

Abstract
Hepatic stellate cells (HSCs) undergo myofibroblastic trans-differentiation in liver fibrogenesis. We previously showed that dual stimulation with three-dimensional type-I collagen and interleukin-1 (IL-1) synergistically induces HSC trans-differentiation in a manner dependent on the activation of matrix met-allopreinase-9 (MMP-9). The present study is aimed to determine the mechanism of MMP-9 activation in this model. The pro-MMP-9-converting activities expressed by trans-differentiating HSCs are characterized as secreted factors that are sensitive to MMP inhibitor and have apparent molecular masses of 50 and 25 kDa. This is in sharp contrast to the pro-MMP-9 activator from mouse and human skin, which is a chymotrypsin-like proteinase. Among multiple MMPs induced in HSCs by the dual stimulation, MMP-13 is most conspicuously up-regulated and meets all criteria as the pro-MMP-9 activator. HSC cultured in three-dimensional type-I collagen, but not in Matrigel, IL-1 induces expression of MMP-13 and its matured form at 50 and 25 kDa, respectively. In vitro reconstitution experiment proves that MMP-13, but not its zymogen, activates pro-MMP-9. Further, short hairpin RNA targeting MMP-13 abolishes pro-MMP-9 activation and HSC trans-differentiation. We further demonstrate that pro-MMP-13 activation is facilitated with a membrane-associated factor, inhibited with tissue inhibitor of metalloproteinase-2, and abolished with short hairpin RNA against MMP-14. Moreover, pro-MMP-13 is also activated by a secreted factor, which is absorbed by gelatin-Sepharose and reconstituted with MMP-9. Thus, IL-1-induced trans-differentiation of HSCs in three-dimensional extracellular matrix is facilitated by an MMP activation cascade (MMP-14 > MMP-13 > MMP-9) and a positive feedback loop of MMP-9 > MMP-13, suggesting their critical roles in liver injury and repair.
Residing in the perisinusoidal space of Disse between the sinusoidal endothelium and hepatocytes, hepatic stellate cells (HSCs)2 are vitamin A-storing and ECM-producing cells (1, 2). In normal liver, HSCs are embraced within loose ECM composed of the basement membrane components mostly produced by these cells themselves (1, 2). In response to liver injury, HSCs undergo trans-differentiation (activation) to myofibroblastic cells that may produce excessive interstitial ECM (3–7). Accompanying the trans-differentiation is the gradual replacement of the basement membrane-like ECM milieu in the space of Disse by fibrillar collagen-rich fibers produced by trans-differentiated, myofibroblastic HSCs. Despite an intensifying need for the development of new therapeutic modalities for cirrhosis and decades of research on HSCs, the key questions still remain as to how HSCs retain their quiescence in normal liver and how the homeostasis is disrupted to provoke activation of HSCs within the perisinusoidal ECM environment. Isolated HSCs undergo spontaneous activation when cultured on plastic (8, 9). Thus, it is conceivable that the basement ECM acquires and maintains HSC at quiescence in normal liver; conversely, ECM degradation is critical to provoke the onset of HSC activation (10). The original observation of the correlation of MMP with liver fibrosis was reported almost three decades ago (11). The temporal association of MMPs with liver fibrosis was further defined in greater detail, and was tied to the initiation of fibrogenesis by the non-parenchymal hepatic cells (12–14). Very recently, MMP-13 deficiency was shown to attenuate liver fibrogenesis (15).
Given the unique ECM-rich architecture in the space of Disse we have designed our study to recapitulate such milieu by culturing isolated HSCs in three-dimensional ECM scaffolds. The resting state of HSCs in normal liver was reproduced by culturing isolated HSCs in three-dimensional basement membrane ECM (Matrigel), and initiation of perisinusoidal fibrosis was simulated by replacing Matrigel with type I collagen. Development of liver fibrosis is closely associated with persistent inflammation (16–18). We speculated that the acute phase cytokines might directly initiate the activation of HSCs. To this end, we tested a role of acute phase cytokines in this three-dimensional culture model. Such work has led to our finding that dual stimulation by IL-1 and type-I collagen results in most conspicuous ECM degradation and HSC trans-differentiation (19). This finding is novel and remarkable in that HSCs have been shown to serve as a major source of multiple MMPs required for ECM degradation and consequent HSC trans-differentiation.
Many secreted MMPs are nearly absent in healthy, resting tissues, although they are expressed in “active” tissues in which ECM breakdown is indispensable, for instance, during development, tumor metastasis, tissue injury, and repair (20). Nascent MMPs are made dormant through the interaction between a conserved cysteine in the N terminus pro-domain and a zinc ion chelated by the conserved histidines within catalytic domains. Activation is normally mediated by cleavage of the prodomain, leading to reorganization of the zinc cysteine interaction, a process called “cysteine switch” (21). Based on in vitro reconstitution with purified proteins, MMP-3 was proposed as a potential pro-MMP-9 activator (22). A similar approach showed that plasmin is an upstream activator of the pro-MMP-3 and down-regulation of TIMP-1 is essentially required (23, 24). On the other hand, transformed mast cells activate pro-MMP-9 by using chymase (25, 26). The role of chymase in activation of pro-MMP-9 is confirmed by mice deficient in a subtype of chymase (27, 28). In human skin, stimulation with tumor necrosis factor-α or IL-1 results in activation of pro-MMP-9 through an unrecognized chymotrypsin-like proteinase tightly attached to skin tissue (29, 30). Nevertheless, how pro-MMP-9 is activated in other organs is largely unknown. We have shown that HSCs activate pro-MMP-9 during their trans-differentiation process in response to a very selective condition of dual stimulation by IL-1 and three-dimensional type-I collagen. Once converted to myofibroblasts, HSCs mostly cease to produce MMPs, thus shifting the equilibrium to accumulation of ECM in fibrosis (19).
In this report we delineated a cascade of pro-MMP-9 activation by HSCs under the dual stimulation with IL-1 and type-I collagen. We started with profiling potential candidates by their temporal correlation with pro-MMP-9 activation. We characterized that the pro-MMP-9-converting activators are soluble factors with molecular masses of 50 and 25 kDa. The converting enzymes are sensitive to inhibitors for MMPs but not serine proteinases. These results promoted us to examine the MMPs expressed by HSCs in the three-dimensional culture. Such analysis pointed to MMP-13 as a candidate that satisfied all criteria as a pro-MMP-9 activator produced by HSCs. We proved this by in vitro reconstitution and RNA inference-mediated knockdown experiments. We also identified MMP-14 and MMP-9 as two independent activators of pro-MMP-13. In support of the roles of these MMPs in HSC trans-differentiation, we demonstrated that HSCs from either MMP-9 knock-out mice or those with MMP-13 knockdown failed to be activated by the dual stimulation. Thus, this study illustrates a complex MMP activation cascade in IL-1 induced trans-differentiation of HSCs within ECM as an example of MMP interactions in tissue injury and repair.
EXPERIMENTAL PROCEDURES
Materials
MMP inhibitor (catalogue number 444247), type-IV collagenase assay kit, pro- and MMP-13 were purchased from Calbiochem. Antibodies for rat MMP-13 (MAB13426), MMP-14 (MAB3317), and TIMP-2 protein (CC1064) were from Chemicon (Temecula, CA). The fluorescein isothiocyanate-conjugated monoclonal antibody against α-SMA was from Sigma-Aldrich. The kit for adenoviral-based shRNA knockdown, including pENTR/U6 plasmid, was purchased from Invitrogen. Type-I collagen was from Cohesion Technologies (Palo Alto, CA), and IL-1α was from R&D Systems (Minneapolis, MN). The pro-MMP-9 was expressed by keratinocytes transduced with lentivirus to express human MMP-9. The proteinase in the conditioned medium was purified by gelatin-conjugated Sepharose 4B as previously described (31).
