most popular polymeric biomaterials in medical use today, was developed. Incorporation of phosphate into the poly(ethylene terephthalate) backbone rendered the co-polymers soluble in chloroform and biodegradable, lowered the Tg, decreased the crystallinity and increased the hydrophilicity. The co-polymers became more hydrolytically labile as the phosphate component (EOP) was increased and the side chains were switched from the ethoxy to the methoxy structure. Detailed physico-chemical and biological characterization was performed on a co-polymer synthesized with an EOP/TC molar feed ratio of 80 : 20. This polymer showed good film-forming property, stability against γ -irradiation, a favorable toxicity profile in vitro and good tissue biocompatibility in the muscular tissue of mice. With interesting physico-chemical and biological properties, this class of biodegradable and versatile poly(ester-phosphoester) co-polymers should find utility in a diverse range of biomedical applications, particularly drug delivery and tissue engineering.
Acknowledgments
This research was partially supported by Guilford Pharmaceuticals and by NIH/R21EB003203. The authors would like to thank the technical assistance of Bart S. Hendriks, David P. Kao (Johns Hopkins University) and Dr. Bing Wang (Guilford Pharmaceuticals Inc.).
References
- 1.Atkins TW, Peacock SJ. J Biomater Sci Polymer Edn. 1996;7:1065. doi: 10.1163/156856296x00552. [DOI] [PubMed] [Google Scholar]
- 2.Kohn J, Langer R. J Am Chem Soc. 1987;109:817. [Google Scholar]
- 3.Ogawa Y. J Biomater Sci Polymer Edn. 1997;8:391. doi: 10.1163/156856297x00173. [DOI] [PubMed] [Google Scholar]
- 4.Gachard I, Bechaouch S, Coutin B, Sekiguchi H. Polym Bull. 1997;38:427. [Google Scholar]
- 5.Dahiyat BI, Hostin E, Posadas EM, Leong KW. J Biomater Sci Polymer Edn. 1993;4:529. doi: 10.1163/156856293x00186. [DOI] [PubMed] [Google Scholar]
- 6.Heller J, Sparer RV, Zentner GM. In: Biodegradable Polymers as Drug Delivery Systems. Chasin M, Langer R, editors. Marcel Dekker; New York, NY: 1990. p. 121. [Google Scholar]
- 7.Leong KW, Domb A, Ron E, Langer R. In: Encyclopedia of Polymer Science Engineering. Kroschwitz JI, editor. Wiley; New York, NY: 1989. [Google Scholar]
- 8.Sen Gupta A, Lopina ST. Mater Sci Forum. 2003;426-4:3261. [Google Scholar]
- 9.Kojima T, Nakano M, Juni K, Inoue S, Yoshida Y. Chem Pharm Bull (Tokyo) 1985;33:5119. doi: 10.1248/cpb.33.5119. [DOI] [PubMed] [Google Scholar]
- 10.Pulapura S, Li C, Kohn J. Biomaterials. 1990;11:666. doi: 10.1016/0142-9612(90)90025-l. [DOI] [PubMed] [Google Scholar]
- 11.Allcock HR. In: Bioerodible Polymers as Drug Delivery Systems. Chasin M, Langer R, editors. Marcel Dekker; New York, NY: 1990. p. 163. [Google Scholar]
- 12.Allcock HR, Pucher SR, Scopelianos AG. Biomaterials. 1994;15:563. doi: 10.1016/0142-9612(94)90205-4. [DOI] [PubMed] [Google Scholar]
- 13.Pitt CG, Chasalow FI, Hibionada YM. J Appl Polym Sci. 1981;26:3779. [Google Scholar]
- 14.Li SM. J Biomed Mater Res. 1999;48:342. doi: 10.1002/(sici)1097-4636(1999)48:3<342::aid-jbm20>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 15.Davis SS, Illum L, Stolnik S. Curr Opin Colloid Interf Sci. 1996;1:660. [Google Scholar]
