

Abstract
Release of endogenous dynorphin opioids within the spinal cord after partial sciatic nerve ligation (pSNL) is known to contribute to the neuropathic pain processes. Using a phosphoselective antibody [κ opioid receptor (KOR-P)] able to detect the serine 369 phosphorylated form of the KOR, we determined possible sites of dynorphin action within the spinal cord after pSNL. KOR-P immunoreactivity (IR) was markedly increased in the L4-L5 spinal dorsal horn of wild-type C57BL/6 mice (7-21 d) after lesion, but not in mice pretreated with the KOR antagonist nor-binaltorphimine (norBNI). In addition, knock-out mice lacking prodynorphin, KOR, or G-protein receptor kinase 3 (GRK3) did not show significant increases in KOR-P IR after pSNL. KOR-P IR was colocalized in both GABAergic neurons and GFAP-positive astrocytes in both ipsilateral and contralateral spinal dorsal horn. Consistent with sustained opioid release, KOR knock-out mice developed significantly increased tactile allodynia and thermal hyperalgesia in both the early (first week) and late (third week) interval after lesion. Similarly, mice pretreated with norBNI showed enhanced hyperalgesia and allodynia during the 3 weeks after pSNL. Because sustained activation of opioid receptors might induce tolerance, we measured the antinociceptive effect of the κ agonist U50,488 using radiant heat applied to the ipsilateral hindpaw, and we found that agonist potency was significantly decreased 7 d after pSNL. In contrast, neither prodynorphin nor GRK3 knock-out mice showed U50,488 tolerance after pSNL. These findings suggest that pSNL induced a sustained release of endogenous prodynorphin-derived opioid peptides that activated an anti-nociceptive KOR system in mouse spinal cord. Thus, endogenous dynorphin had both pronociceptive and antinociceptive actions after nerve injury and induced GRK3-mediated opioid tolerance.
Keywords: κ opioid receptor, opiate, dynorphin, receptor phosphorylation, desensitization, allodynia, hyperalgesia
Introduction
Numerous studies have established that peripheral nerve damage can elicit neuropathic pain characterized by hyperalgesia, during which noxious stimuli are perceived as more painful, and allodynia, during which normally innocuous stimuli elicit pain (Zimmermann, 2001). Although morphine and related analgesics are effective in controlling acute pain, many clinical studies have shown that opioids lack analgesic efficacy in neuropathic pain (Arner and Meyerson, 1988). The basis for the lack of opioid efficacy is not clear, but neuropathic damage has been shown to cause profound reorganization of the nociceptive circuits within the spinal cord and brain, including changes in gene expression and morphology (Mayer et al., 1999; Ossipov et al., 2000; Przewlocki and Przewlocka, 2001).
One of the changes implicated in maintaining neuropathic pain is an induction of dynorphin expression (Wagner et al., 1993; Wang et al., 2001), and considerable evidence now supports the conclusion that enhanced expression of endogenous dynorphin within the spinal cord promotes morphine tolerance (Vanderah et al., 2000) and is pronociceptive (Caudle and Isaac, 1988; Wang et al., 2001). Therefore, a decrease in the anti-allodynic activity of spinal morphine treatment in neuropathic pain may be caused by the elevation of dynorphin across multiple spinal segments after nerve injury (Malan et al., 2000).
The mechanism of the pronociceptive dynorphin effect is not clear, but intrathecal administration of dynorphin A or des-Tyr-dynorphin produces non-opioid receptor-mediated neurotoxicity and neuropathy (Walker et al., 1982; Caudle and Mannes 2000). Pretreatment with the NMDA receptor antagonists dizocilpine (MK-801) and 3S-3α,4aα,6β,8aα-decahydro-6-(phosphonomethyl) -3-isoquinolinecarboxylic acid (LY235959) but not the opioid receptor antagonist naloxone blocks the dynorphin-induced allodynia (Laughlin et al., 1997). Thus, non-opioid effects of dynorphin clearly contribute to the neuropathic pain state.
The dynorphins also act as endogenous agonists at the κ opioid receptors (KORs); however, the increased dynorphin expression in neuropathic pain might also lead to an activation of the intrinsic KORs. Numerous studies have documented antinociceptive effects of intrathecal and systemic administration of selective κ agonists (Nakazawa et al., 1991; Kolesnikov et al., 1996). Thus, the endogenous dynorphin-derived opioids may have both antinociceptive and pronociceptive actions. How sustained activation of opioid receptors by endogenous dynorphins contributes to the neuropathic pain state is not clear. Obara and colleagues (2003) reported that the KOR antagonist nor-binaltorphimine (norBNI) significantly enhanced allodynia and exhibited pronociceptive action after sciatic nerve ligation (SNL). Sustained activation of KOR would be expected to produce tolerance and might shift the balance from antinociceptive to pronociceptive actions of the endogenous dynorphin system, although this has not been established.
The principal goal of the present study was to assess the contribution of endogenous dynorphin activation of KOR on the response induced by partial SNL (pSNL), a widely used neuropathic pain model. We tested the hypothesis that endogenous dynorphin activation of KOR contributed to the behavioral responses observed and asked whether sustained activation of KOR resulted in analgesic tolerance. We report that pSNL resulted in KOR phosphorylation at a site shown to induce receptor desensitization (McLaughlin et al., 2003b). Consistent with this finding, pSNL caused a shift in the agonist dose-response curves characteristic of KOR tolerance. KOR desensitization would be expected to reduce the antinociceptive effects of the endogenous dynorphin-κ opioid system and thus enhance the allodynic and hyperalgesic responses underlying the neuropathy.
Materials and Methods
All protocols with mice were approved by the Institutional Animal Care and Use Committee in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (1996) and guidelines for the International Association for the Study of Pain (Zimmermann, 1983). Mice were inspected regularly by veterinary staff to ensure compliance. Young adult male C57BL/6 mice (Charles River Laboratories, Wilmington, MA) or transgenic mice on a C57BL/6 genetic background were used in these experiments. Transgenic mice having specific disruptions of the genes for KOR (-/-), prodynorphin (-/-), and G-protein receptor kinase 3 GRK3 (-/-) were generated as described (Peppel et al., 1997; Hough et al., 2000; Sharifi et al., 2001). Heterozygous breeding pairs, backcrossed >10 generations onto C57BL/6 backgrounds, were used to generate knockout (-/-) and wild-type littermate controls for this study. Pups were genotyped as described previously (McLaughlin et al., 2003a). Mice used were 16-28 weeks of age and weighed 25-35 gm at the time of the start of the procedures. Knock-out mice showed no discernible differences from wild-type littermates in growth, life span, loco-motor activity, morphine sensitivity, or basal nociceptive response. All mice were housed in groups of two to four in plastic cages using Bed-ACob (The Andersons, Maumee, OH) for home bedding within the Animal Core Facility at the University of Washington and maintained in pathogen-free housing units. The housing rooms were illuminated on a 12 hr light/dark cycle with lights on at 7 A.M.; lab chow and water were available ad libitum.