Determination of the Molecular Weight of Pro-MMP-9-converting Activators
Conditioned medium was prepared by primary rat HSCs cultured in three-dimensional type-I collagen and treated with IL-1α (1 ng/ml) for 5 days. After depletion of endogenous gelatinases by passing through a column of gelatin conjugated Sepharose-4B, the conditioned medium was concentrated by precipitation with ammonia sulfate at 85% saturation. The protein was dissolved in NT buffer (100 mM NaCl and 50 mM Tris at pH 7.5), filtered before loading to Superdex-200 (GE Health Sciences), and resolved by fast-protein liquid chromatography. Fractions were monitored by absorbance at A280, and their molecular weights were calibrated by standard proteins. The pro-MMP-9-converting activities were measured by incubation of 30-μl fractions, 15 μl of purified human pro-MMP-9, and 5 μl of 50 mM CaCl2 at 37 °C for 16 h. Then the reactions were resolved by zymography. Conversion of pro-MMP-9 was measured by the ratio of 82-kDa MMP-9 versus the 92-kDa zymogen. The molecular weight was also measured by resolving the conditioned medium in non-reducing SDS-PAGE. Briefly, the gelatinase-depleted and concentrated conditioned medium was resolved by SDS-PAGE (12.5% w/v) in the cold room. Then the gel was sliced into 20 pieces according to molecular weight. The gel slices were incubated with 1% Triton X-100 in NT buffer to remove residual SDS. After the gel slices were ground in 35 μl of NT buffer, the reactions were made active by adding 15 μl of pro-MMP-9 and 5 μl of CaCl2 followed by incubation at 37 °C for 16 h. Finally, the reactions were resolved by zymography.
Isolation of Mouse HSCs and Culture in Three-dimensional ECM
Isolation and culture of rat HSCs have been previously described (32). MMP-9-deficient and wild-type FVB mice were purchased from Jackson Laboratory (Bar Harbor, ME). Isolation of mouse HSCs was similar to the procedure for isolation of rat HSCs except the following: liver perfusion from the superior vena cava instead of the portal vein, reduced concentrations of Pronase and collagenase, and adjusted speeds for low-speed and ultracentrifugation. Briefly, the mouse liver was subjected to perfusion with 50 ml of 0.4% Pronase for 12 min followed by 0.02% collagenase for 8 min in a circulating water bath at 37 °C. Then the collapsed liver tissue was minced, suspended in 40-ml solution of 10 μg/ml DNase, and shaken for 10 min at 37 °C. After removal of parenchymal cells by centrifugation at 500 ×g for 1 min, non-parenchymal liver cells were collected by additional centrifugation at 1,500 ×g for 8 min. The cell suspension was subjected to gradient centrifugation. The stellate cells were collected from the second top layer between 1.034 and 1.043 g/cm3. After washing, stellate cells were seeded in a culture dish in high glucose Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum, 2 mM L-glutamine, and antibiotics penicillin and streptomycin. Purity of HSCs was judged by comparison of cell number under microscopy of phase contrast and auto-fluorescence excited by UV light. After 2-day recovery, the cells were embedded in three-dimensional ECM as described previously (19).
Cell Treatment and Quantitative RT-PCR
Isolated rat HSCs (day 2) were either plated on culture dishes or embedded in three-dimensional type-I collagen as described previously. Cells were treated with or without IL-1α (1 ng/ml) in Dulbecco’s modified Eagle’s medium with 5% fetal bovine serum for 16 h. Total RNA was extracted using TRIzol reagent according to the manufacturer’s instructions (Invitrogen). First strand cDNA was produced using First-Strand cDNA Synthesis by SuperScript II reverse transcriptase with random primers. Two micrograms of total RNA was used for each reverse transcription reaction mixture (20 μl). Real-time PCR was carried out using an ABI Prism 7900HT (Applied Biosystems). 10-μl reactions were carried out in a 384-well PCR plate using the following final concentrations: 1 μmol each of forward and reverse primers, 1× SYBR Green master mix (qPCR Mastermix Plus for SYBR Green I, Eurogentec), and 5 ng of cDNA. For each condition triplicates were used to minimize the variation. Cycling conditions were as follows: initial step (50 °C for 2 min), hot activation (95 °C for 10 min), amplification (95 °C for 15 s and 60 °C for 1 min) repeated 40 times, and quantification with a single fluorescence measurement. Data were analyzed using ABI Prism SDS 2.1 software. The relative gene expression was calculated by the ΔΔCt method. Briefly, the resultant mRNA was normalized to its own GAPDH. Final results were expressed as n-fold difference in gene expression relative to GAPDH mRNA and calibrator as follows: n-fold = 2–(ΔCt sample – ΔCt GAPDH), where ΔCt values of the sample and the calibrator were determined by subtracting the average Ct value of the transcript under investigation from the average Ct value of the GAPDH gene for each sample. For rat MMP-9 the forward primer has the sequence CAG ACC AAG GGT ACA GCC TGT, and the reverse primer is AGC GCA TGG CCG AAC TC. For rat GAPDH the forward and backward primers are CCT GGA GAA ACC TGC CAA GTA T and CTC GGC CGC CTG CTT. The sequence information for the primers of other MMPs and TIMPs will be released upon request.
Immunofluorescence Staining
After being fixed with cold methanol for 20 min, the cells in three-dimensional ECM were permeabilized by 0.15% Triton X-100 in phosphate-buffered saline for 15 min. Blocked with 5% nonfat milk in phosphate-buffered saline, the cells were incubated with fluorescein iso-thiocyanate-conjugated monoclonal antibody against α-SMA (1:100, Sigma). Nuclei were stained by 4′,6-diamidino-2-phen-ylindole (1 μg/ml) for 5 min. Images were captured by a Nikon Eclipse TE2000-U, a digital camera from Roper Scientific Photometrics, and Metamorph software.
Knockdown of MMP-13 and MMP-14
The shRNA for rat MMP-13 and -14 was designed according to the BLOCK-iT RNAi Designer (Invitrogen). The inserts for shRNA targeting of the N-terminal region of rat MMP-13 have the following sequences (construct AB): forward, 5′-CACCGCAGAGCACTACTTGAA-ATCACGAATGATTTCAAGTAGTGCTCTGC-3′ and backward, 5′-AAAAGCAGAGCACTACTTGAAATCATTCGTG-ATTTCAAGTAGTGCTCTGC-3′. Another set of shRNA against the catalytic region of rat MMP-13 (construct CD) has the following sequences: forward, 5′-CACCGCCAGAA-CTTCCCAACCATGTCGAAACATGGTTGGGAAGTTC-TGGC-3′ and backward, 5′-AAAAGCCAGAACTTCCCAA-CCATGTTTCGACATGGTTGGGAAGTTCTGGC-3′. For shRNA against rat MMP-14, the construct AB has the following sequence: forward, CACCGTCTAGGAATCACATTCCGT-TCCTTTCGAAAAAGGAACGGAATGTGATTCCTAGA; backward, AAAATCTAGGAATCACATTCCGTTCCTTTTT-CGAAAGGAACGGAATGTGATTCCTAGAC. Construct CD for MMP-14 shRNA has the following sequence: forward, CAC-CGTCATGATCTTGTTTGCTGAGGGTTTCGAAAAACCC-TCAGCAAACAAGATCATGA; backward, AAAATCATGAT-CTTGTTTGCTGAGGGTTTTTCGAAACCCTCAGCAAA-CAAGATCATGAC. A control vector that expresses shRNA for lacZ was also constructed with the insert sequence according to the manufacturer’s instruction. Both the plasmid (U6 promoter)- and adenovirus-based knockdown procedures have been applied. For plasmid-mediated knockdown, rat HSCs were plated in a 6-cm dish and transfected with pENTR/U6-shRNA for 3–4 h using LipofectAMINE (Invitrogen). For viral-based delivery, HSCs on a culture dish were infected by adeno-virus at a multiplicity of infection of 100. The transfected or transduced cells were seeded in three-dimensional type-I collagen and stimulated with or without IL-1 (1 ng/ml) in 5% fetal bovine serum for 3 days. Knockdowns of MMP-13 and MMP-14 were confirmed by Western blot and real-time quantitative PCR, respectively.
