

Abstract
Subfamily II of the solute-carrier (Slc)39a family contains three highly conserved members (ZIPs 1 to 3) that share a twelve amino acid signature sequence present in the putative fourth transmembrane domain and function as zinc transporters in transfected cells. The physiological significance of this genetic redundancy is unknown. Herein, we report that the complete elimination of all three of these Zip genes, by targeted mutagenesis and cross-breeding mice, causes no overt phenotypic effect. When fed a zinc-adequate diet, several indicators of zinc status were indistinguishable between wild-type and triple-knockout mice; including embryonic morphogenesis and growth, alkaline phosphatase activity in the embryo, and ZIP4 protein in the visceral yolk sac and initial rates (30 min) of accumulation/retention of 67Zn in liver and pancreas. When fed a zinc-deficient diet, embryonic membrane-bound alkaline phosphatase activity was reduced to a much greater extent and 80% of the embryos in the triple-knock mice developed abnormally compared to 12% of the embryos in wild-type mice. During zinc deficiency, the accumulation/retention (3 hr) of 67Zn in the liver and pancreas of weanlings was significantly impaired in the triple-knockout mice compared to wild-type mice. Thus, none of these three mammalian Zip genes apparently plays a critical role in zinc homeostasis when zinc is replete, but they play important, non-compensatory roles when this metal is deficient.
Keywords: pregnancy, stable zinc isotope, triple knockout mice, zinc deficiency, zinc homeostasis
Introduction
The maintenance of zinc homeostasis is critical, and multiple genes have evolved to modulate the storage, efflux and uptake of this essential metal in response to its availability. In mice it is estimated that 28 different genes may contribute to zinc homeostasis. Two superfamilies of mammalian zinc transporters have been identified that belong to the solute carrier (Slc)30a and the Slc39a families (16; 32; 41). Slc30a members, named ZnTs, function in zinc efflux and compartmentalization and are cation diffusion proteins (32). Members of the Slc39a family, named ZIPs, function in the uptake of zinc and other metals (12; 16; 41). In mice and humans there are 14 members of the ZIP family most of which can be grouped into one of two subfamilies named subfamily II (3 members) and LIV-1 (9 members). Many of these zinc transporters are expressed in a tissue-specific manner and in specific cellular localizations. In addition, they can display specific changes in cellular localization and stability in response to zinc deficiency or excess (4; 11; 22; 44).
Mutations in many of the members of the ZnT family and a few of the ZIP family members have begun to suggest their physiological functions. The mouse Znt1 gene is essential during early development and may be involved in the transfer of zinc into the conceptus (1), whereas the ZnT3 gene is expressed in neurons and is important for the acquisition of zinc in synaptic vesicles (3). Mutations in the mouse Znt4 gene and human Znt2 gene have been associated with a loss or reduction, respectively, of zinc in milk (2; 18). Znt5-knockout mice survive but have multiple abnormalities in muscle and bone (20), probably due to impaired transport of zinc into the secretory compartment (36). Znt7-knockout mice also fail to thrive, have low body fat and show diminished acquisition and distribution of dietary zinc (17) perhaps also reflecting impaired zinc transport into the Golgi apparatus (25). Less is known about the genetics of mammalian Zip genes. Mutations in human Zip4 cause the rare and fatal recessive disease acrodermatitis enteropathica (27; 43) and the mouse Zip4 gene is essential for early embryonic development and may be involved in the transfer of zinc via the visceral yolk sac into the embryo and later in uptake of dietary zinc (10).
Our previous studies of three members of the ZIP subfamily II (8) revealed that targeted deletion of mouse ZIP1, 2 or 3 did not cause overt signs of zinc deficiency but did lead to an impaired ability to adapt to zinc deficiency (5; 6; 33). Analyses of single and double-knockout mice with no functional Zip1 and/or Zip3 suggested that they contribute essentially additively to the degree of sensitivity to zinc deficiency during pregnancy (6). Analysis of the patterns of expression of these genes in mice revealed co-expression in intestinal stromal cells, nephric-tubular epithelial cells, pancreatic ductal epithelial cells, and hepatocytes surrounding the central vein, as well as some unique cell-specific expression patterns (5; 6). This suggested that these zinc transporters might exert some compensatory functions in the redistribution and/or retention of zinc rather than its acquisition from the diet (6; 8). In contrast, the expression patterns of Zip2 were found to be remarkably cell-type specific in peri-central hepatocytes, keratinocytes and immature dendritic cells (33), but the loss of function of this gene also rendered mice more sensitive to zinc deficiency during pregnancy.
Herein, we examined the effects of a complete loss of function of the ZIP subfamily II genes in mice. Remarkably, although these genes are highly conserved and related to one another, the entire subfamily II is dispensable when dietary zinc is replete. Consistent with the conclusions from previous studies, the functions of these genes is apparently required only during periods of zinc deficiency where they function in the accumulation/retention and distribution of dietary zinc. Compensatory functions among members of the Slc39a subfamily II are minimal, consistent with the concept that they evolved independent cell-specific functions in zinc homeostasis in mice.
Materials and Methods
Animal Care and Use
Experiments with mice were performed in accordance with the guidelines from the National Institutes of Health for the care and use of animals and were approved by the Institutional Animal Care and Use Committee. Mouse diets were purchased from Harlan Teklad (Teklad.com) and were identical except for zinc levels, which were as follows: zinc-deficient, 1 ppm zinc; zinc-adequate, 50 ppm zinc.
The effects of zinc deficiency during pregnancy on morphogenesis and growth of the embryo were determined as described in detail previously (5; 6; 9; 33). Female mice were mated (d1 = vaginal plug) and provided free access to zinc-adequate feed and deionized distilled water until d8 of pregnancy. Mice were then placed in pairs in cages with stainless steel false bottoms, and the diet was changed to the zinc-deficient feed. On day 14 of pregnancy, the embryos were collected and examined for gross morphological defects as described previously (5; 10), and as presented in the legend to Figure 2 herein.
Figure 2. Effects of dietary zinc deficiency during pregnancy on embryonic growth and morphogenesis in triple-knock and wild-type mice.