Surgical procedures-neuropathic pain model. The pSNL model of neuropathic pain was used as described previously (Seltzer et al., 1990). The right sciatic nerve was exposed at the gluteal region, and approximately one-third to one-half the diameter of the nerve was tightly ligated with 7-0 silk suture (Surgical Specialties Corporation, Reading, PA).
Behavior tests for allodynia and hyperalgesia. The mice were habituated to handling and testing equipment at least 20-30 min before experiments. Threshold for tactile allodynia was measured with a series of von Frey filaments (Semmes-Weinstein monofilaments, Stoelting, Wood Dale, IL). The mice stood on a metal mesh covered with a plastic dome. The plantar surface of the hindpaws was touched with different von Frey filaments with a bending force from 0.166 to 3.63 gm until the threshold that induced paw withdrawal was found. According to the manufacturer's specifications, a 1 gm filament induces a force of ∼9.8 mN. Unresponsive mice received a maximal score of 3.63 gm. Responsiveness to the von Frey filament is usually described as allodynia (Bennett and Xie, 1988) because unlesioned mice do not respond with paw withdrawal after touch with filaments of these strengths (see Fig. 2); however, electrophysiological measures were not made to confirm that A-β fibers had been activated. Thus the response measured might also have been caused by mechanical hyperalgesia.
Figure 2.

Response thresholds of the ipsilateral (A) and contralateral (B) hindpaw to usually innocuous tactile (von Frey hair) stimuli in wild-type (WT) mice (n = 16), KOR (-/-) mice (n = 6), and prodynorphin (-/-) mice after pSNL (n = 10) during 21 d after partial sciatic nerve ligation. A, After pSNL, both wild-type and prodynorphin (-/-) mice developed significantly (*p<0.05) greater allodynia on the ipsilateral side as evident from the decreased thresholds to tactile stimulation compared with mice receiving sham-ligation surgeries (baseline). One week after pSNL, prodynorphin (-/-) mice showed significantly less allodynia compared with WT mice after pSNL. KOR (-/-) mice developed significantly more allodynia when compared with WT mice after pSNL throughout the test period. On day 21 after pSNL, norBNI (10 mg/kg, i.p.) administered to wild-type mice significantly decreased response threshold (#p < 0.05). In contrast, KOR (-/-) mice and prodynorphin (-/-) mice were not significantly affected by norBNI treatment. B, The contralateral hindpaw of wild-type mice showed a significant (p < 0.05) decrease in response threshold during the 21 d after pSNL compared with sham-ligated mice (baseline). The allodynic response of KOR (-/-) mice was even greater than that of wild-type mice as evident from the significantly lower response thresholds. Prodynorphin (-/-) mice after pSNL showed significantly less allodynia compared with WT mice after pSNL.
To assess thermal sensitivity (hyperalgesia), paw withdrawal latencies to a radiant heat stimulus were measured using the paw flick test apparatus (IITC Life Science, Woodland Hills, CA) (Hargreaves et al., 1988). Paw withdrawal latency was determined as the average of three measurements per paw. The stimulus intensity was adjusted to give 6-8 sec withdrawal latency in the normal mouse (baseline). The cutoff time in the absence of response was 15 sec to prevent tissue damage.
Immunohistochemical staining in the spinal cord. Adult mice were anesthetized with halothane (Sigma, St. Louis, MO) and intracardially perfused with a fresh solution of 4% paraformaldehyde in phosphate buffer (PB) (0.1 m sodium phosphate, pH 7.4). The lumbar spinal cords were removed and postfixed in the same fixative for 1-2 hr. The tissue was then sunk in solution of 30% (w/v) sucrose in PB at 4°C over 24 hr. Transverse sections (40 μm) of the spinal cords were cut on a sliding microtome and placed in PB until the tissue was processed for immunohistochemistry. Free-floating spinal cord sections were rinsed in 0.1 m Tris buffer, pH 7.4, for 15 min, in 0.1 m Tris buffered saline (TBS), pH 7.4, for 15 min, blocked using 2% avidin in TBS for 30 min, rinsed in TBS for 30 min, blocked using 2% biotin for 30 min, and finally rinsed in TBS for 30 min. The tissue sections were then incubated in 20 μg/ml affinity-purified rabbit anti-KOR-phosphoselective (KOR-P) antiserum [primary antibody (Ab)] (McLaughlin et al., 2003b) for 36 hr at 4°C. All antibodies were diluted in a solution containing 0.1% Triton X-100 and 4% normal goat serum in 0.1 m TBS. The tissue sections were rinsed in TBS for 60 min and incubated in biotinylated goat anti-rabbit IgG (secondary Ab) diluted 1:300 for 1 hr at 37°C. The tissue sections were rinsed with TBS for 60 min and incubated in avidin d-fluorescein diluted 1:300 for 1 hr at 37°C. The sections were rinsed in TBS for 10 min, rinsed in TB for 20 min, and then mounted on gelatin-coated slides and viewed with a Nikon Eclipse E600 fluorescence microscope (Tokyo, Japan) or an MRC 600 confocal microscope (Bio-Rad, Hercules, CA) located in the W. M. Keck Imaging Facility at the University of Washington.
For double staining of KOR-P and glial fibrillary acidic protein (GFAP), KOR-P and GABA, and KOR-P and neurokinin-1 receptor (NK1), the primary antibodies were the mixture of 20 μg/ml rabbit anti-KOR-P antiserum and guinea pig anti-GFAP antiserum (diluted 1:1000) (Chemicon, Temecula, CA), 20 μg/ml rabbit anti-KOR-P antiserum and guinea pig anti-GABA antiserum (diluted 1:1000) (Chemicon), or 20 μg/ml rabbit anti-KOR-P antiserum and guinea pig anti-NK1 antiserum (diluted 1:1000) (Chemicon). The secondary antibodies were the mixture of biotinylated goat anti-rabbit IgG and donkey anti-guinea pig IgG rhodamine conjugate (1:250; Jackson ImmunoResearch, West Grove, PA). For control sections, primary antibodies were preincubated with their cognate peptide or no primary Ab was used. In either case, no specific staining was observed. Relative staining intensities were analyzed using NIH Image version 1.62 software (National Institutes of Health, Bethesda, MD). Values were expressed as ratios of labeling intensity over background.
Chemicals. NorBNI was obtained from the National Institute on Drug Abuse drug supply program (National Institutes of Health). U50,488 was purchased from Sigma.