Preparation of Crude Membrane Fractions and Assay of Pro-MMP-13-converting Enzyme
HSCs were seeded in three-dimensional type-I collagen treated with or without IL-1 for 24 h. After washing with NT buffer the gel lattice was collected and collapsed by centrifugation at 14,000 × g. The pellet was suspended in NT buffer and homogenized by Dounce homogenizer. After repeating wash with NT buffer, the pellet was collected by centrifugation at 14,000 ×g. The membrane fractions and pro-MMP-13 or conditioned medium were mixed together with or without TIMP-2, and the mixture was incubated at 37 °C for the defined time. Conversion of pro-MMP-13 was measured by Western blot.
RESULTS
Expression and Activation of Pro-MMP-9 in Trans-differentiation of HSCs
First, we assessed temporal correlation between IL-1-induced HSC trans-differentiation and pro-MMP-9 expression/activation in the three-dimensional ECM culture. HSCs were isolated from rat liver and recovered by culture on plastic for 2 days. At this point, HSCs still appear quiescent as judged by ample vitamin A droplets as measured by autofluorescence and typical star-shaped morphology. Then the cells were embedded in three-dimensional type-I collagen and cultured in Dulbecco’s modified Eagle’s medium with 5% fetal bovine serum with or without IL-1α. At the indicated time intervals, the cells were immunostained for α-SMA, and the conditioned medium was resolved by gelatinolytic zymography. As shown in Fig. 1A, under IL-1 stimulation HSCs were morphologically converted to myofibroblastic cells, as evident by filamentous actin fibers. Vitamin A droplets, as measured by autofluorescence under UV, were depleted by the treatment (data not shown). Pro-MMP-9 (105 kDa for rodents) appeared on day 2 without activation. On day 3, activation of pro-MMP-9 (95 kDa) began to occur, and this became more pronounced on days 4 and 5 (Fig. 1B). This temporal pattern of MMP-9 expression/activation coincides with initiation and progression of HSC trans-differentiation (Fig. 1A) and degradation of collagen lattices, which indicates a possible causative relationship between MMP-mediated ECM remodeling and cellular activation (Fig. 1C).
FIGURE 1. Temporal correlation of IL-1-induced MMP-9 expression/activation with trans-differentiation of HSCs in three-dimensional type-I collagen.

A, isolated rat HSCs were embedded in three-dimensional type-I collagen and treated with IL-1α (0.5 ng/ml). Cells were fixed at the indicated time points for immunofluorescence staining of α-SMA and 4′,6-diamidino-2-phenylindole. B, MMP-9 in the conditioned medium was resolved by zymography. C, global ECM degradation was monitored by collapse of type-I collagen lattices.
Genetic and Pharmacological Evidence Demonstrates an Essential Role of MMP-9 in Trans-differentiation of HSCs in Three-dimensional ECM
Previously we showed that exogenous TIMP-1 or depletion of gelatinases attenuated IL-1-induced trans-differentiation of HSCs in three-dimensional type-I collagen. In light of closely associated induction and activation of pro-MMP-9 in this cellular activation process, our results suggested the critical role of MMP-9 in trans-differentiation of HSC in three-dimensional ECM (19). However, TIMP-1 also inhibits other MMPs, and gelatin affinity depletion may remove other gelatinases such as MMP-2. To directly test the role of MMP-9 in activation of HSCs in three-dimensional ECM, we isolated HSCs from MMP-9 null and genetically matched wild-type mice. Isolated mouse HSCs were highly pure (>96%), because almost all the cells identified by phase-contrast microscopy had characteristic transient emission of UV-excited autofluorescence as evidence of intracellular vitamin A storage (Fig. 2A, lower panels). The HSCs were plated on plastic or embedded in type-I collagen followed by treatment with or without IL-1 for 4 days. Cultured on plastic, HSCs from wild-type and MMP-9 knock-out mice exhibited similar extent of activation, demonstrating MMP-9 independence for cellular activation in this “ECM-free” and artificial setting (Fig. 2B). In fact, no MMP-9 was induced by HSCs cultured on plastic (data not shown). In contrast, cultured in three-dimensional type-I collagen, the wild-type but not MMP-9-deficient cells underwent activation in response to IL-1 treatment as determined by the expression of α-SMA stress fibers.
FIGURE 2. Genetic and pharmacological evidence demonstrates the essential role of MMP-9 in IL-1-induced trans-differentiation of HSCs in three-dimensional collagen.

A, HSCs isolated from wild-type and MMP-9 knock-out (KO) mice were examined by microscopy under phase contrast and UV for autofluorescence (vitamin A droplets). B, the cells were embedded in type-I collagen or cultured on plastic. After 5 days of culture with or without IL-1α, the cells were stained for α-SMA. C, rat HSCs were embedded in three-dimensional type-I collagen and stimulated with IL-1α in the presence or absence of MMP inhibitor. After 5-day culture, the cells were stained for α-SMA. D, type-IV collagenase activity was measured by peptide cleavage assay. E, MMP inhibitor abolished the global breakdown of the reconstituted interstitial collagen gel.
Then, we tested the effect of a hydroxamate-type MMP inhibitor on pro-MMP-9 conversion and trans-differentiation. Isolated rat HSCs were cultured in three-dimensional type-I collagen, treated with IL-1 together with the MMP inhibitor. As shown in Fig. 2C, the MMP inhibitor abolished the IL-1-induced HSC activation in three-dimensional type-I collagen as demonstrated by reduced stress fibers and exhibited ruffled actin rings around cells, a typical phenotype of HSC in three-dimensional collagen. In contrast, the compound was without effect on activation of HSCs grown on plastic, demonstrating the lack of cellular toxicity of the inhibitor (data not shown). The type-IV collagenase activities in the conditioned medium, as determined by a fluorogenic substrate, were significantly abrogated by the inhibitor, supporting its efficacy (Fig. 2D). Conversion of pro-MMP-9 as well as global degradation of the collagen gel were clearly prevented by the inhibitor, indicating an interstitial collagenase may also be involved in degradation of type-I collagen and cell activation (Fig. 2E). Thus, these results demonstrate that activation of pro-MMP-9 is likely mediated by another MMP and that active MMP-9 plays an unequivocal role in cellular activation induced by IL-1 and three-dimensional type I collagen.
Pro-MMP-9-converting Activators Are Secreted with Molecular Masses of 50 and 25 kDa
First, we attempted to determine if the converting enzymes are secreted. To this end, the conditioned medium derived from the three-dimensional culture of HSCs in type-I collagen and exposed to IL-1 was run through gelatin-conjugated Sepharose-4B column to deplete endogenous MMP-9. Then, the conditioned medium was incubated with purified human pro-MMP-9 for 16 h at 37 °C to assess the conversion of pro-MMP-9. As shown in Fig. 3A, the converting activity was evident in the conditioned medium, demonstrating a soluble form of the putative pro-MMP-9 activator released by HSCs. Then, we estimated the molecular weight of the converting enzymes. The conditioned medium was concentrated by precipitation with ammonia sulfate and resolved by gel filtration (Superdex-200). The protein profile was monitored by A280 absorbance, and the pro-MMP-9-converting activity was assayed by incubation of the fractions with purified human pro-MMP-9 followed by zymography. The molecular weight of the fractions was calibrated by standard proteins. The converting activities were eluted in fractions of a major peak of ~50 kDa and a minor one at 25 kDa (Fig. 3B). Because the 50-kDa molecule may represent a dimmer of the 25-kDa converting enzyme, we resolved the converting activity by a non-denaturing SDS-PAGE, and slices were analyzed for the pro-MMP-9-converting activities. The converting activities were not only preserved in the SDS- PAGE but also gave the similar molecular mass of a major peak at ~50 kDa and a minor one at ~25 kDa (Fig. 3C). These results indicate a monomeric status of the 50-kDa converting enzyme secreted by HSCs in response to IL-1 and type-I collagen.