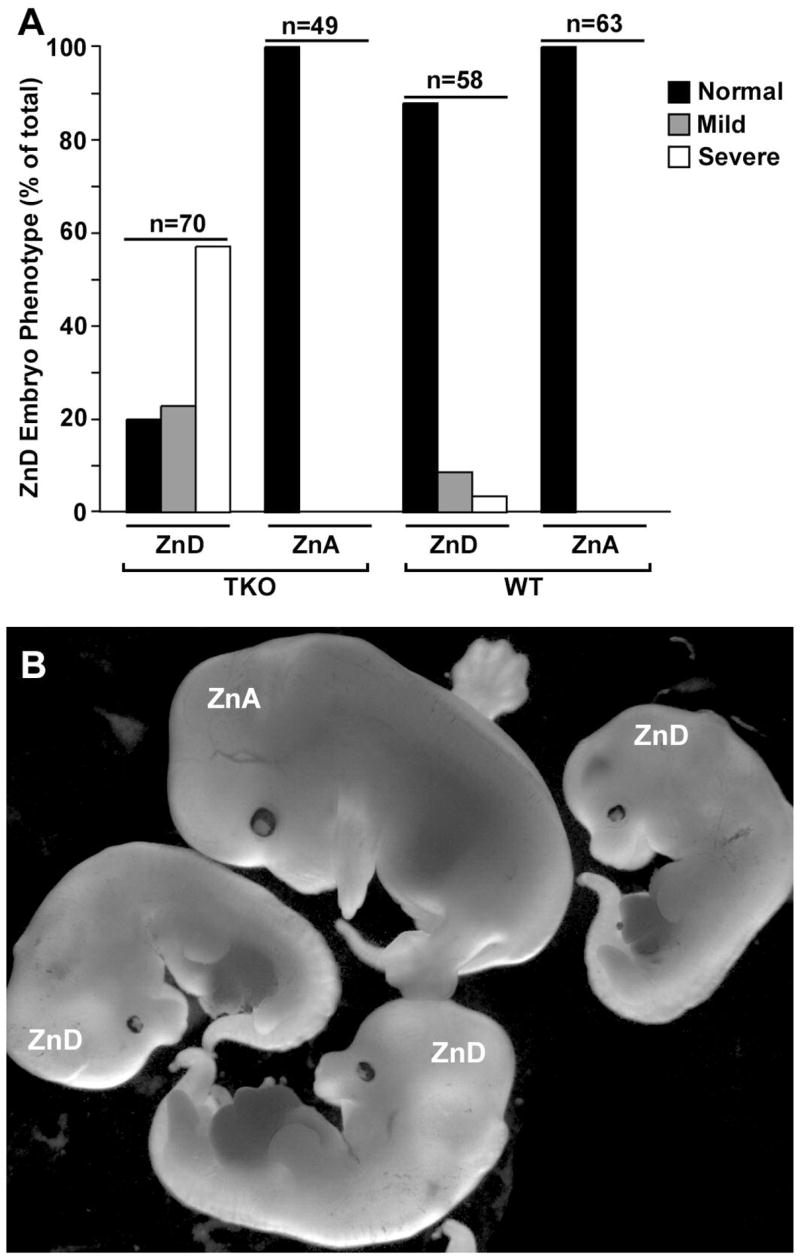
Mice were fed a zinc-adequate (ZnA) or zinc-deficient (ZnD) diet beginning on day 8 of pregnancy and the morphology of embryos was examined on day 14. Part A: Embryos were divided into three groups based on morphology; normal, mildly affected and severely affected. Embryos of normal size, with well-formed forelimb digits and normal gross features were called Normal. Embryos that were smaller and had delayed formation of the forelimb digits but were otherwise apparently normal were called Mild, whereas those that were very small, had abnormal forelimbs and hind limbs as well as craniofacial abnormalities were called Severe. An example of a normal zinc-adequate triple-knockout embryo (ZnA) and several severely zinc-deficient (ZnD) triple-knockout embryos are shown in Part B.
The effect of zinc deficiency on the accumulation/retention of zinc was examined in weaned mice. Newly weaned mice (< 5 days post weaning, 6 mice per group) were fed the zinc-adequate or the zinc-deficient diet for one week. Food was withheld overnight and mice were then given an oral gavage containing the stable isotope 67Zn (26; 30). The gavage solution was prepared as follows: Zinc-deficient feed was dissolved in deionized water (1g feed in 2 ml water) and the slurry was adjusted to 100 ppm 67Zn. 67Zn (>94% enriched, 67 zinc oxide) from Trace Sciences International Corporation (isotopetrace.com) was dissolved in 3 drops of concentrated HCl and then diluted to a final concentration of 5 mg/ml in distilled water. This stock solution was diluted 1:50 into the food slurry just before use. The pH of the gavage solution was neutral. Mice were given an oral gavage (100 μl food slurry containing 10 μg 67Zn) and tissues (liver, pancreas) were harvested at the indicated times after the gavage, rinsed in PBS and prepared for elemental analysis as described below.
Zip1, Zip2 and Zip3 Targeting Vector Construction
Bacterial artificial chromosomes that contain these mouse Zip genes from the 129/SvJ mouse strain were obtained from Incyte Genomics (incyte.com) and sequenced by Bruce Roe (University of Oklahoma).
The methods of construction and final structures of each targeting vector have been described in detail previously (5; 6; 33). Each targeting vector had the coding region of the EGFP cDNA, from the vector pEGFPKT1loxneo was introduced immediately downstream of the start codon. A loxP-flanked MC1-Neo cassette was immediately downstream of the EGFP cDNA and the targeting vector was flanked by herpes simplex virus thymidine kinase and diphtheria toxin negative selectable markers.
Targeted Disruption of Zip Genes in Embryonic Stem Cells
Targeting vectors were linearized within the vector backbone and electroporated into RW4 embryonic stem (ES) cells. Colonies were selected with 300 μg/ml G418 (cellgro.com) and 2 μM gancyclovir (roche-applied-science.com). Homologous recombinants were screened for by Southern blot hybridization as described in detail previously for each knockout allele (5; 6; 33). Positive clones were subsequently screened with a Y-chromosome-specific probe derived from pY2 (28), and a Neo cDNA to detect the presence of a single -targeted MC1-Neo cassette.
Generation of Zip-Knockout Mice
Chimeric mice were generated by microinjection of ES cell clones into 3.5-day-old C57BL/6 blastocysts, followed by transfer to pseudopregnant CD-1 foster mothers. Resulting chimeric mice were crossed with C57BL/6 females (harlan.com). Germline transmission was confirmed by PCR from tail DNA of agouti offspring.
The PCR screens for the targeted Zip alleles each utilized a set of three primers that directed amplification of the wild-type and the targeted alleles. The details of those primer sequences and where they anneal have been described previously (5; 6; 33). For the Zip1 targeted allele the PCR product for the mutant allele was 433 bp, and the product for the wild-type allele was 328 bp. For the Zip2 targeted allele the PCR product for the mutant allele was 319 bp, and that for the wild-type allele was 403 bp. For the Zip3 targeted allele the PCR product for the mutant allele was 326 bp whereas the product for the wild-type allele was 461 bp.
Agouti offspring heterozygous for the knockout allele were crossed to generate homozygous knockout mice. To remove the loxP-flanked MC1-Neo cassette, knockout mice were mated with commercially available Cre-expressing transgenic mice (strain name: B6.FVB-TgN (EIIa-cre) C5379 Lmgd from JAX.org) which express Cre in all tissues. Zip-knockout mice that lacked the Neo cassette were crossed to generate homozygous knockout mice and wild-type mice.
Generation of Zip1, Zip2, Zip3 Triple-Knockout Mice
Homozygous ZipI and Zip3 mice were cross-bred, and then offspring were back-crossed to generate homozygous double-knockout mice and wild-type mice as described (6). These double-knockout mice and wild-type mice were used to create working colonies. The double-knockout mice were then crossed with homozygous Zip2-knockout mice yielding offspring heterozygous for each knockout allele. These mice were then crossed and offspring homozygous for two of the knockout alleles and heterozygous for the third knockout allele were crossed. Offspring homozygous for all three knockout alleles were identified and crossed to create a working colony. Triple-knockout mice were compared with wild-type mice generated during creation of the double-knockout strain. All of the knockout lines and the wild-type line were therefore on a similar mixed genetic background.