Statistical analysis. All data were analyzed by repeated measures ANOVA (Prism4, Graph-Pad, San Diego, CA). Significant results demonstrated by ANOVA were analyzed further with Tukey test or Student's t test for significant pair-wise comparisons. Response data are presented as means ± SEM of the animal treatment group, with significance set at p < 0.05. EC50 values and 95% confidence intervals (CIs) were derived using Prism4 software. Transgenic mouse studies were done using paired wild-type and knock-out littermates; the investigator performing the behavioral or anatomical studies was blind to genotype. Although data were statistically analyzed using the littermate pairings, graphical presentations of wild-type groups were merged because no statistical differences were observed.
Results
Behavioral manifestations of neuropathic pain: thermal hyperalgesia and tactile allodynia
Prodynorphin (-/-) and wild-type (+/+) littermate controls displayed similar response latencies after noxious thermal stimulation in the radiant heat test (Fig. 1). After pSNL, both prodynorphin (-/-) and littermate control (+/+) animals appeared healthy, with well groomed coats, but they guarded the injury-side hindpaw. Initially, both groups showed significantly increased sensitivity of the ipsilateral hindpaw to thermal stimulation compared with the values before lesion (Fig. 1A) (p < 0.05). After pSNL, wild-type mice showed a progressive reduction in the mean paw withdrawal latency with maximum at day 9 after pSNL of 4.43 sec (Fig. 1A). Paw withdrawal latencies recovered slowly but did not return to baseline latencies even 21 d after pSNL (Fig. 1A). As reported previously (Wang et al., 2001), the prodynorphin (-/-) mice showed significantly less hyperalgesia than wild-type mice 6 d after the lesion, and we found that the difference between prodynorphin (-/-) and (+/+) mice persisted at least 21 d after the pSNL. Sham surgery did not induce thermal hyperalgesia in either prodynorphin (-/-) or (+/+) during the 21 d testing (data not shown). Except at day 1 after pSNL, mice showed no significant changes (p < 0.05) in paw withdrawal latencies in the contralateral, unlesioned side (Fig. 1B).
Figure 1.

Response latencies of the ipsilateral (A) and contralateral (B) hindpaws to noxious thermal (radiant heat applied to the hindpaw) stimuli for wild-type (WT) mice (n = 16), KOR (-/-) mice (n = 6), and prodynorphin (-/-) mice (n = 10) during 21 d after pSNL. A, After pSNL, both WT and knock-out mice developed significant hyperalgesia as evident from the significantly decreased thermal latencies compared with before pSNL. WT mice receiving sham ligation did not significantly change response latencies from baseline when tested on the same repeated measures schedule (data not shown). KOR (-/-) mice developed significantly greater hyperalgesia when compared with WT mice after pSNL. One week after pSNL, prodynorphin (-/-) mice showed significantly less hyperalgesia compared with WT mice. At day 21, administration of norBNI (10 mg/kg, i.p.) significantly reduced the response latency of WT mice (p < 0.05) but did not affect the responses of KOR (-/-) mice or prodynorphin (-/-) mice after pSNL. B, Except on the first day after pSNL, the contralateral hindpaw of neither WT nor prodynorphin (-/-) mice showed any reduction in thermallatency. The contralateral hindpaw of the KOR(-/-) mice showed significantly more hyperalgesia compared with WT mice after pSNL. Data represent means ± SEM; *p < 0.05, using one-way ANOVA followed by either Tukey or t test as appropriate.
To define the contribution of the κ opioid receptor to the hyperalgesia response after lesion, we measured the behavioral responses of mice lacking κ opioid receptors [KOR(-/-)] and littermate wild-type mice (Fig. 1). Consistent with the effects of the selective κ receptor antagonist norBNI on pSNL-induced allodynia reported previously (Obara et al., 2003), we found that KOR gene deletion enhanced the hyperalgesic response after pSNL (Fig. 1A). The ipsilateral paw withdrawal latency changes after pSNL of KOR(-/-) were significantly larger than for wild-type controls. In addition, we found that the hyperalgesic responses of KOR(-/-) mice during the 21 d after ligation were not significantly different from wild-type mice pretreated with norBNI (10 mg/kg, i.p.; given every 3 d) (data not shown). The contralateral hindpaw of the KOR(-/-) mice after pSNL developed significantly more hyperalgesia than that of the wild-type mice but still significantly less hyperalgesia than the ipsilateral hindpaw (Fig. 1B). The hyperalgesia of the contralateral hindpaw of the KOR(-/-) mice resolved after 2 weeks.
To assess whether the difference between the wild-type and KOR(-/-) responses was caused by sustained activation of KOR by dynorphin, we administered norBNI (10 mg/kg) on day 21 after pSNL. As shown in Figure 1A, norBNI given on day 21 significantly reduced the response latency in wild-type mice but not in KOR(-/-) mice. As expected, the response latencies of ligated mice lacking prodynorphin were not significantly affected by norBNI given 21 d after pSNL (Fig. 1A). These results suggest that in wild-type mice, prodynorphin-derived opioids continued to be released to activate KOR and produce an antinociceptive effect for at least 3 weeks after pSNL.
Partial sciatic nerve ligation induced tactile allodynia
Similar effects on tactile allodynia were observed using the von Frey assay (Fig. 2). Before the pSNL, prodynorphin (-/-), KOR(-/-), and wild-type mice showed no response to the maximal von Frey hair that was applied (3.63 gm). After the pSNL, both prodynorphin (-/-) and wild-type mice showed enhanced sensitivity that was not significantly different until day 6, when the prodynorphin (-/-) mice showed an accelerated recovery toward baseline (Fig. 2A). The tactile allodynia of wild-type mice attenuated in the subsequent days to a mean threshold of 2.16 gm at day 21 (Fig. 2A). On days 7-12 after pSNL, prodynorphin (-/-) mice showed significantly less allodynia compared with wild-type mice (Fig. 2A) (p < 0.05). Unlike the hyperalgesic response, significant tactile allodynia manifested on the contralateral side as well (Fig. 2B). The tactile allodynia on the contralateral side resolved after 2 weeks for both the prodynorphin (-/-) and (+/+) mice, and the (-/-) mice showed significantly less allodynia than the wild-type mice.
KOR(-/-) mice showed enhanced allodynia on the ipsilateral and contralateral side compared with wild-type littermate controls (Fig. 2A). The allodynic responses of KOR(-/-) mice during the 21 d after pSNL were not significantly different from wild-type mice pretreated with norBNI before pSNL (data not shown). Because KOR gene deletion and norBNI pretreatment produced the same effect, the actions of both experimental manipulations are likely to be caused by a specific disruption of κ receptor-mediated response. As seen in the hyperalgesia assay, norBNI treatment on day 21 significantly enhanced the allodynic response of wild-type mice but not KOR(-/-) or prodynorphin (-/-) mice (Fig. 2A). Thus, pSNL was likely to have induced a sustained anti-allodynic effect by dynorphin activation of KOR in addition to its previously documented pro-allodynic effect mediated by non-opioid mechanisms.