FIGURE 3. The pro-MMP-9-converting activities secreted by HSCs have molecular masses of 50 and 25 kDa.
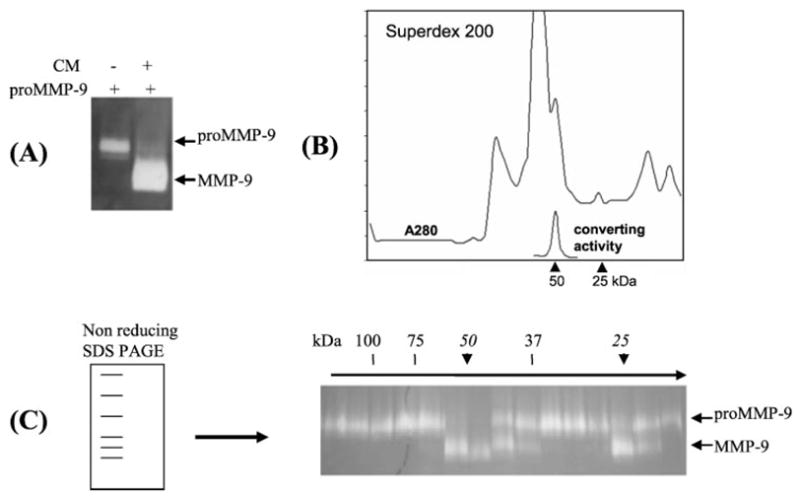
A, after depletion of endogenous gelatinases, the conditioned medium by HSCs cultured in collagen with IL-1α for 5 days was incubated with or without purified human pro-MMP-9. After 16 h the reactions were resolved by zymography. B, the conditioned medium was resolved by Superdex-200, and fractions were assayed by incubation with purified pro-MMP-9 followed by zymography. C, the conditioned medium was resolved by non-reducing SDS-PAGE followed by slicing of the gel into 20 pieces according to molecular weight. After incubation of the slices with pro-MMP-9, the converting activities were resolved by zymography. The converting activities at 50 and 25 kDa are indicated by arrows.
Pro-MMP-9-converting Enzymes from HSCs and Human Skin Are Drastically Different
Our previous report and preceding experiment, showing the inhibition of pro-MMP-9 activation by TIMP-1 and MMP inhibitor, demonstrated the potential role of MMP in pro-MMP-9 activation. We then addressed this notion in a cell-free system. The endogenous gelatinases in the conditioned medium were depleted, and the resulting medium was incubated with purified human pro-MMP-9 together with the MMP inhibitor or chymostatin, an inhibitor for chymotrypsin-like proteinases. Result showed pro-MMP-9 was converted by soluble factors in the conditioned medium. Further, the pro-MMP-9-converting activities were inhibited by the MMP inhibitor at 100 nM while the conversion was totally resistant to chymostatin (Fig. 4A), demonstrating again the activators are most likely to be MMPs. In contrary, the converting activities derived from human skin extract were insensitive to the MMP inhibitor but completely abrogated by chymostatin (Fig. 4B). Similarly, the extracts from mouse skin also showed the capacity to activate pro-MMP-9 and the inhibition by chymostatin but not by MMP inhibitors.3 Thus, these results reveal that HSCs and skin have tissue-specific, differential mechanisms for pro-MMP-9 activation.
FIGURE 4. HSCs and skin use different mechanisms to activate pro-MMP-9.

A, after depletion of endogenous gelatinases, the conditioned medium by HSCs in collagen with IL-1 was incubated with human pro-MMP-9 in the presence or absence of proteinase inhibitors as indicated. After incubation for 16 h the reactions were resolved by zymography. B, normal human skin was sequentially extracted by 1% Triton X-100, 6 M urea, and 2 M NaCl. After dialysis the final extract was incubated with pro-MMP-9 and the proteinase inhibitors. After 16 h the reactions were resolved by zymography.
HSCs Produce Multiple MMPs in Response to the Dual Stimulation of IL-1 and Type-I Collagen
We then profiled the expression of the MMP family by HSCs under the dual stimulation of IL-1α and type-I collagen using the Affymetrix DNA microarray analysis. Among 800 genes up-regulated by the cells in gel and in response to IL-1α, we noticed five MMPs: MMP-3, MMP-9, MMP-10, MMP-12, and MMP-13 (Fig. 5A). Among them, MMP-13 induction was the highest with a 1,300-fold increment, followed by MMP-9, which was up-regulated by 110-fold. The messenger of membrane-type MMP-14 was constitutively expressed at a high level without significant increase in response to IL-1. Because the converting enzymes are secreted factors, the membrane-type MT-MMP is unlikely a direct activator for pro-MMP-9. MMP-23 is down-regulated, whereas other MMPs are absent from the stellate cells. We then confirmed the DNA microarray data by real-time quantitative RT-PCR analysis. As shown in Fig. 5B, these MMPs were not induced by IL-1 when the cells were cultured on plastic but were explosively up-regulated in three-dimensional type-I collagen with the cytokine. MMP-13 was up-regulated by 800-fold, confirming the microarray result.
FIGURE 5. MMPs and TIMPs expressed by HSCs in response to IL-1 and type-I collagen.

A, isolated rat HSCs were embedded in three-dimensional type-I collagen followed by a treatment with IL-1α for 16 h. RNA was extracted and converted to cDNA. Expression of MMPs and TIMPs was screened by an Affymetrix DNA microarray. IL-1α-induced expression of MMPs is presented as -fold changes to the basal levels. “A” stands for the absence. B, HSCs were cultured either on plastic or in three-dimensional type-I collagen with or without IL-1 for 16 h. The mRNA levels of MMPs are measured by quantitative RT-PCR.
A Tight Association of MMP-13 with Pro-MMP-9-converting Activities
Conspicuous induction of MMP-13 mRNA shown above was quite intriguing in light of the molecular mass of this MMP (52 kDa for pro-MMP-13 and 48 kDa for MMP-13 as predicated) that approximates the size of the converting activities demonstrated by gel filtration and SDS-PAGE. To this end, we performed immunoblot analysis of the conditioned medium and demonstrated that the appearance of active MMP-13 (48 kDa) and truncated form (25 kDa) on day 2 (Fig. 6A), preceding the conversion of pro-MMP-9 (Fig. 1B). The 25-kDa MMP-13 is presumably the catalytic domain because of its recognition by the monoclonal antibody for the hinge region and its activity as shown in the next section. We then asked whether the lacking of pro-MMP-9 activation by the cells in three-dimensional Matrigel is due to absence of MMP-13 expression or activation. As shown in the Fig. 6B, no expression of MMP-13 was detected in three-dimensional Matrigel culture, again demonstrating a potential causative role of MMP-13 in activation of pro-MMP-9.
FIGURE 6. Lack of pro-MMP-9 activation in Matrigel is attributed to the absence of MMP-13.

A, isolated rat HSCs were either seeded on plastic or embedded in type-I collagen or Matrigel, and then stimulated with IL-1α. Conditioned medium was sampled at the indicated intervals. MMP-13 expression/ activation was measured by Western blot with a monoclonal antibody that recognizes the hinge region of MMP-13. B, the conditioned medium from HSCs cultured for 5 days in the defined conditions was resolved by zymography.
Direct Activation of Pro-MMP-9 by Active MMP-13
We then tested if MMP-13 can directly convert pro-MMP-9 in vitro. Purified pro-MMP-13 and active MMP-13 (catalytic domain) were incubated with purified human pro-MMP-9 in the presence of 5 mM CaCl2 in a buffer containing 100 mM NaCl and 50 mM Tris at pH 7.5. After 16-h incubation the reactions were resolved by zymography. As shown in Fig. 7A, the active MMP-13, but not its zymogen (pro-MMP-13), was able to convert pro-MMP-9 to its active form. We also examined the inhibitory effects of the MMP inhibitor-I and chymostatin. As expected, MMP inhibitor-I efficiently blocked MMP-13-mediated pro-MMP-9 activation, whereas chymostatin was without the effect (Fig. 7B). Conversely, chymostatin, but not MMP inhibitor-I, readily abolished cathepsin-G-mediated activation of pro-MMP-9 (data not shown).