RT-PCR Amplification of Zip Transcripts
Total RNA (5 μg) isolated from mouse liver using TRIzol according to the manufacturer’s instructions (invitrogen.com), was reverse transcribed using Superscript III Reverse Transcriptase (RT) (invitrogen.com) and then amplified using Taq polymerase or LA-Taq polymerase (TAKARABio.com). Zip1 and Zip2 mRNAs were amplified for 30 cycles to yield 1000 bp and 406 bp products, respectively, under reaction conditions described in detail previously (8). Zip3 mRNA was amplified using Zip3 (S) primer (5′-GTTCTTCTTCATGCTGCTGGGCTCCCTGCT-3′) and Zip3 (AS) primer (5′-GCACCAGGAACAGCACCTTCAGC-3′) for 35 cycles to generate an 884 bp product. GAPDH was amplified using GAPDH (S) primer (5′-TCACGGCAAATTCAACGGCACAGTCAAGGC-3′) and GAPDH (AS) primer (5′-CAGCACCAGTGGATGCAGGGATGATGTTCT-3′). Negative control reactions, in which the RT was omitted, were performed in parallel.
Membrane Bound Alkaline Phosphatase Assay
The method of membrane preparation and the alkaline phosphatase assay have been described in detail previously (35; 36). In brief, VYS and embryos (6 each) were collected on day 14 of pregnancy from wild-type and triple-knockout mice fed a zinc-adequate or zinc-deficient diet beginning of day 8. Samples were homogenized and membranes were recovered from the post-nuclear supernatant by centrifugation (100,000 × g for 15 min at 4°C). The membrane pellet was suspended in buffer (10 mM Tris-HCl, pH 7.5, 0.5 mM MgCl2, and 0.1% Triton X-100) and frozen at −70 °C until assayed. Membrane proteins (5 μg) were assayed for alkaline phosphatase activity using 2 mg/ml p-nitrophenyl phosphate in 1 M diethanolamine buffer, pH 9.8, containing 0.5 mM MgCl2. p-Nitrophenol release was measured by the absorbance at 405 nm. Antarctic phosphatase was used as a standard.
Western Blot Analysis
Membrane proteins from the VYS, prepared for the alkaline phosphatase assay described above, were also examined by Western blotting. Membrane proteins (40 μg) were heated in 1X SDS-sample buffer at 37°C for 10 min, resolved on a 10% SDS-polyacrylamide gel and transferred to polyvinylidine difluoride membranes. Membranes were blocked overnight and incubated with primary antibody as described previously (7; 19; 44). Immunoreactive bands were visualized using ECL Plus Western Blotting Detection System with Hyperfilm ECL (amershambiosciences.com). Anti-peptide antibodies against ZIP4 and ZIP1 have been generated and characterized as described previously (9; 19).
Trace metal determination
Elemental profiling via inductively-coupled plasma mass-spectrometry (ICP-MS) was performed for Na, Mg, P, K, Ca, Fe, Co, Cu, Zn, As, Se, and Mo as described in detail previously (33). In addition, the stable isotopes of zinc, 67Zn and 66 Zn were measured in each sample, as indicated. The natural ratio of these zinc isotopes is 0.146. Mouse pancreata and livers (n = 4 to 6 per group) were dried in a vacuum oven, digested in concentrated HNO3 and analyzed on an Elan DRCe ICP-MS (PerkinElmer). Methane was used as a collision cell gas to measure iron. Gallium and Indium were used as internal standards. National Institute of Standards and Technology traceable single element ICP standards (ultrasci.com) were used to make up the calibration standards.
Statistics
Experimental data were evaluated by Student’s t-test. Data are presented as means ± standard deviation (S.D.) and differences between groups are presented in the appropriate figure legends. Differences were considered significant at P < 0.05.
Results
Creation of mice with no functional Slc39a subfamily II genes (Zip1, 2 and 3 triple-knockout mice)
Zip1 and/or Zip3-knockout mice as well as Zip2-knockout mice were created as described previously (5; 6; 33). In each case, the targeting construct fused the initiator methionine codon with the open reading frame of the enhanced green fluorescent protein (EGFP) reporter.
Homozygous Zip1, Zip3 double-knockout mice were crossed with homozygous ZIP2-knockout mice and the offspring were genotyped and then back-crossed to ultimately yield homozygous triple-knock mice. Wild-type mice, generated while creating Zip1, Zip3 double-knockout mice and Zip2-knockout mice, were crossed to yield a working colony. Successful creation of homozygous triple-knockout mice was verified by genotyping PCR which showed amplification of only the knockout alleles in these mice (Fig. 1A) and by RT-PCR showing that these gene transcripts are absent in the adult liver of triple-knockout mice (Fig. 1B). These three Zip genes are expressed in a cell-specific pattern in the adult liver (6; 33).
Figure 1. Confirmation of the genotype of triple-knockout mice with targeted deletions of the Zip1, Zip2 and Zip3 genes. Part A.
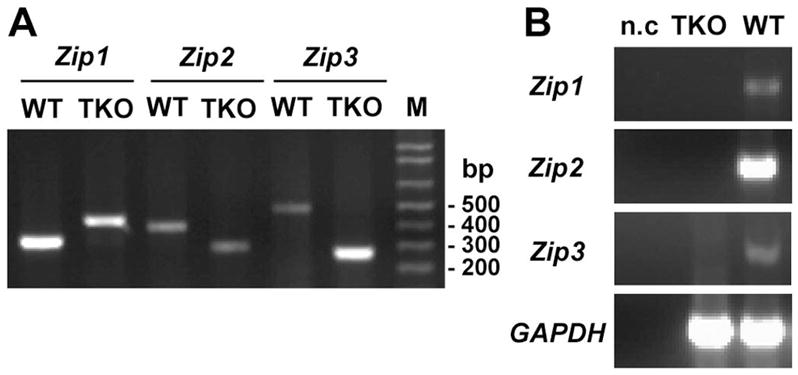
PCR genotyping of wild-type (WT) and knockout alleles for Zip1, Zip2 and Zip3 genes in triple-knockout (TKO) mice. PCR reactions contained three primers each that directed the amplification of both wild-type and knockout alleles of these Zip genes as described in Methods. Details of the targeting of each of theses genes have been published previously (5; 6; 33). M: DNA ladder. Part B. RT-PCR detection of Zip1, Zip2 and Zip3 mRNAs in total liver RNA from wild-type and triple-knockout mice. GAPDH served as an internal control for the RT-PCR, and n.c. indicates samples in which the reverse transcriptase was omitted from the reaction.
Mice with no functional Zip1, 2 or 3 genes appeared normal when fed a zinc-adequate diet. There was no difference between wild-type and triple-knockout mice with regard to growth rate, pregnancy rate, litter size or embryo growth and development (Fig. 2 and data not shown).
The triple-knockout mice showed no overt signs of zinc deficiency. On day 14 of pregnancy, the amount of membrane bound alkaline phosphatase activity in the embryo (Fig 3A), a zinc-dependent enzyme that is sensitive to zinc deficiency (36), and the abundance of ZIP4 protein in the visceral yolk sac (Fig 3C), a sensitive indicator of zinc deficiency (7; 9; 44), were indistinguishable between the triple-knockout and wild-type mice.