KOR activation increased KOR phosphorylation in wild-type mice spinal cord
We next asked whether activated κ opioid receptors could be visualized in the mouse spinal cord after sciatic nerve ligation. Phosphorylation of serine-369 in KOR by G-protein receptor kinase initiates receptor desensitization (Appleyard et al., 1997), and this phosphorylation event may also be an effective marker of κ opioid receptor activation caused by endogenous dynorphin release. We previously generated and characterized a rabbit polyclonal antibody raised against the C-terminal domain of KOR having phospho-serine 369 (McLaughlin et al., 2003b). The affinity-purified Ab (KOR-P) reacted more intensely with KOR expressed in human embryonic kidney 293 or AtT20 cells after treatment with κ agonist, and KOR-P also reacted more intensely with KOR isolated from brains of mice treated in vivo with κ agonist (McLaughlin et al., 2003b, 2004).
To assess the utility of this Ab in detecting KOR activation in the spinal cord, mice were injected with the κ agonist U50,488 (50 mg/kg, i.p.), with U50,488 plus norBNI (10 mg/kg, i.p.), or with saline 30 min before they were anesthetized with halothane and perfused with paraformaldehyde. For wild-type mice treated with U50,488, KOR-P staining in lumbar spinal cord (L4-L5) increased significantly (Fig. 3A, D) compared with tissue from mice pretreated with saline (Fig. 3B, E). The increased KOR-P staining was located primarily in laminas I-III of the spinal cord. Using NIH Image quantitation, KOR-P labeling intensity was 44 ± 13% (n = 5) greater than in dorsal horn of untreated mice. KOR-P staining in wild-type mice pretreated with both norBNI and U50,488 was not significantly different from untreated, control mice (Fig. 3C,F). As an important specificity control, KOR(-/-) mice did not show specific KOR-P staining after U50,488 treatment (Fig. 3G).
Figure 3.
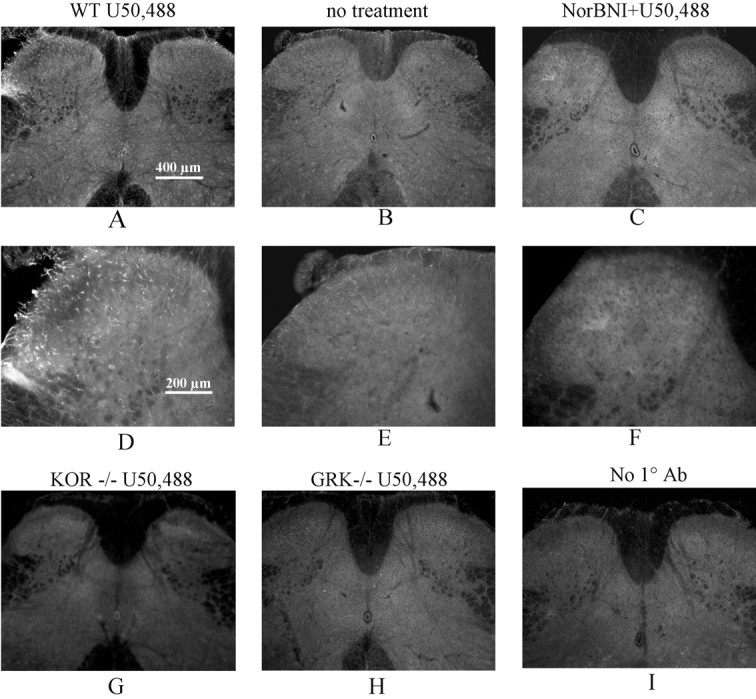
κ Opioid receptor activation increased the intensity of KOR-P Ab staining in the spinal cord dorsal horn. Injection of 50 mg/kg U50,488 30 min before perfusion caused a significant increase in KOR-P IR (A, D) compared with that of saline injection (B, E) in the dorsal horn of the mouse lumbar spinal cord. This increase in KOR-P Ab staining was blocked by 10 mg/kg norBNI pretreatment (C,F) in wild-type mice and was not evident in mice lacking κ opioid receptors (KOR-/-) (G) or GRK3 (GRK-/-) (H). Negative controls showed the lack of specific staining in the absence of primary Ab (I). Scale bars: A-C, G-I, 400 μm; D-F, 200 μm. The results shown are representative images taken from more than three independent replications.
On the basis of our previous work (McLaughlin et al., 2004), we tested the hypothesis that the increase in KOR-P labeling was caused by phosphorylation by GRK3, the enzyme shown to mediate homologous KOR desensitization and analgesic tolerance. Mice lacking GRK3 did not show specific increases in KOR-P labeling in spinal cord after U50,488 treatment (Fig. 3H). Additional controls showed that there was no KOR-P labeling in the absence of primary Ab (Fig. 3I) or after the primary Ab was preabsorbed with the immunogen (data not shown). These results support the conclusion that KOR-P immunolabeling specifically detected agonist-activated κ opioid receptors expressed in spinal cord that had been phosphorylated on serine 369 by GRK3. These findings are consistent with our previous characterization of the specificity of this Ab in transfected cells (McLaughlin et al., 2003b) and in mouse brain after U50,488 treatment (McLaughlin et al., 2004).
pSNL activated KOR and increased KOR phosphorylation in the lumbar spinal cord dorsal horn of wild-type mice
To assess the effects of endogenous dynorphin released after nerve damage, κ receptor phosphorylation was determined by KOR-P labeling of spinal cord tissue 7 d after pSNL. As shown in Figure 4, KOR-P staining was significantly increased in tissue from pSNL mice (Fig. 4A,B) compared with spinal cord tissue from wild-type mice having the sham-ligation surgery (Fig. 4C,D). Image intensity quantitation showed that pSNL increased KOR-P labeling intensity 52 ± 0.1% over sham-ligated staining in dorsal horn (n = 5 different animals in each group). KOR-P IR remained elevated even 21 d after pSNL (data not shown).
Figure 4.