FIGURE 7. Active MMP-13, but not its zymogen, activates pro-MMP-9.
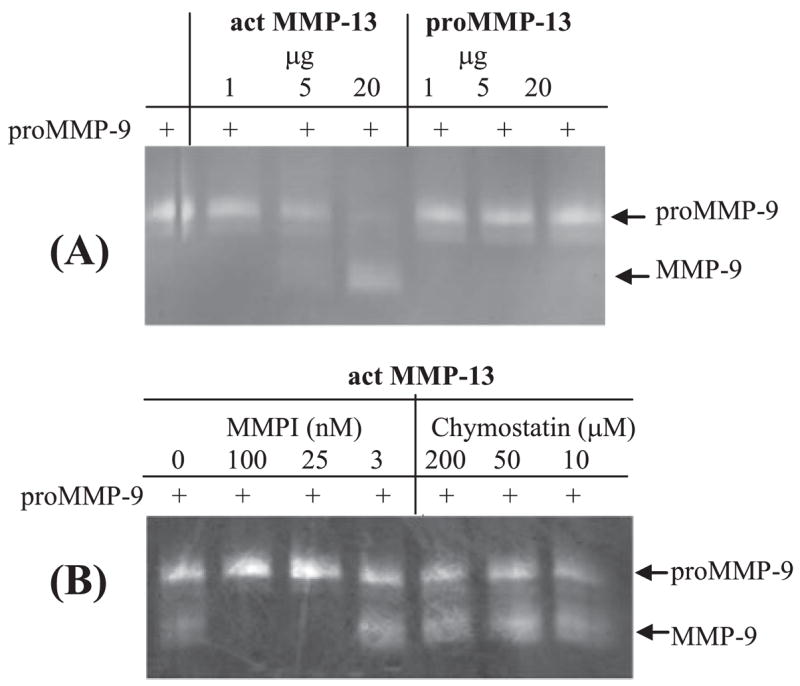
A, purified active MMP-13 and pro-MMP-13, at the indicated dosages, were incubated with purified pro-MMP-9 for 16 h followed by zymography. B, active MMP-13 was incubated with pro-MMP-9 in the presence or absence of the MMP inhibitor or chymostatin for 16 h followed by zymography.
Knockdown of MMP-13 in HSCs Abolishes Pro-MMP-9 Activation
To prove MMP-13 as an HSC-derived pro-MMP-9 activator, we tested whether knockdown of MMP-13 by RNA interference prevents IL-1-induced pro-MMP-9 activation by HSCs cultured in three-dimensional type I collagen. Two small interference RNA designed to target MMP-13 were generated from shRNA. Expression of shRNA is driven by U6 promoter in the context of either a plasmid or adenovirus. An shRNA against LacZ was used as a control. Isolated rat HSCs were transfected by the plasmids, cultured in three-dimensional type-I collagen, and treated by IL-1α. After 4-day culture the conditioned medium was analyzed for pro-MMP-9 activation by zymography and for MMP-13 expression by Western blot. As shown in Fig. 8, both shRNAs (AB for the pro-peptide domain and CD for the catalytic domain) effectively suppressed the expression of MMP-13 and coordinately prevented pro-MMP-9 conversion without affecting expression of the zymo-gen itself. Specificity of the knockdown was also demonstrated by the absence of the effects of shRNAs on the constitutive expression and activation of pro-MMP-2. To determine whether HSC trans-differentiation is inhibited as a consequence MMP13 knockdown, we expressed the shRNA for MMP-13 or lacZ by an adenoviral vector. Primary HSCs were transduced with the adenovirus followed by culture in three-dimensional type-I collagen and treatment with IL-1. As expected, the viral-mediated expression of shRNA thoroughly knocked down MMP-13 and totally abolished pro-MMP-9 maturation, whereas the expression of the zymogen was not affected (Fig. 8, C and D). The global degradation of the three-dimensional type-I collagen, a major substrate for MMP-13, was also abrogated by the shRNA against MMP-13 as an evidence of functional consequence of MMP-13 knockdown (insets of Fig. 8E). Concurrently, HSC activation as assessed by filamentous α-SMA was clearly attenuated by MMP-13 knock-down. These results unequivocally demonstrate the pivotal importance of a cascade of MMPs and ECM degradation in trans-differentiation of HSCs in the ECM milieu.
FIGURE 8. Knockdown of MMP-13 abolishes HSC-mediated pro-MMP-9 activation.

A, isolated rat HSCs were transfected with plasmids (pENTR/U6) that delivered shRNA against MMP-13 and lacZ. After 4-h transfection, the cells were embedded in type-I collagen and stimulated with IL-1α. Expression and activation of pro-MMP-9 was analyzed by zymography. B, down-regulation of MMP-13 was measured by Western blot. C and D, transduction of the cells in three-dimensional culture with adenovirus encoding shRNA for MMP-13 abolishes activation of pro-MMP-9 without any effect on the expression of the zymogen. E, phenotypes of the cells transduced by adenovirus encoding shRNA for MMP-13 and lacZ. Inhibition of collagen breakdown by shRNA for MMP-13 is shown by the insets.
MMP-14-mediated Activation of Pro-MMP-13
Con-canavalin A-stimulated fibroblasts express MT1-MMP and activate pro-MMP-13 (33). Thus, we examined the potential role of MT1-MMP in pro-MMP-13 activation in our three-dimensional ECM culture model. Results from both DNA Microarray and quantitative RT-PCR showed the constitutive expression of MMP-14 mRNA and the absence of IL-1 regulation (Fig. 5). However, Western blot analysis clearly demonstrated induction of the MMP-14 protein in response to IL-1, indicating a possible post-transcriptional regulation (Fig. 9A). Thereafter, we tested if TIMP-2, a preferential tissue inhibitor for MMP-14, could inhibit pro-MMP-13 activation. Exogenous TIMP-2 was added to the three-dimensional culture, and it indeed inhibited maturation of pro-MMP-13 in a dose-dependent manner (Fig. 9B). In a perfect agreement, activation of pro-MMP-9 was also inhibited by TIMP-2, proving again the causative relationship between MMP-13 and MMP-9. To determine if pro-MMP-13 can be directly processed by membrane-bound proteinase, pro-MMP-13 was incubated with the membrane fractions derived from the three-dimensional culture stimulated with or without IL-1. Western blotting revealed that pro-MMP-13 was activated only with the membrane fraction of IL-1-stimulated HSCs, but not unstimulated HSCs, in three-dimensional culture, which is consistent with the change of MMP-14 protein level (Fig. 9, A and C). Further, the membrane fraction-mediated activation of pro-MMP-13 was inhibited with TIMP-2 (Fig. 9D). Finally, we knocked down MMP-14 by adenovirally expressing shRNA, targeting two regions of the proteinase. HSCs were transduced with adenovirus encoding shRNA for MMP-14 (constructs AB and CD) or lacZ. The cells were embedded in collagen and stimulated with IL-1. Quantitative RT-PCR demonstrated substantial down-regulation of MMP-14 mRNA (Fig. 9E) with shRNA, and activation of pro-MMP-13 was attenuated by this knockdown. There was residual pro-MMP-13 activation, and this most likely reflected the remaining MMP-14 protein following the shRNA treatment (Fig. 9F). Taken together, these results demonstrate an essential role of MMP-14 in processing of pro-MMP-13 in our three-dimensional culture model.
FIGURE 9. Activation of pro-MMP-13 is mediated by membrane type MMP-14.