Figure 3. Effects of dietary zinc deficiency during pregnancy on embryonic membrane bound alkaline phosphatase activity and ZIP4 protein abundance in triple-knock and wild-type mice.
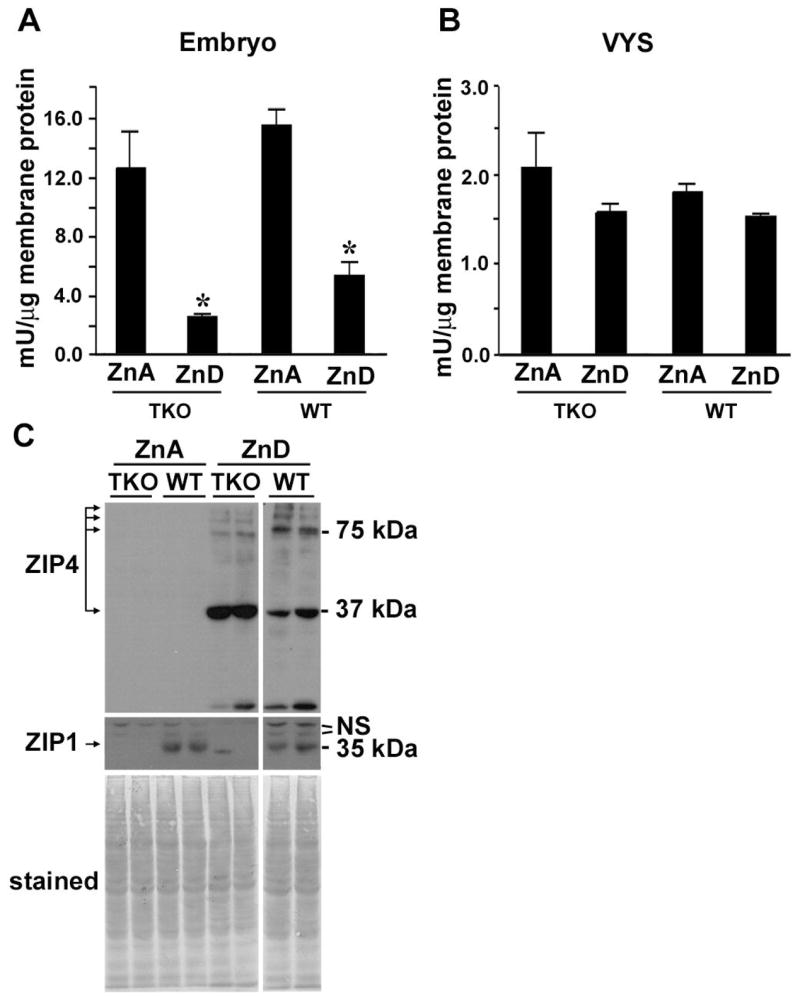
Wild-type (WT) and triple-knockout (TKO) embryos (Part A) and their visceral yolk sacs (VYS: Part B) were collected (6 per group) on day 14 of pregnancy from mice fed a zinc-adequate (ZnA) or zinc-deficient (ZnD) diet beginning of day 8. Membranes were isolated from each tissue sample and assayed for alkaline phosphatase activity. The data are expressed as the mean ± S.D. from six determinations each. *, Signifies a significant difference between ZnD TKO and WT (P < 0.001). Part C: Western blot detection of ZIP4 in membrane proteins from the day 14 visceral yolk sacs of wild-type and triple-knockout embryos from samples used in Part A. Several ZIP4 peptides are detected with a prominent band at ~37 kDa and another at ~75 kDa, as reported previously (44). ZIP1 was also detected by Western blotting of these same membrane preparations (middle panel), as described previously (44), and the proteins transferred to the membrane were detected by staining with Coomassie Blue (bottom panel). N.S. indicates non-specific bands. The minor band in lane 5 (TKO ZnD) of the ZIP1 blot is a spurious artifact that was not reproducible.
The initial accumulation/retention of exogenous zinc in the liver and pancreas were examined by giving mice an oral gavage containing the stable zinc isotope 67Zn mixed into a slurry of zinc-deficient feed (100 ppm final Zn content). Liver and pancreas were harvested 30 min after the gavage and the ratio of 67Zn to 66Zn was determined using inductively coupled plasma mass spectrometry (ICP-MS) (33). The natural ratio of these isotopes is 0.146. As shown in Figure 4A, the initial rate of accumulation/retention of zinc was the same in triple-knockout and wild-type mice.
Figure 4. Effects of dietary zinc status on the accumulation/retention of an oral gavage of 67Zn in the liver and pancreas of triple-knockout and wild-type mice.
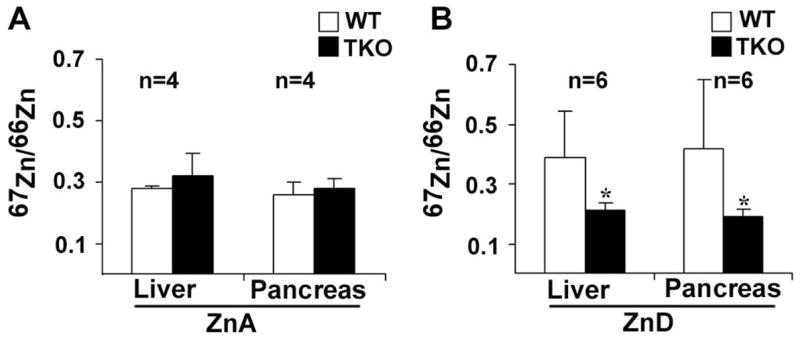
Newly Weaned wild-type (WT) and triple-knockout (TKO) mice were fed a zinc-adequate (ZnA) or zinc-deficient (ZnD) diet for 7 days. Food was withheld overnight and the next morning the mice (6 per group) were given an oral gavage containing 67Zn mixed into a slurry of zinc deficient feed (10 μg zinc in 100 μl feed). This was considered a physiologically relevant dose of zinc. Part A: The liver and pancreas were harvested 30 minutes after the gavage to detect the initial accumulation/retention of 67Zn in mice fed the zinc-adequate diet. Part B: The liver and pancreas were harvested 3 hr after the gavage to monitor the accumulation/retention of zinc in mice fed the zinc-deficient diet. Tissues were dehydrated, and multiple trace metals were measured by ICP-MS. The ratio of the stable isotopes 67Zn/66Zn in each sample was determined. The natural ratio of these isotopes of zinc is 0.146. Data are expressed as the mean ± S.D. from 6 determinations. *, Signifies a significant difference between ZnD TKO and WT (liver, P = 0.014; pancreas, P = 0.029).
Effects of zinc deficiency during pregnancy on morphogenesis, membrane bound alkaline phosphatase activity and ZIP4 protein abundance in triple-knockout and wild-type mice
Our previous studies showed that the functions of ZIPs1, 2 and 3 individually are not evident when dietary zinc is replete, but become apparent when zinc is limiting. Therefore, we examined the effects of dietary zinc deficiency during pregnancy in triple-knockout and wild-type mice (Fig 2A). Mice were fed a zinc-adequate or zinc-deficient diet beginning of day 8 of pregnancy and embryos were examined on d14 for morphological defects (Fig 2B). Embryos in wild-type mice were relatively resistant to these conditions of zinc-deficiency and about 14% showed developmental defects when examined. These included growth retardation, abnormal limb development and cranio-facial abnormalities. Only a small percentage of wild-type embryos were severely affected (see the legend to Fig 4). In contrast about 80% of the embryos in the triple-knockout mice were abnormal on day 14 and about 60% were severely affected (Fig 2B).