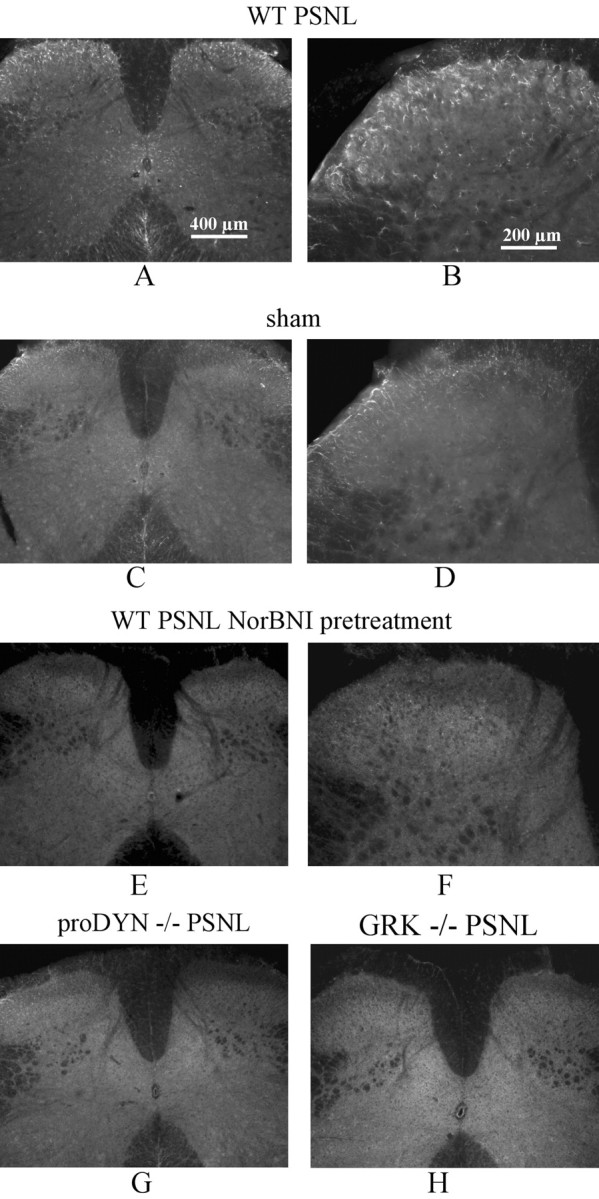
pSNL resulted in an increased intensity of KOR-P Ab staining in the lumbar spinal cord dorsal horn in wild-type mice. All mice were perfused and fixed on day 7 after pSNL or sham-ligation surgery. A substantial increase in immunofluorescence intensity of KOR-P Ab staining was evident in wild-type pSNL mice (A, B) but not in sham-ligated mice (C, D) in the lumbar spinal cord, mainly in laminas I-III and X. No apparent differences were evident between the ipsilateral and contralateral sides (A). B shows a higher-power image of the same dorsal horn as in A. The increase in KOR-P staining was blocked by 10 mg/kg norBNI pretreatment before pSNL (E, F) and was not evident in mice lacking prodynorphin (proDYN-/-) (G) or GRK 3 (GRK-/-) (H). Scale bars: A, C, E, G, H, 400 μm; B, D, F, 200 μm. The results shown are representative images taken from more than three independent replications.
The selective KOR antagonist norBNI has the additional advantage of producing very prolonged KOR antagonism in mice (Horan et al., 1992). We confirmed that after norBNI (10 mg/kg, i.p.) the analgesic effects of U50,488 in the thermal nociception assay were blocked for >1 week (data not shown). Pretreatment with norBNI (10 mg/kg, i.p.) before pSNL blocked the increase in KOR-P staining (Fig. 4E,F). Image intensity quantitation showed that there was no significant change (p > 0.05; n = 5).
Furthermore, the increase in KOR-P labeling was not evident in mice lacking prodynorphin (Fig. 4G) or in mice lacking GRK (Fig. 4H). These findings are consistent with the results after U50,488 treatment and suggest that endogenous prodynorphin-derived opioids were released after the sciatic nerve damage to activate κ opioid receptors. Furthermore, the results are consistent with the hypothesis that agonist-activated KORs in spinal cord were substrates for GRK3, which specifically phosphorylated KOR-serine 369. Image quantitation of labeling intensities after pSNL confirmed that neither GRK(-/-) nor prodynorphin (-/-) mice showed significant changes in KOR-P labeling compared with matched, unligated controls (data not shown).
Different cell types were phosphorylated after pSNL in spinal cord of wild-type mice
To define the cell types expressing the enhanced KOR-P labeling after pSNL, we double labeled with KOR-P and antibodies against markers expressed in different cell types in spinal dorsal horn. GABA is the principal inhibitory neurotransmitter in the superficial laminas of the dorsal horn and has an important role in the regulation of nociceptive transmission (Todd and Sullivan, 1990; Somers and Clemente, 2002). Confocal microscopy of spinal cord sections double labeled with KOR-P (Fig. 5A,D) and GABA (Fig. 5B,E) antibodies revealed colocalization of KOR-P and GABA immunoreactivities (Fig. 5C,F) in the dorsal horn of the lumbar spinal cord.
Figure 5.
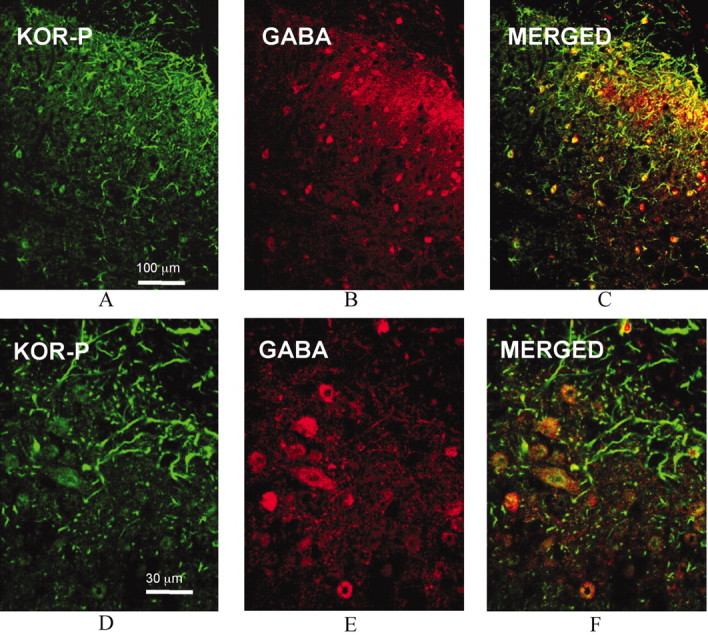
Comparison of immunolocalization of KOR-P and GABA IR in the dorsal horn of wild-type mouse lumbar spinal cord on day 7 after pSNL shows partial overlap. Representative sections show that KOR-P and GABA Ab staining were mainly in laminas I-III (A, B). KOR-P (A, D, green) and GABA immunoreactivity (B, E, red) colocalized in neurons evident in the dorsal horn (C, F, yellow). These results suggest GABAergic neurons expressed KOR that was phosphorylated after pSNL. Bottom panels (D-F) were higher magnification of the same confocal images shown in the top panels (A-C).