A, expression of MMP-14 by HSCs in three-dimensional type-I collagen with or without IL-1 for 2 days was measured by Western blot. B, TIMP-2 was added to the three-dimensional culture for 3 days. Inhibition of activation of pro-MMP-13 and pro-MMP-9 with TIMP-2 was detected by Western blot and zymography, respectively. The maturated forms and presumable catalytic domains of these MMPs are indicated. C, crude membrane fractions were prepared from HSCs in three-dimensional collagen with or without IL-1 for 2 days. The fractions were incubated with pro-MMP-13 for the indicated time followed by Western blot analysis for MMP-13. D, the membrane fractions from the three-dimensional culture with IL-1 were incubated with pro-MMP-13 together with TIMP-2. After incubation, activation of pro-MMP-13 was measured by Western blot analysis. E, knockdown of MMP-14 by HSCs was carried out by transduction with adenovirus encoding shRNA for MMP-14 or lacZ as a control. The efficiency of down-regulation of the MMP-14 mRNA was measured by quantitative RT-PCR. F, inhibition of MMP-13 activation by down-regulation of MMP-14 was assessed by Western blot analysis.
Positive Feedback Activation of Pro-MMP-13 with MMP-9
In addition to testing the membrane fractions for activation of pro-MMP-13, we also examined the activity in the conditioned medium. The conditioned medium from the 2-day culture mostly contained pro-MMP-13. The conditioned medium was then incubated for additional time in a cell-free system. To our surprise, the endogenous pro-MMP-13 was spontaneously matured during this incubation with soluble factors in the conditioned medium, and this activation was inhibited by MMP inhibitor at 200 nM (Fig. 10A). We then depleted the gelatinases in the conditioned medium using the gelatin beads and assayed for pro-MMP-13 activation. As shown in Fig. 10B, depletion of gelatinases, mostly MMP-9 in this preparation, resulted in abrogation of the processing of pro-MMP-13, suggesting the role of MMP-9 in pro-MMP-13 activation. To prove such notion we added back active human MMP-9 to the gelatinase-depleted conditioned medium. This restored the processing of pro-MMP-13 in the medium (Fig. 10C). Thus, these data reveal a positive feedback loop of MMP-9 in activating MMP-13 in the three-dimensional culture.
FIGURE 10. A positive feedback loop of pro-MMP-13 activation with MMP-9 and an MMP activation cascade.

A, the conditioned medium from HSCs in three-dimensional collagen with IL-1 for 2 days was collected, and the mixture was incubated for additional time with or without MMP inhibitor. Activation of the endogenous pro-MMP-13 was measured by Western blot analysis. B, gelatinases in the conditioned medium were depleted by gelatin-Sepharose 4B. The original and depleted conditioned media were incubated for additional time and resolved by zymography for gelatinases and Western blot for MMP-13. C, active human MMP-9 was added back to the conditioned medium depleted of gelatinases. After incubation, MMP-13 was resolved by Western blot. D, a proposed model for MMP activation cascade by HSCs in three-dimensional ECM in response to IL-1 stimulation is illustrated.
DISCUSSION
A major impetus for advancement of research on liver fibrogenesis has been the establishment of methods to isolate and thereafter to identify HSCs as ECM-producing cells (8, 34). Upon culture on plastic, quiescent, vitamin A-storing HSCs undergo spontaneous trans-differentiation to myofibroblastic cells (35). Such in vitro activation is also accelerated by the addition of Kupffer cell-conditioned medium or soluble factors identified to stimulate HSCs. Of these factors, transforming growth factor-β (36, 37) and platelet-derived growth factor (38) are the two most potent mediators to activate HSCs on plastic. However, HSCs in vivo reside in the three-dimensional ECM milieu, and fewer studies have experimentally reproduced this microenvironment to examine the mechanisms of HSC trans-differentiation. Incorporation of this three-dimensional ECM milieu into HSC culture has led to our recent discovery that HSCs serve as a major site for both expression and activation of pro-MMP-9, which in turn facilitates IL-1-induced HSC trans-differentiation in three-dimensional collagen. In this report we delineated an MMP activation cascade consisting of activation of pro-MMP-13 with a membrane-type MMP (MMP-14), which in turn processes pro-MMP-9. Further, we identified a positive feedback loop of MMP-9 to activate pro-MMP-13. Through the use of HSCs from MMP-9 knock-out mice, shRNA to knockdown MMPs, and MMP inhibitor we unequivocally demonstrated the role of MMP in trans-differentiation of HSCs in the three-dimensional ECM culture. A working model for IL-1-induced cascade of MMP activation in trans-differentiation of HSCs in ECM is illustrated in Fig. 10D.
A proposal that incriminates MMPs in liver injury and fibrogenesis may not be necessarily groundless. A clear rational is that, if particular ECM components maintain the phenotype of quiescent HSCs in normal liver, matrix breakdown initiated by MMPs should contribute to HSC activation. Indeed, evidence from both clinical and animal studies exists that documents up-regulation of MMPs in many types of liver injury (39). For instance, induction of MMP-3 (stromelysin-1) is detectable as early as 6 h after CCl4 administration in rats and peaks at 48 –72 h (40). A more comprehensive study reveals correlation of MMPs in liver injury. After a single dose of CCl4, MMP-13, MMP-2, MMP-9, MT1-MMP, MMP-3, and MMP-10 were all increased, with peak expression coinciding with induction of inflammatory cytokines (41). Additionally, immunostaining of the liver reveals localization of MMP-9 in the “scaring” areas of active fibrogenesis, indicating that HSCs may be an important source of MMP-9 besides macrophages (14). In rat hepatic fibrosis induced by bile duct ligation, activities of MMP-2 and MMP-9 increase 2 days after bile duct ligation, reach maximal levels at day 10, and remain high throughout the 30-day study period, suggesting that sustained tissue damage and inflammation due to chronic cholestasis maintain induction of MMPs (13). In addition to the documentation of MMP in liver fibrosis, evidence is ample for the association of MMP with fibrosis in lung (15, 42–44). In our three-dimensional type I collagen culture model, the pro-inflammatory cytokine IL-1, but not growth factors such as transforming growth factor-β or platelet-derived growth factor, induced and activated pro-MMP-9. Such acute phase cytokine-induced massive expression and activation of multiple MMPs within a selective ECM have a profound implication in the early phase of tissue destruction and activation of HSCs, presumably, in part, through degradation of the ECM in the space of Disse.
The identity of physiological converting enzymes for pro-MMP-9 still remains elusive. Most cells cultured in vitro can produce pro-MMP-9 in response to cytokines or growth factors, but maturation (activation) of the zymogen rarely occurs by normal cells. For instance, chondrocytes from osteoarthritis, but not normal tissue, produce active MMP-9 in an MMP-13-dependent manner (45). In human and mouse skin, chymotrypsin-like proteinase is responsible for maturation of pro-MMP-9, whereas isolated skin cells are unable to convert pro-MMP-9 (29, 31). To find pro-MMP-9 activator in the highly stringent context of our culture model, we first characterized the activator as a secreted pro-teinase sensitive to an inhibitor for MMPs. This inhibition is in line with our previous finding that TIMP-1 prevents activation of pro-MMP-9 in the identical model, indicating another MMP as an activator of pro-MMP-9 (19). We then determined the molecular mass of the converting enzyme, which has a major component approximating 50 kDa with a minor one of ~25 kDa. These sizes are identical to those of MMP-13 (~50 kDa) and its catalytic domain (~25 kDa). The notion of the 25-kDa band being the catalytic domain is based on the results that the band was detected by Western blot using a monoclonal antibody against the hinge region of MMP-13 and that actual pro-MMP-9-converting activity was demonstrated in this fraction. We then globally surveyed to determine which MMPs are differentially up-regulated in our three-dimensional culture model by DNA microarray analysis. This analysis, with the confirmation by real-time RT-PCR, indicated MMP-13 as a primary candidate for pro-MMP-9 activator. Finally, our reconstitution and knockdown experiments for MMP-13 proved that this notion is true.