Measurements of membrane bound alkaline phosphatase activity in the embryo revealed that zinc deficiency caused a large reduction in the wild-type and triple-knockout embryos (Fig 3A). However, triple-knockout embryos retained less than half the amount of enzyme activity (2.5 mU/μg membrane protein) relative to that in wild-type embryos (5.2 mU/μg membrane protein). The visceral yolk sac contains only low amounts of membrane bound alkaline phosphatase activity that were not dramatically reduced by zinc deficiency (Fig 3B). Taken together these results show that triple-knockout mice are very sensitive to dietary zinc deficiency during pregnancy.
Mouse ZIP4 plays a critical role in zinc homeostasis during development (10), and this protein is induced during periods of zinc deficiency in the visceral yolk sac. Western blot analysis of membranes from the visceral yolk sac demonstrated that ZIP4 is dramatically induced to the same extent during zinc deficiency in triple-knockout and wild-type mice (Fig 3C). Thus, this response to zinc deficiency is not impaired in the triple-knockout and ZIP4 cannot compensate for the loss of function of ZIPs 1, 2 and 3.
Triple-knockout mice show impaired accumulation/retention of oral 67Zn in the liver and pancreas only when fed a zinc-deficient diet
The above results suggest that triple-knockout mice may be impaired in the ability to accumulate zinc during periods of zinc deficiency. To directly test this possibility, weanling mice were fed a zinc-deficient diet for 7 days and then given an oral gavage (100 μl) containing 100 ppm 67Zn mixed into a slurry of zinc-deficient feed. Liver and pancreas were harvested 3 hr after the gavage and the ratio of 67Zn to 66Zn was determined using ICP-MS (Fig 4B). Wild-type mice accumulated/retained twice as much 67Zn in the liver and pancreas than triple-knockout mice within 3 hr. By 6 hrs the accumulation/retention of 67Zn in the liver and pancreas had increased in the triple-knockout mice but was highly variable between individuals (data not shown). These results are consistent with the concept that ZIP1, 2 and 3 function during periods of zinc deficiency to facilitate the accumulation/retention of zinc in the liver and pancreas.
Triple-knockout mice do not accumulate hepatic iron during zinc deficiency
Our previous studies demonstrated that Zip2-knockout mice fail to accumulate iron in the liver during periods of zinc deficiency. Whether ZIP1 and ZIP3 also play a role in iron metabolism in the liver is unknown. To explore this possibility, liver was collected from day 14 pregnant mice that had been fed the zinc-adequate or zinc-deficient diet beginning of day 8 of pregnancy. Livers were dehydrated and subjected to elemental analysis by ICP-MS (Fig 5). Iron content in the liver was indistinguishable in triple-knockout and wild-type mice fed the zinc-adequate diet, and during zinc deficiency, iron content doubled in the liver of wild-type mice but remained unchanged in the triple-knockout mice (Fig 5A & C). These findings are similar to those reported in Zip2-knockout mice (33). Interestingly, zinc content in the liver was not clearly different between wild-type and triple-knockout mice and the loss of zinc from the liver was not apparently exacerbated in the triple-knockout mice during zinc deficiency. However, it should be noted that only a small proportion (< 20%) of total cellular zinc appears to be labile and a further loss of total zinc content leads to lethality.
Figure 5. Accumulation of hepatic iron during zinc deficiency in wild-type but not in triple-knockout mice.
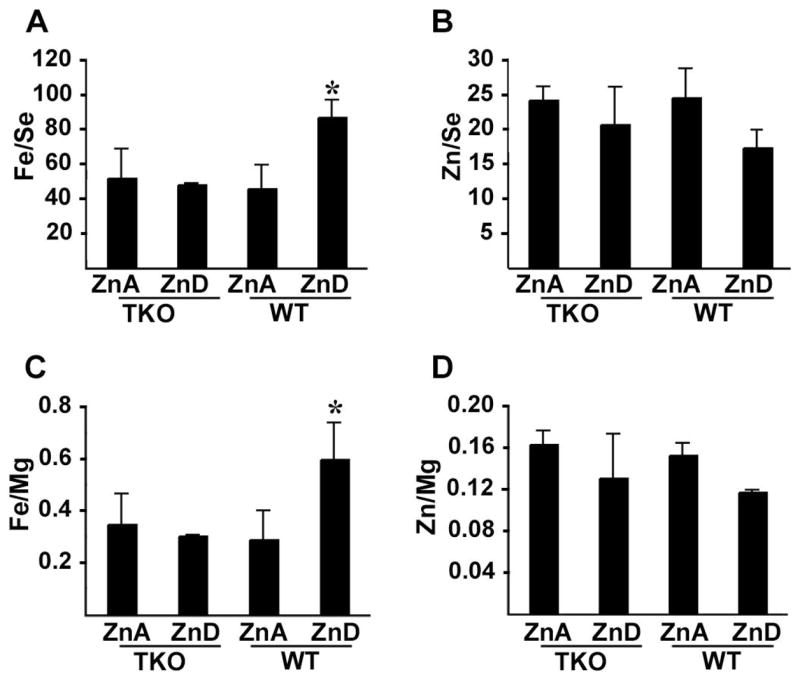
Pregnant wild-type (WT) and triple-knockout (TKO) mice were fed a zinc deficient (ZnD) or zinc adequate (ZnA) diet beginning on day 8 of pregnancy and the maternal liver was harvested on day 14. The liver (6 per group) was dehydrated, and multiple trace metals were measured by ICP-MS. Data for iron (Parts A & C) and zinc (Parts B & D) are shown and are expressed as the mean of the ratios to either selenium (A & B) or magnesium (C & D) ± S.D. from 6 determinations. No elements assayed in addition to zinc and iron showed reproducibly significant changes in these samples. *, Signifies a significant difference between ZnD TKO and WT (Fe/Se, P < 0.001; Fe/Mg, P <0.01).
Discussion
The evolution of two large gene families of zinc transporters in mammals is consistent with the complexity of essential metal homeostasis between different tissues and organs as well as within specific microenvironments both intra- and extracellular. There is a need to acquire the metal from varied diets, distribute the metal within the body and accumulate, store or eliminate the metal in a dynamic and often cell-specific manner. Mutations in many (7/10) of the ZnT gene family (Slc30a) of zinc transporters have been shown to cause overt phenotypes indicative of tissue-specific defects in zinc homeostasis in mice or humans (21; 22; 32). In contrast, to date only mutations in Zip4, among the nine Slc39a LIV-1 subfamily members, have been associated with zinc deficient phenotypes in mice and humans (10; 43). The effects of mutations in other members of the LIV-1 family of zinc transporters in animals have not been reported, although many studies suggest that these genes play important roles in many fundamental cellular processes (38; 40).