In Figure 5, C and F, merged images demonstrated other structures labeled by KOR-P that did not express GABA immunoreactivity. Because these KOR-P-labeled structures resembled astrocytes, we next double labeled sections with KOR-P and GFAP antibodies. GFAP is a selective marker for astrocytes (Ma and Quirion, 2002). Confocal microscopy of mice spinal cord sections revealed substantial colocalization of KOR-P (Fig. 6A,D) and GFAP (Fig. 6B,E) immunoreactivities (Fig. 6C,F) in the dorsal horn of the lumbar spinal cord. Expression of KOR by spinal cord astrocytes is consistent with previous results from other brain regions (Bunn et al., 1985; Stiene-Martin et al., 1998). These results suggest that κ opioid receptors expressed in both glial cells (astrocytes) and GABAergic neurons in wild-type mouse spinal cord were phosphorylated after pSNL.
Figure 6.
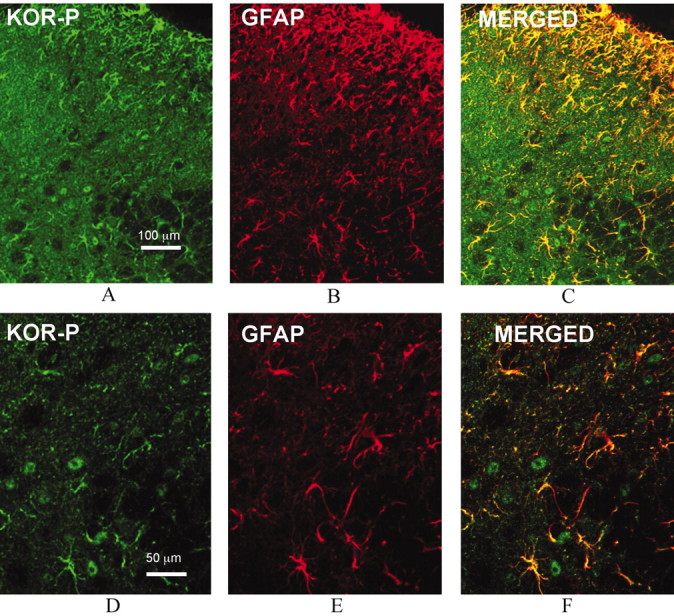
Comparison of immunolocalization of KOR-P and GFAP IR in the dorsal horn of wild-type mouse lumbar spinal cord on day 7 after pSNL shows partial overlap. Representative images show that KOR-P and GFAP Ab staining were mainly in laminas I-III (A, B). KOR-P (A, D, green) and GFAP immunoreactivity (B, E, red) were coexpressed in the dorsal horn (C, F, yellow). Bottom panel is higher-magnification confocal images of the same images shown in the top panel.
Dual labeling with KOR-P and NK1 antibodies after pSNL in wild-type mouse spinal cord
Because the hyperalgesic effects of pSNL were restricted primarily to the lesioned side of the mouse, we were surprised that the change in KOR-P labeling seemed bilateral (Fig. 4). To confirm that the lesions produced in our study were consistent with previous anatomical studies, we assessed changes in NK1 receptor immunoreactivity after pSNL. NK1 receptors are activated in chronic sciatic nerve injury (Cumberbatch et al., 1998), and NK1 receptor immunoreactivity markedly increased on the ipsilateral spinal dorsal horn (Goff et al., 1998). Double labeling with KOR-P and NK1 Ab in wild-type mice 7 d after surgery showed that NK1 receptor immunoreactivity was increased only on the ipsilateral side of the spinal cord, whereas in adjacent sections, KOR-P immunoreactivity increased on both the ipsilateral and contralateral sides of the spinal cord (data not shown). The bilateral changes in KOR-P staining were consistent with the changes in allodynic response on both the ipsilateral and contralateral paws after pSNL (Fig. 2). In addition, the bilateral activation of KOR is consistent with bilateral descending activation of spinal dynorphin systems (Vanderah et al., 2001).
Changes in antinociceptive responses of wild-type and knock-out mice to pharmacological activation of κ receptors after pSNL
Phosphorylation of KOR by GRK3 was shown previously to induce receptor desensitization in cell lines and analgesic tolerance in vivo (Appleyard et al., 1997; McLaughlin et al., 2004), and the increased KOR-P staining suggested that the κ receptors may have become phosphorylated and desensitized after pSNL. To assess whether phosphorylated KORs in spinal cord were less sensitive to opioids after pSNL, we measured the analgesic effects of the κ agonist U50,488 in the radiant heat, paw withdrawal test. U50,488 produced a dose-dependent antinociception in sham-lesioned mice with an EC50 of 15 mg/kg (95% CI, 8.8-28.1) compared with an EC50 of 37 mg/kg (95% CI, 31.2-45.1) for wild-type mice 7 d after pSNL (Fig. 7A).
Figure 7.

Dose-response curves showing the antinociceptive effect of theκ agonist U50,488 in wild type (WT), prodynorphin (-/-), and GRK3 (-/-) mice after sham-ligation surgery or after pSNL in the Hargreave's radiant heat test. Different doses of U50,488 (5, 10, 25, 50, or 75 mg/kg) were administered intraperitoneally to mice 30 min before their paw withdrawal response latencies were measured. Maximum possible effect (%MPE) was calculated as 100 × (latency-baseline)/(cutoff-baseline); cutoff was 15 sec in this experiment. A, U50,488 was significantly less effective in wild-type mice after pSNL (p < 0.05) than in WT mice after sham-ligation surgery (n = 10 for each group). B, pSNL did not affect the U50,488 dose-response curves for either the prodynorphin (-/-) or the GRK3 (-/-) mice (p>0.05) (n=5 for each group);however, the prodynorphin (-/-) mice were significantly less sensitive than the GRK3 (-/-) mice to U50,488. C, Response latencies of the ipsilateral hindpaw to noxious thermal (radiant heat applied to the hindpaw) stimuli in wild-type (WT) mice after pSNL (n = 16) and in GRK3 (-/-) mice after pSNL (n = 5). GRK3 (-/-) mice after pSNL developed partially significantly less hyperalgesia compared with wild-type mice after pSNL. At day 21, norBNI(10 mg/kg, i.p.) was injected in WT and GRK3 (-/-) mice after pSNL. GRK3 (-/-) mice after pSNL developed a significant decrease in latency (#p < 0.05). Consistent with results shown in Figure 1 A, wild-type mice showed a significant decrease in response latency as well. Results are shown to illustrate that GRK3 (-/-) and prodynorphin (-/-) mice both show a decrease in response latencies after pSNL. Thus, the shift in U50,488 EC50 shown in A cannot result simply from a stronger nociceptive stimulus.