- 16.Edlund U, Albertsson AC. Adv Polym Sci. 2002;67:229. [Google Scholar]
- 17.Richards M, Dahiyat B, Arm DM, Brown P, Leong KW. J Biomed Mater Res. 1991;25:1151. doi: 10.1002/jbm.820250908. [DOI] [PubMed] [Google Scholar]
- 18.Zhao Z, Wang J, Mao HQ, Leong KW. Adv Drug Deliv Rev. 2003;55:483. doi: 10.1016/s0169-409x(03)00040-1. [DOI] [PubMed] [Google Scholar]
- 19.Leong KW, Mao HQ, Zhuo RX. Chin J Polym Sci. 1995;13:289. [Google Scholar]
- 20.Penczek S, Pretula J, Kaluzynski K. Polish J Chem. 2001;75:1171. [Google Scholar]
- 21.Penczek S, Lapienis G, Kaluzynski K, Nyk A. Polish J Chem. 1994;68:2129. [Google Scholar]
- 22.Penczek S, Klosinski P, Narebska A, Wodzki R. Makromol Chem Macromol Symp. 1991;48-49:1. [Google Scholar]
- 23.Mao HQ, Dang W, Shipanova-Kadiyala I, Zhao Z, Cline D, Leong KW. Proceedings of the American Institute of Chemical Engineers Topical Conference: Biomaterials Carriers, Drug Delivery Scaffolds, Tissue Engineering; 1997. p. 193. [Google Scholar]
- 24.Chaubal MV, Su G, Spicer E, Dang WB, Branham KE, English JP, Zhao Z. J Biomater Sci Polymer Edn. 2003;14:45. doi: 10.1163/15685620360511137. [DOI] [PubMed] [Google Scholar]
- 25.Harper E, Dang W, Lapidus RG, Garver RI., Jr Clin Cancer Res. 1999;5:4242. [PubMed] [Google Scholar]
- 26.Tunstall A, Eberhart RC, Prager MD. J Biomed Mater Res. 1995;29:1193. doi: 10.1002/jbm.820291006. [DOI] [PubMed] [Google Scholar]
- 27.Aronoff MS. J Biomater Appl. 1995;9:205. doi: 10.1177/088532829500900303. [DOI] [PubMed] [Google Scholar]
- 28.Lyoo WS, Lee SG, Ha WS, Lee J, Kim JH. Polym Bull. 1999;42:9. [Google Scholar]
- 29.Brosse JC, Derouet D, Fontaine L, Chairatanathavorn S. Makromol Chem. 1989;190:2339. [Google Scholar]
- 30.Wan ACA, Mao HQ, Wang S, Phua SH, Lee GP, Pan J, Lu S, Wang J, Leong KW. J Biomed Mater Res Appl Biomater. 2004;70B:91. doi: 10.1002/jbm.b.30022. [DOI] [PubMed] [Google Scholar]
- 31.Brandrup J, Immergut EH, editors. Polymer Handbook. Wiley-Interscience; New York, NY: 1975. [Google Scholar]
- 32.Cleek RL, Rege AA, Denner LA, Eskin SG, Mikos AG. J Biomed Mater Res. 1997;35:525. doi: 10.1002/(sici)1097-4636(19970615)35:4<525::aid-jbm12>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 33.Uchida T, Yoshida K, Ninomiya A, Goto S. Chem Pharm Bull (Tokyo) 1995;43:1569. doi: 10.1248/cpb.43.1569. [DOI] [PubMed] [Google Scholar]
- 34.Sullu Y, Oge I, Erkan D, Ariturk N, Mohajeri F. Acta Ophthalmol Scand. 1998;76:96. doi: 10.1034/j.1600-0420.1998.760118.x. [DOI] [PubMed] [Google Scholar]
- 35.Kato Y, Numaga J, Kato S, Kaburaki T, Kawashima H, Fujino Y. Clin Exp Ophthalmol. 2001;29:335. doi: 10.1046/j.1442-9071.2001.00445.x. [DOI] [PubMed] [Google Scholar]
- 36.Jaffe GJ, Yang CS, Wang XC, Cousins SW, Gallemore RP, Ashton P. Ophthalmology. 1998;105:46. doi: 10.1016/s0161-6420(98)91176-9. [DOI] [PubMed] [Google Scholar]
- 37.Pearson PA, Jaffe GJ, Martin DF, Cordahi GJ, Grossniklaus H, Schmeisser ET, Ashton P. Arch Ophthalmol. 1996;114:311. doi: 10.1001/archopht.1996.01100130307014. [DOI] [PubMed] [Google Scholar]
- 38.Gilger BC, Malok E, Stewart T, Horohov D, Ashton P, Smith T, Jaffe GJ, Allen JB. Vet Immunol Immunopathol. 2000;76:239. doi: 10.1016/s0165-2427(00)00219-1. [DOI] [PubMed] [Google Scholar]