Studies reported herein focused on the effects of mutating three members of Slc39a subfamily II called Zip 1, 2 and 3. These genes have been well conserved during evolution. Mouse and human ZIP1 share 93% amino acid sequence identity (8). Structural (amino acid sequence) relatedness groups these three proteins into subfamily II (12). Remarkably, a complete loss of function of the entire subfamily II of genes in mice did not lead to overt signs of zinc deficiency based on several criteria. However, subfamily II genes were found to be important during periods of zinc deficiency in mice. The high degree of conservation of these genes suggests that their roles in protecting against zinc deficiency are also important in humans, particularly during pregnancy.
Our previous studies showed that knocking out the Zip1, Zip2 or Zip3 genes rendered mice more sensitive to the effects of zinc deficiency during pregnancy (5; 6; 33) Under the same experimental conditions employed herein about 45% of the Zip1-knockout embryos and 36% of the Zip3-knockout embryos developed abnormally. In contrast, 91% of the embryos were abnormal in the double-knockout mice (Zip1 and Zip3) under these conditions. Zip1 and 3 genes appear to function in an additive manner in this model of zinc deficiency (6). Studies of Zip2-knockout mice under identical experimental conditions of zinc deficiency revealed that 57% of the Zip2-knockout embryos developed abnormally (33). This suggested that ZIP2 might have a more profound influence on zinc homeostasis than either ZIP1 or ZIP3 alone. Surprisingly, mice lacking all three of these Zip genes were not more sensitive to zinc deficiency than the Zip1-3-double-knockout mice. Thus, members of Slc39a subfamily II appear to have evolved unique non-compensatory functions during zinc deficiency. We previously reported that ZIP2 plays a role in iron accumulation/retention in the liver during zinc deficiency; Zip2- knockout mice do not accumulate hepatic iron under these conditions (33). Whether ZIP1 and ZIP3 play roles in hepatic iron accumulation is unknown, but as shown herein deletion of Zip1 and Zip3 did not modify this Zip2-knockout phenotype. These results are consistent with the concept that these three zinc transporters have evolved unique tissue- and cell-specific functions.
Results obtained studying triple-knockout mice fed a zinc-adequate diet revealed that these genes are not essential for the rapid uptake and/or distribution of zinc in the body. In contrast, studies of these mice fed a zinc-deficient diet suggested a significant attenuation in the accumulation/retention of zinc in liver and pancreas. The amount of 67Zn accumulated in the pancreas and liver within 30 minutes in zinc-replete mice was twice that of the natural abundance of this isotope in these tissues. 67Zn represents 4.2% of the total natural Zn isotopes (64Zn> 66Zn = 68Zn ≫ 67Zn in relative abundance). In these zinc-replete mice an increase of 4.2% in total cellular zinc was detected within 30 min of the gavage. This is a reasonable approximation of a physiologically relevant amount. In the zinc-deficient mice, by three hours after the gavage triple-knockout mice had accumulated only a small increase in total zinc in the liver (1.6% increase) and pancreas (1.1% increase) whereas the wild-type mice had accumulated 6.8% more zinc in the liver and 7.6% more zinc in the pancreas. Although these studies do not reveal the exact reason for these differences (uptake, distribution, retention) in zinc metabolism, they clearly reveal that these Zip genes play an important role(s) in zinc homeostasis during periods of zinc deficiency.
The physiological mechanisms of action of these, and most other ZIPs remain to be determined. We hypothesize that on an organismal level, subfamily II members function in zinc homeostasis by controlling the distribution and/or retention of zinc in specific cells and organs. Zip1 and Zip3 are actively expressed in the lamina propria of the intestine and are thus unlikely to be important in the uptake of dietary zinc from the intestinal lumen (6). They may instead function in the distribution of zinc once it is acquired from the diet and transported through enterocytes. All three of these Zip genes are expressed in liver surrounding the central vein in a remarkable gradient pattern. Zip2 is expressed at high levels exclusively in a single layer of peri-central hepatocytes (33). Zip1 and Zip3 are also expressed in these cells as well as in a gradient that diminishes away from the central vein in deeper layers of hepatocytes (5; 6). This pattern of expression suggests that these proteins function in the retention of zinc in the liver by preventing its escape into the central vein or that they function to collect zinc from the venous blood back into the liver. The cellular localization of these proteins in hepatocytes remains to be determined. These genetic studies indicate that these three zinc transporters are not essential for the rapid accumulation/retention of zinc in the liver when zinc is replete, that their functions are not interdependent, and that they function in the accumulation/retention of zinc in the liver (and pancreas) during periods of zinc deficiency. Zip2 appears to have a unique role in iron metabolism in the liver although the mechanism is not understood (33).
Studies of ZIP1 and ZIP3 in transfected cells suggest that these proteins are recruited to the cell surface under zinc-deficient culture conditions (42) and in vivo studies of Zip1 expression and protein abundance do not suggest significant changes during zinc deficiency in the liver, intestine and visceral yolk sac (Fig. 3C) (44), but studies of the cellular localization of these proteins in vivo have yielded conflicting results. ZIP1 can display different localization patterns in adherent versus non-adherent cells, being vesicular in epithelial cell types and apical in K562 cells (31). Perhaps in vivo the localization of this protein reflects the microenvironment of each cell-type. When zinc levels are low these proteins may be moved to the cell surface. However, in the human prostate, which is very rich in zinc, ZIP1 plays a role in zinc uptake by normal cells and diminished expression of this gene has been associated with prostate cancer (13). In contrast, over-expression of Zip1 inhibits growth of malignant PC-3 cells (14). There is little information on the role of zinc in the mouse prostate gland but as yet we have seen no evidence for prostate or other tumors in the triple-knockout mouse colony. Whether these mice may be more prone to the induction of cancer remains to be examined. ZIP1 has also been reported to induce an osteogenic phenotype in transfected mesenchymal stem cells (37). We have noted no difference in the bone zinc content of triple-knockout versus wild-type mice and no gross abnormalities in bone formation in the triple-knockout mice (J. Geiser and G.K. Andrews, unpublished observations).
ZIP3 has been proposed to play a major role in zinc uptake by mammary epithelial cells (23; 24) and even in the maintenance of viable mammary cells (24), but neonatal pups nursing from triple-knockout females showed no signs of zinc deficiency, unlike the lethal milk mutant phenotypes (34) or the phenotypes noted during dietary zinc deficiency while nursing. Perhaps ZIP3 functions transiently during lactation but its absence and the absence of the all subfamily II members does not lead to obvious phenotypic abnormalities in mice when zinc-replete. ZIP3 does not appear to play a pivotal physiological role in the survival of functional secretory mammary epithelial cells in mice.
The functions of ZIP2 are clearly unique from those of ZIP1 and ZIP3 as is its pattern of expression. ZIP2 influences calcium and iron metabolism in zinc deficient embryos and liver, respectively (33). ZIP14 (39) has recently been reported mediate transport of non- transferrin bound iron into hepatocytes (29) although this finding has recently been challenged (15). Our studies reveal that ZIP2 plays a unique role in iron accumulation in the liver. Whether ZIP2 can transport iron is under investigation.