The present study also identified the upstream activators for pro-MMP-13 in the three-dimensional culture model. Through a systematic approach we found that pro-MMP-13 is activated by two mechanisms, one by the membrane-type MMP (MMP-14) and another by secreted MMP-9. First, we demonstrated a membrane-associated pro-MMP-13-converting activity and its inhibition by TIMP-2. Although the mRNA of MMP-14 is constitutively expressed, IL-1 appears to regulate the proteinase at post-transcriptional level as demonstrated by its increased protein level. The extent of knockdown of MMP-14 with shRNA correlates perfectly with abrogation of pro-MMP-13 maturation. To our surprise, we also identified the presence of an additional pro-MMP-13 activator in the soluble faction. Depletion of gelatinases in the fraction abolished its activity, which led us to examine two gelatinases, MMP-9 and MMP-2. Although MMP-9 and MMP-2 were both presented in the medium, the quantity of the latter was minimal, and the reconstitution experiment proved that MMP-9 is the soluble pro-MMP-13 activator.
Global ECM degradation is commonly mediated by collaborative actions of many MMPs. Indeed, our three-dimensional ECM culture model is shown to undergo collapse and degradation of the matrix through a cascade of MMP activation (MMP-14 > MMP-13 > MMP-9 plus MMP-9 > MMP13). Other MMPs induced by the three-dimensional system, such as MMP-3, MMP-10, and MMP-12 (Fig. 5), may also participate in ECM degradation. To degrade the interstitial collagens such as type-I collagen, MMP-13 needs to make an initial cleavage of the triple-helix collagen resulting in denature of the collagen. This is usually followed by further degradation by gelatinases such as MMP-9. Accumulating evidence also exists for the role of MMP-14 as a non-canonical collagenase (46, 47). Indeed, collagen breakdown in our three-dimensional culture model was prevented by knockdown of MMP-14.
In three-dimensional Matrigel, IL-1 induced the expression of pro-MMP-9 but not its activation. This correlated with the lack of MMP-13 expression. Only when type I collagen was introduced, did MMP-13 induction and activation ensue, leading to pro-MMP-9 activation. Central questions remain as to what signaling is induced by IL-1 to up-regulate both MMP-9 and MMP-13 in type I collagen and how Matrigel prevents pro-MMP-13 but not pro-MMP-9 induction in response to IL-1. To address these questions, we sought to identify IL-1 signaling differentially activated by HSCs in these two types of ECM. Two major regulators known for MMP-9 transcription are NF-κB and AP-1 (29, 48 –50). Although type I collagen can transduce signals via integrin or discoidin domain receptors and induce pro-MMP-2 maturation (51–53), such signal is not sufficient to induce MMP-9 or MMP-13. Thus induction of MMP-9 and -13 appears to require additional signals that are rendered by IL-1.
In general, proliferation and differentiation of mammalian cells are best studied in the three-dimensional ECM milieu. Although in-depth investigation of cell differentiation in three-dimensional ECM is lacking, emerging reports have demonstrated the role of ECM remodeling in cell fate determination (mostly conducted by culture cells on top of ECM). The effects of MMP on cell fate decision may derive from the loss of ECM-mediated mechanical support or from ligands released from the matrices or by ectodomain shedding. For instance, MMP-9 that is constitutively expressed at a higher level in bone marrow cells is responsible for a release of a soluble kit ligand, permitting the differentiation of endothe-lial and hematopoietic stem cells (54). A plasma kallikrein-dependent plasminogen activation cascade was shown to be essential for adipocyte differentiation through ECM degradation (55). Epithelial mesenchymal transition is being considered as an additional mechanism of tissue fibrogenesis. Recently, MMP-3 was shown to initiate epithelial mesenchymal transition through activation of Rac and generation of reactive oxygen species (56). Our study of the trans-differentiation of HSCs in three-dimensional ECM opens an avenue to better understand the molecular mechanisms underlying regulation of MMP expression and activation as well as biological significance of these processes in HSC phenotypic alteration. Our future work will focus on the intracellular signals concomitantly exerted by the dual stimulation of IL-1 and ECM in trans-differentiation of HSCs. Equally, we have to address how the ECM degradation provokes the trans-differentiation of the HSCs. And finally, we will determinate the role of MMP and IL-1 in liver fibrogenesis in animal models.
Acknowledgments
We thank Jiaohong Wang for isolation of HSCs.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants DK069418 and AR051558, and Robert and May Wright Foundation (to Y. P. H.) and by NIH Grants P50 AA11999 and R24AA12885 (to H. T.).
The abbreviations used are: HSCs, hepatic stellate cells; ECM, extracellular matrix; MMP, matrix metalloproteinase; shRNA, short hairpin RNA; IL-1, interleukin-1; RT, reverse transcription; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; TIMP, tissue inhibitor of metalloproteinase; α-SMA, α-smooth muscle actin.
Y.-P. Han, unpublished data.
References
- 1.Wake K. Int Rev Cytol. 1980;66:303–353. doi: 10.1016/s0074-7696(08)61977-4. [DOI] [PubMed] [Google Scholar]
- 2.Geerts A. Semin Liver Dis. 2001;21:311–335. doi: 10.1055/s-2001-17550. [DOI] [PubMed] [Google Scholar]
- 3.Friedman SL, Roll FJ, Boyles J, Bissell DM. Proc Natl Acad Sci U S A. 1985;82:8681–8685. doi: 10.1073/pnas.82.24.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jezequel AM, Mancini R, Rinaldesi ML, Macarri G, Venturini C, Orlandi F. J Hepatol. 1987;5:174–181. doi: 10.1016/s0168-8278(87)80570-6. [DOI] [PubMed] [Google Scholar]
- 5.Minato Y, Hasumura Y, Takeuchi J. Hepatology. 1983;3:559–566. doi: 10.1002/hep.1840030414. [DOI] [PubMed] [Google Scholar]
- 6.Okanoue T, Burbige EJ, French SW. Arch Pathol Lab Med. 1983;107:459–463. [PubMed] [Google Scholar]
- 7.Takahara T, Kojima T, Miyabayashi C, Inoue K, Sasaki H, Muragaki Y, Ooshima A. Lab Invest. 1988;59:509–521. [PubMed] [Google Scholar]
- 8.de Leeuw AM, McCarthy SP, Geerts A, Knook DL. Hepatology. 1984;4:392–403. doi: 10.1002/hep.1840040307. [DOI] [PubMed] [Google Scholar]
- 9.Friedman SL, Rockey DC, McGuire RF, Maher JJ, Boyles JK, Yamasaki G. Hepatology. 1992;15:234–243. doi: 10.1002/hep.1840150211. [DOI] [PubMed] [Google Scholar]
- 10.Han YP. J Gastroenterol Hepatol. 2006;21(Suppl 3):S88–S91. doi: 10.1111/j.1440-1746.2006.04586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okazaki I, Maruyama K. Nature. 1974;252:49–50. doi: 10.1038/252049a0. [DOI] [PubMed] [Google Scholar]
- 12.Milani S, Herbst H, Schuppan D, Grappone C, Pellegrini G, Pinzani M, Casini A, Calabro A, Ciancio G, Stefanini F. Am J Pathol. 1994;144:528–537. [PMC free article] [PubMed] [Google Scholar]