The rightward shift in the U50,488 dose-response curve evident in Figure 7A is consistent with opioid tolerance, but whether the reduction in potency was caused by receptor tolerance or by heightened pain sensitivity after pSNL was not clear. pSNL-induced hyperalgesia might require a more analgesic drug to overcome the greater nociceptive stimulus. To resolve this question, U50,488 dose-response curves for mice lacking either GRK3 or prodynorphin genes were obtained after sham or pSNL. Sciatic nerve ligation induced hyperalgesia for both prodynorphin (-/-) (Fig. 1A) and GRK3 (-/-) (Fig. 7C) mice; however, neither group of knock-out mice showed enhanced KOR-P labeling (Fig. 4). After pSNL, neither the prodynorphin (-/-) nor the GRK3 (-/-) dose-response curves were significantly different from sham-ligated animals (Fig. 7B). Neither knock-out genotype demonstrated a rightward shift in the U50,488 antinociceptive dose-response curve after pSNL; thus neither showed U50,488 tolerance despite being hyperalgesic. The important aspect of this result is that the analgesic tolerance evident in Figure 7A is not an artifactual consequence of the shift in baseline nociception evident in Figure 7C and thus was likely to have been caused by KOR desensitization evoked by dynorphin release and subsequent GRK3 phosphorylation of KOR.
The U50,488 EC50 in prodynorphin (-/-) mice was 14 mg/kg [9.0-23.4, 95% CI], which was not significantly different from sham-ligated wild-type mice (Fig. 7A). The U50,488 EC50 of the GRK3 (-/-) mice was 6 mg/kg (5.1-7.2 CI), which was significantly less than sham-ligated wild-type mice. The basis of the greater sensitivity of the GRK3 (-/-) mice to U50,488 is not known but may suggest that normal, unlesioned animals have a basal level of GRK3-mediated desensitization caused by normal receptor activation. Consistent with the interpretation that GRK3 (-/-) mice had a larger fraction of functional (nondesensitized) KOR, pSNL produced significantly less hyperalgesia in GRK3 (-/-) mice than in wild-type mice (Fig. 7C).
Consistent with the results using the radiant heat assay, the sensitivity of wild-type mice to U50,488 was significantly less in the von Frey allodynia assay than that of GRK3 (-/-) or prodynorphin (-/-) mice. Sham-lesioned, wild-type, and knockout mice do not show allodynia (data not shown), but all three showed allodynia after pSNL. U50,488 dose-response curves yielded EC50 and 95% CI values of 26 (17.7-38.5) mg/kg for wild-type, 16 (15.9-17.3) mg/kg for prodynorphin (-/-), and 12 (6.7-22.3) mg/kg for GRK3 (-/-) mice. The lower potency of U50,488 in the wild-type mice further supports the conclusion that KOR tolerance was induced after pSNL and supports the suggestion that tolerance reduced the sensitivity of neuropathic pain to κ analgesics.
Discussion
The data from the present study support the hypothesis that pSNL activates the endogenous κ opioid system in mouse spinal cord and induces opioid receptor tolerance. As shown by the results using prodynorphin knock-out mice, endogenous dynorphins have complex effects on nociceptive circuits. Activation of non-opioid systems enhances neuropathic pain, whereas simultaneous activation of κ opioid systems has analgesic effects. The balance between the antinociceptive and pronociceptive effects of dynorphins may be controlled by receptor desensitization mechanisms. κ receptor phosphorylation after sustained release of endogenous dynorphins was mediated by GRK3 and resulted in receptor tolerance. Although the principal opiate analgesics used therapeutically are thought to act at the μ opioid receptor, the reduction in KOR sensitivity evident in the pSNL model suggests that tolerance to endogenous opioids may contribute to the lower sensitivity to opiate analgesics during neuropathic pain.
In this study, we used immunohistochemistry and a newly generated phosphospecific Ab able to detect the GRK3-phosphorylated form of KOR to directly determine the sites of endogenous dynorphin action within the spinal cord. The KOR-P Ab was generated against a sequence within the C-terminal domain of the κ opioid receptor shown previously to be phosphorylated after agonist activation of the receptor; mutation of serine 369 to alanine blocked homologous desensitization of KOR mediated by GRK3 and β-arrestin (Appleyard et al., 1997). The specificity of this phosphoselective Ab was demonstrated previously (McLaughlin et al., 2003b), and injection of mice with κ agonist was found to produce both analgesic tolerance and coincident KOR phosphorylation detected by increased KOR-P staining (McLaughlin et al., 2004). Results from the present study extend those observations by demonstrating that the sustained release of endogenous dynorphins during chronic pain may also increase KOR phosphorylation as detected by increased KOR-P staining. The increased KOR-P staining and analgesic tolerance evident in pSNL mice are consistent with the hypothesis that pSNL induces endogenous dynorphin release, KOR activation, GRK3-mediated phosphorylation of KOR, and homologous desensitization of KOR.
The results presented in this study suggest that endogenous dynorphins act on both GABAergic neurons and GFAP-positive astrocytes in spinal cord. This is a novel means to identify targets of endogenous opioid action. Previous studies have shown that dynorphin immunoreactivity is present to a large extent in the superficial layers of the dorsal horn, and dynorphin peptide levels have been shown to increase bilaterally after traumatic injury (Cox et al. 1985; Wagner et al., 1993). The bilateral increase in peptide levels is consistent with the bilateral increase in KOR-P immunoreactivity evident in this study. We do not identify the sites of dynorphin release within the spinal cord in the present study; however, previous work by Duggan and coworkers (Riley et al., 1996) demonstrated that dynorphin A(1-8) released by chronic joint inflammation could be detected by an Ab-coated probe within rat spinal cord. Interestingly, released peptide was detected in both laminas I and IV-V by this method, despite the concentration of dynorphin immunoreactivity in the superficial layers of dorsal horn. These results are consistent with the broad distribution of KOR-P labeling observed in the present study. The dimensions of the dynorphin synapse and diffusion range from sites of release to sites of action in the spinal cord are not yet know, but we estimated previously that dynorphin released in the dentate gyrus of the guinea pig hippocampus may diffuse ∼50-100 μm from sites of release to sites of action (Drake et al., 1994). The principal concentration of KOR immunolabeling is in laminas I and II of the rat dorsal horn, and this overlaps with the majority of the preprodynorphin-immunoreactive fibers (Arvidsson et al., 1995). The apparent discrepancy between where the dynorphin peptides are concentrated and where the activated receptors detected by KOR-P staining is very intriguing; however, the sites of dynorphin release and the location of the functional targets are not fully understood and require further study. It is important to acknowledge that KOR-P labeling is likely to detect only a fraction of the pool of functional and activated receptor. Detection depends on the sensitivity of the probe and the fraction of activated receptors that were phosphorylated after activation. These values have not yet been determined.