Perspectives and Significance
The molecular mechanisms regulating zinc homeostasis in mammals are important to understand because zinc deficiency is a world-wide problem that affects hundreds of thousands of children, in particular. A deficiency of zinc causes a multitude of negative physiological effects during periods of rapid growth and differentiation. It is thought that the 14 members of the solute-carrier (Slc)39a family are critical for the uptake of zinc into cells, but the physiological functions of most remain obscure. Herein we report studies of three highly conserved members (ZIPs 1 to 3) that compose subfamily II. These studies show that this family of genes has evolved to function during periods of dietary zinc deficiency but are dispensable when dietary zinc is replete. They play important, non-compensatory roles consistent with their tissue-specific patterns of expression. Although much remains to be learned about the structure, function and regulation of the mammalian ZIP proteins, our studies show that mutations in these Zip genes can modify sensitivity to the stress of dietary zinc deficiency and could therefore be important in human health.
Acknowledgments
We thank Drs. Mario Capecchi and Richard Palmiter for gene targeting vectors and members of the Transgenic and Gene-Targeting Facility at the KU Medical Center for generating chimeric mice. We also wish to thank Dr. Bruce Roe at the University of Oklahoma for BAC sequencing.
Grants: This study was funded, in part, by NIH grant DK059369 to G.K.A. T.K. was supported, in part, by a fellowship from the Kanae Foundation for the Promotion of Medical Science.
References
- 1.Andrews GK, Wang H, Dey SK, Palmiter RD. The mouse zinc transporter 1 gene provides an essential function during early embryonic development. Genesis. 2004;40:74–81. doi: 10.1002/gene.20067. [DOI] [PubMed] [Google Scholar]
- 2.Chowanadisai W, Lonnerdal B, Kelleher SL. Identification of a mutation in SLC30A2 (ZnT-2) in women with low milk zinc concentration that results in transient neonatal zinc deficiency. J Biol Chem. 2006;281:39699–39707. doi: 10.1074/jbc.M605821200. [DOI] [PubMed] [Google Scholar]
- 3.Cole TB, Robbins CA, Wenzel HJ, Schwartzkroin PA, Palmiter RD. Seizures and neuronal damage in mice lacking vesicular zinc. Epilepsy Res. 2000;39:153–169. doi: 10.1016/s0920-1211(99)00121-7. [DOI] [PubMed] [Google Scholar]
- 4.Cousins RJ, Liuzzi JP, Lichten LA. Mammalian zinc transport, trafficking, and signals. J Biol Chem. 2006;281:24085–24089. doi: 10.1074/jbc.R600011200. [DOI] [PubMed] [Google Scholar]
- 5.Dufner-Beattie J, Huang ZL, Geiser J, Xu W, Andrews GK. Generation and Characterization of Mice Lacking the Zinc Uptake Transporter ZIP3. Mol Cell Biol. 2005;25:5607–5615. doi: 10.1128/MCB.25.13.5607-5615.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dufner-Beattie J, Huang ZL, Geiser J, Xu W, Andrews GK. Mouse ZIP1 and ZIP3 genes together are essential for adaptation to dietary zinc deficiency during pregnancy. Genesis. 2006;44:239–251. doi: 10.1002/dvg.20211. [DOI] [PubMed] [Google Scholar]
- 7.Dufner-Beattie J, Kuo YM, Gitschier J, Andrews GK. The adaptive response to dietary zinc in mice involves the differential cellular localization and zinc-regulation of the zinc transporters ZIP4 and ZIP5. J Biol Chem. 2004;279:49082–49090. doi: 10.1074/jbc.M409962200. [DOI] [PubMed] [Google Scholar]
- 8.Dufner-Beattie J, Langmade SJ, Wang F, Eide D, Andrews GK. Structure, function, and regulation of a subfamily of mouse zinc transporter genes. J Biol Chem. 2003;278:50142–50150. doi: 10.1074/jbc.M304163200. [DOI] [PubMed] [Google Scholar]
- 9.Dufner-Beattie J, Wang F, Kuo YM, Gitschier J, Eide D, Andrews GK. The Acrodermatitis Enteropathica Gene ZIP4 Encodes a Tissue-specific, Zinc-regulated Zinc Transporter in Mice. J Biol Chem. 2003;278:33474–33481. doi: 10.1074/jbc.M305000200. [DOI] [PubMed] [Google Scholar]
- 10.Dufner-Beattie J, Weaver BP, Geiser J, Bilgen M, Larson M, Xu W, Andrews GK. The mouse acrodermatitis gene Slc39a4 (ZIP4) is essential for development and heterozygosity causes hypersensitivity to zinc deficiency. Hum Mol Genet. 2007;16:1391–1399. doi: 10.1093/hmg/ddm088. [DOI] [PubMed] [Google Scholar]
- 11.Eide DJ. Zinc transporters and the cellular trafficking of zinc. Biochim Biophys Acta. 2006;1763:711–722. doi: 10.1016/j.bbamcr.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 12.Eng BH, Guerinot ML, Eide D, Saier MH., Jr Sequence analyses and phylogenetic characterization of the ZIP family of metal ion transport proteins. J Membr Biol. 1998;166:1–7. doi: 10.1007/s002329900442. [DOI] [PubMed] [Google Scholar]
- 13.Franklin RB, Feng P, Milon B, Desouki MM, Singh KK, Kajdacsy-Balla A, Bagasra O, Costello LC. hZIP1 zinc uptake transporter down regulation and zinc depletion in prostate cancer. Mol Cancer. 2005;4:32–45. doi: 10.1186/1476-4598-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Franklin RB, Ma J, Zou J, Guan Z, Kukoyi BI, Feng P, Costello LC. Human ZIP1 is a major zinc uptake transporter for the accumulation of zinc in prostate cells. J Inorg Biochem. 2003;96:435–442. doi: 10.1016/s0162-0134(03)00249-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Girijashanker K, He L, Soleimani M, Reed JM, Liu Z, Wang B, Li H, Dalton TP, Nebert DW. Slc39a14 gene encodes ZIP14, a metal/bicarbonate symporter: similarities to the ZIP8 transporter. Mol Pharmacol. 2008 doi: 10.1124/mol.107.043588. In Press: PM:18270315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guerinot ML. The ZIP family of metal transporters. Biochim Biophys Acta. 2000;1465:190–198. doi: 10.1016/s0005-2736(00)00138-3. [DOI] [PubMed] [Google Scholar]
- 17.Huang L, Yu YY, Kirschke CP, Gertz ER, Lloyd KK. Znt7 (Slc30a7)-deficient mice display reduced body zinc status and body fat accumulation. J Biol Chem. 2007;282:37053–37063. doi: 10.1074/jbc.M706631200. [DOI] [PubMed] [Google Scholar]
- 18.Huang LP, Gitschier J. A novel gene involved in zinc transport is deficient in the lethal milk mouse. Nature Genet. 1997;17:292–297. doi: 10.1038/ng1197-292. [DOI] [PubMed] [Google Scholar]