- 13.Kossakowska AE, Edwards DR, Lee SS, Urbanski LS, Stabbler AL, Zhang CL, Phillips BW, Zhang Y, Urbanski SJ. Am J Pathol. 1998;153:1895–1902. doi: 10.1016/S0002-9440(10)65703-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knittel T, Mehde M, Kobold D, Saile B, Dinter C, Ramadori G. J Hepatol. 1999;30:48–60. doi: 10.1016/s0168-8278(99)80007-5. [DOI] [PubMed] [Google Scholar]
- 15.Uchinami H, Seki E, Brenner DA, D’Armiento J. Hepatology. 2006;44:420–429. doi: 10.1002/hep.21268. [DOI] [PubMed] [Google Scholar]
- 16.Friedman SL. Semin Liver Dis. 1999;19:129–140. doi: 10.1055/s-2007-1007105. [DOI] [PubMed] [Google Scholar]
- 17.Gressner AM, Bachem MG. Semin Liver Dis. 1990;10:30–46. doi: 10.1055/s-2008-1040455. [DOI] [PubMed] [Google Scholar]
- 18.Tsukada S, Parsons CJ, Rippe RA. Clin Chim Acta. 2005;364:33–60. doi: 10.1016/j.cca.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 19.Han YP, Zhou L, Wang J, Xiong S, Garner WL, French SW, Tsukamoto H. J Biol Chem. 2004;279:4820–4828. doi: 10.1074/jbc.M310999200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parks WC. Wound Repair Regen. 1999;7:423–432. doi: 10.1046/j.1524-475x.1999.00423.x. [DOI] [PubMed] [Google Scholar]
- 21.Springman EB, Angleton EL, Birkedal-Hansen H, Van Wart HE. Proc Natl Acad Sci U S A. 1990;87:364–368. doi: 10.1073/pnas.87.1.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogata Y, Enghild JJ, Nagase H. J Biol Chem. 1992;267:3581–3584. [PubMed] [Google Scholar]
- 23.Ramos-DeSimone N, Hahn-Dantona E, Sipley J, Nagase H, French DL, Quigley JP. J Biol Chem. 1999;274:13066–13076. doi: 10.1074/jbc.274.19.13066. [DOI] [PubMed] [Google Scholar]
- 24.Hahn-Dantona E, Ramos-DeSimone N, Sipley J, Nagase H, French DL, Quigley JP. Ann N Y Acad Sci. 1999;878:372–387. doi: 10.1111/j.1749-6632.1999.tb07696.x. [DOI] [PubMed] [Google Scholar]
- 25.Fang KC, Raymond WW, Lazarus SC, Caughey GH. J Clin Invest. 1996;97:1589–1596. doi: 10.1172/JCI118583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang KC, Raymond WW, Blount JL, Caughey GH. J Biol Chem. 1997;272:25628–25635. doi: 10.1074/jbc.272.41.25628. [DOI] [PubMed] [Google Scholar]
- 27.Coussens LM, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, Caughey GH, Hanahan D. Genes Dev. 1999;13:1382–1397. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tchougounova E, Lundequist A, Fajardo I, Winberg JO, Abrink M, Pejler G. J Biol Chem. 2005;280:9291–9296. doi: 10.1074/jbc.M410396200. [DOI] [PubMed] [Google Scholar]
- 29.Han YP, Tuan TL, Hughes M, Wu H, Garner WL. J Biol Chem. 2001;276:22341–22350. doi: 10.1074/jbc.M010839200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han YP, Downey S, Garner WL. Surgery. 2005;138:932–939. doi: 10.1016/j.surg.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han YP, Nien YD, Garner WL. J Biol Chem. 2002;277:27319–27327. doi: 10.1074/jbc.M202842200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsukamoto H, Cheng S, Blaner WS. Am J Physiol. 1996;270:G581–G586. doi: 10.1152/ajpgi.1996.270.4.G581. [DOI] [PubMed] [Google Scholar]
- 33.Knauper V, Will H, Lopez-Otin C, Smith B, Atkinson SJ, Stanton H, Hembry RM, Murphy G. J Biol Chem. 1996;271:17124–17131. doi: 10.1074/jbc.271.29.17124. [DOI] [PubMed] [Google Scholar]
- 34.Friedman SL, Roll FJ. Anal Biochem. 1987;161:207–218. doi: 10.1016/0003-2697(87)90673-7. [DOI] [PubMed] [Google Scholar]
- 35.Geerts A, Vrijsen R, Rauterberg J, Burt A, Schellinck P, Wisse E. J Hepatol. 1989;9:59–68. doi: 10.1016/0168-8278(89)90076-7. [DOI] [PubMed] [Google Scholar]
- 36.Bachem MG, Meyer D, Melchior R, Sell KM, Gressner AM. J Clin Invest. 1992;89:19–27. doi: 10.1172/JCI115561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsuoka M, Pham NT, Tsukamoto H. Liver. 1989;9:71–78. doi: 10.1111/j.1600-0676.1989.tb00382.x. [DOI] [PubMed] [Google Scholar]
- 38.Friedman SL, Arthur MJ. J Clin Invest. 1989;84:1780–1785. doi: 10.1172/JCI114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benyon RC, Arthur MJ. Semin Liver Dis. 2001;21:373–384. doi: 10.1055/s-2001-17552. [DOI] [PubMed] [Google Scholar]
- 40.Herbst H, Heinrichs O, Schuppan D, Milani S, Stein H. Virchows Arch B Cell Pathol Incl Mol Pathol. 1991;60:295–300. doi: 10.1007/BF02899560. [DOI] [PubMed] [Google Scholar]
- 41.Knittel T, Mehde M, Grundmann A, Saile B, Scharf JG, Ramadori G. Histochem Cell Biol. 2000;113:443–453. doi: 10.1007/s004180000150. [DOI] [PubMed] [Google Scholar]
- 42.Watanabe T, Niioka M, Ishikawa A, Hozawa S, Arai M, Maruyama K, Okada A, Okazaki I. J Hepatol. 2001;35:465–473. doi: 10.1016/s0168-8278(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 43.Lemjabbar H, Gosset P, Lechapt-Zalcman E, Franco-Montoya ML, Wallaert B, Harf A, Lafuma C. Am J Respir Cell Mol Biol. 1999;20:903–913. doi: 10.1165/ajrcmb.20.5.3260. [DOI] [PubMed] [Google Scholar]
- 44.Corbel M, Theret N, Caulet-Maugendre S, Germain N, Lagente V, Clement B, Boichot E. Inflamm Res. 2001;50:129–135. doi: 10.1007/s000110050736. [DOI] [PubMed] [Google Scholar]
- 45.Dreier R, Grassel S, Fuchs S, Schaumburger J, Bruckner P. Exp Cell Res. 2004;297:303–312. doi: 10.1016/j.yexcr.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 46.d’Ortho MP, Will H, Atkinson S, Butler G, Messent A, Gavrilovic J, Smith B, Timpl R, Zardi L, Murphy G. Eur J Biochem. 1997;250:751–757. doi: 10.1111/j.1432-1033.1997.00751.x. [DOI] [PubMed] [Google Scholar]
- 47.Itoh Y, Ito N, Nagase H, Evans RD, Bird SA, Seiki M. Mol Biol Cell. 2006;17:5390–5399. doi: 10.1091/mbc.E06-08-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sato H, Kita M, Seiki M. J Biol Chem. 1993;268:23460–23468. [PubMed] [Google Scholar]
- 49.Gum R, Lengyel E, Juarez J, Chen JH, Sato H, Seiki M, Boyd D. J Biol Chem. 1996;271:10672–10680. doi: 10.1074/jbc.271.18.10672. [DOI] [PubMed] [Google Scholar]
- 50.Bond M, Fabunmi RP, Baker AH, Newby AC. FEBS Lett. 1998;435:29–34. doi: 10.1016/s0014-5793(98)01034-5. [DOI] [PubMed] [Google Scholar]
- 51.Ikeda K, Wang LH, Torres R, Zhao H, Olaso E, Eng FJ, Labrador P, Klein R, Lovett D, Yancopoulos GD, Friedman SL, Lin HC. J Biol Chem. 2002;277:19206–19212. doi: 10.1074/jbc.M201078200. [DOI] [PubMed] [Google Scholar]
- 52.Johnson JD, Edman JC, Rutter WJ. Proc Natl Acad Sci U S A. 1993;90:5677–5681. doi: 10.1073/pnas.90.12.5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Han YP, Tuan TL, Wu H, Hughes M, Garner WL. J Cell Sci. 2001;114:131–139. doi: 10.1242/jcs.114.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heissig B, Hattori K, Dias S, Friedrich M, Ferris B, Hackett NR, Crystal RG, Besmer P, Lyden D, Moore MA, Werb Z, Rafii S. Cell. 2002;109:625–637. doi: 10.1016/s0092-8674(02)00754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Selvarajan S, Lund LR, Takeuchi T, Craik CS, Werb Z. Nat Cell Biol. 2001;3:267–275. doi: 10.1038/35060059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, Werb Z, Bissell MJ. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]