- 19.Huang ZL, Dufner-Beattie J, Andrews GK. Expression and Regulation of SLC39A Family Zinc Transporters in the Developing Mouse Intestine. Dev Biol. 2006;295:571–579. doi: 10.1016/j.ydbio.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 20.Inoue K, Matsuda K, Itoh M, Kawaguchi H, Tomoike H, Aoyagi T, Nagai R, Hori M, Nakamura Y, Tanaka T. Osteopenia and male-specific sudden cardiac death in mice lacking a zinc transporter gene, Znt5. Hum Mol Genet. 2002;11:1775–1784. doi: 10.1093/hmg/11.15.1775. [DOI] [PubMed] [Google Scholar]
- 21.Kambe T, Weaver BP, Andrews GK. The Genetics of Essential Metal Homeostasis during Development. Genesis. 2008 doi: 10.1002/dvg.20382. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kambe T, Yamaguchi-Iwai Y, Sasaki R, Nagao M. Overview of mammalian zinc transporters. Cell Mol Life Sci. 2004;61:49–68. doi: 10.1007/s00018-003-3148-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelleher SL, Lonnerdal B. Zn transporter levels and localization change throughout lactation in rat mammary gland and are regulated by Zn in mammary cells. J Nutr. 2003;133:3378–3385. doi: 10.1093/jn/133.11.3378. [DOI] [PubMed] [Google Scholar]
- 24.Kelleher SL, Lonnerdal B. Zip3 plays a major role in zinc uptake into mammary epithelial cells and is regulated by prolactin. Am J Physiol Cell Physiol. 2005;288:C1042–C1047. doi: 10.1152/ajpcell.00471.2004. [DOI] [PubMed] [Google Scholar]
- 25.Kirschke CP, Huang LP. ZnT7, a novel mammalian zinc transporter, accumulates zinc in the Golgi apparatus. J Biol Chem. 2003;278:4096–4102. doi: 10.1074/jbc.M207644200. [DOI] [PubMed] [Google Scholar]
- 26.Krebs NF, Miller LV, Naake VL, Lei S, Westcott JE, Fennessey PV, Hambidge KM. The use of stable isotope techniques to assess zinc metabolism. J Nutr Biochem. 1995;6:292–301. [Google Scholar]
- 27.Kury S, Dreno B, Bezieau S, Giraudet S, Kharfi M, Kamoun R, Moisan JP. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:239–240. doi: 10.1038/ng913. [DOI] [PubMed] [Google Scholar]
- 28.Lamar EE, Palmer E. Y-encoded, species-specific DNA in mice: evidence that the Y chromosome exists in two polymorphic forms in inbred strains. Cell. 1984;37:171–177. doi: 10.1016/0092-8674(84)90312-x. [DOI] [PubMed] [Google Scholar]
- 29.Liuzzi JP, Aydemir F, Nam H, Knutson MD, Cousins RJ. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc Natl Acad Sci U S A. 2006;103:13612–13617. doi: 10.1073/pnas.0606424103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller LV, Krebs NF, Hambidge KM. Human zinc metabolism: Advances in the modeling of stable isotope data. Adv Exp Med Biol. 1998;445:253–269. doi: 10.1007/978-1-4899-1959-5_16. [DOI] [PubMed] [Google Scholar]
- 31.Milon B, Dhermy D, Pountney D, Bourgeois M, Beaumont C. Differential subcellular localization of hZip1 in adherent and non- adherent cells. FEBS Lett. 2001;507:241–246. doi: 10.1016/s0014-5793(01)02950-7. [DOI] [PubMed] [Google Scholar]
- 32.Palmiter RD, Huang L. Efflux and compartmentalization of zinc by members of the SLC30 family of solute carriers. Pflugers Arch - Eur J Physiol. 2004;447:744–751. doi: 10.1007/s00424-003-1070-7. [DOI] [PubMed] [Google Scholar]
- 33.Peters JL, Dufner-Beattie J, Xu W, Geiser J, Lahner B, Salt DE, Andrews GK. Targeting of the mouse Slc39a2 (Zip2) gene reveals highly cell-specific patterns of expression, and unique functions in zinc, iron and calcium homeostasis. Genesis. 2007;45:339–352. doi: 10.1002/dvg.20297. [DOI] [PubMed] [Google Scholar]
- 34.Piletz JE, Lonnerdal B, Hurley LS, Berry W, Ganschow RE, Herschman HR. Zinc and copper in milk and tissues of nursing lethal milk mutant mice. J Nutr. 1987;117:83–90. doi: 10.1093/jn/117.1.83. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki T, Ishihara K, Migaki H, Ishihara K, Nagao M, Yamaguchi-Iwai Y, Kambe T. Two different zinc transport complexes of cation diffusion facilitator proteins localized in the secretory pathway operate to activate alkaline phosphatases in vertebrate cells. J Biol Chem. 2005;280:30956–30962. doi: 10.1074/jbc.M506902200. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki T, Ishihara K, Migaki H, Matsuura W, Kohda A, Okumura K, Nagao M, Yamaguchi-Iwai Y, Kambe T. Zinc transporters, ZnT5 and ZnT7, are required for the activation of alkaline phosphatases, zinc-requiring enzymes that are glycosylphosphatidylinositol-anchored to the cytoplasmic membrane. J Biol Chem. 2005;280:637–643. doi: 10.1074/jbc.M411247200. [DOI] [PubMed] [Google Scholar]
- 37.Tang Z, Sahu SN, Khadeer MA, Bai G, Franklin RB, Gupta A. Overexpression of the ZIP1 zinc transporter induces an osteogenic phenotype in mesenchymal stem cells. Bone. 2006;38:181–198. doi: 10.1016/j.bone.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 38.Taylor KM, Hiscox S, Nicholson RI. Zinc transporter LIV-1: a link between cellular development and cancer progression. Trends Endocrinol Metab. 2004;15:461–463. doi: 10.1016/j.tem.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 39.Taylor KM, Morgan HE, Johnson A, Nicholson RI. Structure-function analysis of a novel member of the LIV-1 subfamily of zinc transporters, ZIP14. FEBS Lett. 2005;579:427–432. doi: 10.1016/j.febslet.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 40.Taylor KM, Morgan HE, Smart K, Zahari NM, Pumford S, Ellis IO, Robertson JF, Nicholson RI. The emerging role of the LIV-1 subfamily of zinc transporters in breast cancer. Mol Med. 2007;13:396–406. doi: 10.2119/2007-00040.Taylor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taylor KM, Nicholson RI. The LZT proteins; the LIV-1 subfamily of zinc transporters. Biochimica et Biophysica Acta. 2003;1611:16–30. doi: 10.1016/s0005-2736(03)00048-8. [DOI] [PubMed] [Google Scholar]
- 42.Wang F, Dufner-Beattie J, Kim BE, Petris MJ, Andrews G, Eide DJ. Zinc-stimulated endocytosis controls activity of the mouse ZIP1 and ZIP3 zinc uptake transporters. J Biol Chem. 2004;279:24631–24639. doi: 10.1074/jbc.M400680200. [DOI] [PubMed] [Google Scholar]
- 43.Wang K, Zhou B, Kuo YM, Zemansky J, Gitschier J. A novel member of a zinc transporter family is defective in acrodermatitis enteropathica. Am J Hum Genet. 2002;71:66–73. doi: 10.1086/341125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weaver BP, Dufner-Beattie J, Kambe T, Andrews GK. Novel zinc-responsive post-transcriptional mechanisms reciprocally regulate expression of the mouse Slc39a4 and Slc39a5 zinc transporters (Zip4 and Zip5) Biol Chem. 2007;388:1301–1312. doi: 10.1515/BC.2007.149. [DOI] [PMC free article] [PubMed] [Google Scholar]