Expression of KOR on GABAergic neurons is consistent with previous electrophysiological studies showing that κ receptor activation by dynorphin A suppresses GABA-A chloride currents (Li et al., 2003), presumably by presynaptically inhibiting GABA release. GABA is the principal inhibitory neurotransmitter in the superficial laminas of the dorsal horn and has an important role in the regulation of nociceptive transmission. Local application of the GABAA receptor antagonist bicuculline to the spinal cord in the rat produces behavioral signs of tactile allodynia (Yaksh, 1989; Burgess et al., 2002) and can cause low-threshold mechanical stimuli to produce a flexion withdrawal reflex (Sivilotti and Woolf, 1994). Thus, dynorphin-induced inhibition of GABA release would be predicted to produce allodynia and not antinociception. Other studies have demonstrated that activation of KOR in spinal cord results in presynaptic inhibition of excitatory transmission at low dynorphin A concentrations (Randic et al., 1995). Suppression of excitatory input would seem to be a more plausible mechanism of dynorphin-mediated antinociceptive actions, but further work would be required to define the effects of endogenous dynorphins on this system.
Activation of KOR expressed by spinal cord astrocytes is also very interesting. The role of non-neuronal cells during chronic pain is gaining increasing appreciation (Watkins and Maier, 2003). Spinal nerve ligation was found to activate p38 mitogen-activated kinase to induce a microglial cell proliferation that enhanced the neuropathic pain response (Jin et al., 2003). Our results demonstrating dynorphin activation of KOR expressed by astrocytes further supports the hypothesis that activation of non-neuronal cells has an important role in the neuropathic pain response. For years, glial cells were considered only to have supportive and nutritive functions in the CNS. Glial cells are known to release many kinds of neurotransmitters and respond to neurotransmitters with changes in membrane potential (Barres, 1991). How the activation of KOR in astrocytes affects the neuropathic response is not yet clear, but activation of CNS glial cells has been implicated in the pathogenesis of several neurodegenerative diseases.
Our results with prodynorphin knock-out mice confirm previous findings by Porreca and colleagues (Ossipov et al., 2003). These results suggest that the endogenous dynorphins have an important pronociceptive role in the neuropathic response to nerve injury. Similarly, our results are consistent with previous work by Przewlocka and colleagues (Obara et al., 2003), who showed that pharmacological antagonism of κ receptors potentiated the allodynic response. The results presented in our study extend these findings by identifying the sites of endogenous opioid actions and suggesting a role for endogenous opioid-induced receptor tolerance in neuropathic pain. A consistent image emerges supporting the conclusion that endogenous dynorphins have both pronociceptive and antinociceptive actions after nerve injury.
Footnotes
The work was supported by United States Public Health Service Grants DA16898 and DA15916 (C.C.) from the National Institute on Drug Abuse. M. P. was supported by a fellowship from the German Research Foundation. R.J.L. and M.G.C. are investigators of the Howard Hughes Medical Institute. Dr. Uwe Hochgeschwender provided the prodynorphin knock-out mice; Joe Novak performed the mouse genotyping.
Correspondence should be addressed to Dr. Charles Chavkin, Department of Pharmacology, Box 357280, University of Washington School of Medicine, Seattle, WA 98195-7280. E-mail: cchavkin@u.washington.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/244576-09$15.00/0
M.X. and M.P. contributed equally to this work.
References
- Appleyard SM, Patterson TA, Jin W, Chavkin C (1997) Agonist-induced phosphorylation of the kappa-opioid receptor. J Neuroses 69: 2405-2412. [DOI] [PubMed] [Google Scholar]
- Arner S, Meyerson BA (1988) Lack of analgesic effect of opioids on neuropathic and idiopathic forms of pain. Pain 33: 11-23. [DOI] [PubMed] [Google Scholar]
- Arvidsson U, Riedl M, Chakrabarti S, Vulchanova L, Lee JH, Nakano AH, Lin X, Loh HH, Law PY, Wessendorf MW, Elde R (1995) The kappa-opioid receptor is primarily postsynaptic: combined immunohistochemical localization of the receptor and endogenous opioids. Proc Natl Acad Sci USA 92: 5062-5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA (1991) Glial ion channels. Curr Opin Neurobiol 1: 354-359. [DOI] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK (1988) A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33: 87-107. [DOI] [PubMed] [Google Scholar]
- Bunn SJ, Hanley MR, Wilkin GP (1985) Evidence for a kappa-opioid receptor on pituitary astrocytes: an autoradiographic study. Neurosci Lett 55: 317-323. [DOI] [PubMed] [Google Scholar]
- Burgess SE, Gardell LR, Ossipov MH, Malan Jr TP, Vanderah TW, Lai J, Porreca F, Wang Z, Brennan MB, Hochgeschwender U, Hruby VJ (2002) Time-dependent descending facilitation from the rostral ventromedial medulla maintains, but does not initiate, neuropathic pain. J Neurosci 22: 5129-5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudle RM, Isaac L (1988) A novel interaction between dynorphin (1-13) and an N-methyl-d-aspartate site. Brain Res 443: 329-332. [DOI] [PubMed] [Google Scholar]
- Caudle RM, Mannes AJ (2000) Dynorphin: friend or foe? Pain 87: 235-239. [DOI] [PubMed] [Google Scholar]
- Cox BM, Molineaux CJ, Jacobs TP, Rosenberger JG, Faden AI (1985) Effects of traumatic injury on dynorphin immunoreactivity in spinal cord. Neuropeptides 5: 571-574. [DOI] [PubMed] [Google Scholar]
- Cumberbatch MJ, Carlson E, Wyatt A, Boyce S, Hill RG, Rupniak NM (1998) Reversal of behavioural and electrophysiological correlates of experimental peripheral neuropathy by the NK1 receptor antagonist GR205171 in rats. Neuropharmacology 37: 1535-1543. [DOI] [PubMed] [Google Scholar]
- Drake CT, Terman GW, Simmons ML, Milner TA, Kunkel DD, Schwartzkroin PA, Chavkin C (1994) Dynorphin opioids present in dentate granule cells may function as retrograde inhibitory neurotransmitters. J Neurosci 14: 3736-3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff JR, Burkey AR, Goff DJ, Jasmin L (1998) Reorganization of the spinal dorsal horn in models of chronic pain: correlation with behaviour. Neuroscience 82: 559-574. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988) A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32: 77-88. [DOI] [PubMed] [Google Scholar]
- Horan P, Taylor J, Yamamura HI, Porreca F (1992) Extremely long-lasting antagonistic actions of nor-binaltorphimine (nor-BNI) in the mouse tail-flick test. J Pharmacol Exp Ther 260: 1237-1243. [PubMed] [Google Scholar]
- Hough LB, Nalwalk JW, Chen Y, Schuller A, Zhu Y, Zhang J, Menge WM, Leurs R, Timmerman H, Pintar JE (2000) Improgan, a cimetidine analog, induces morphine-like antinociception in opioid receptor-knockout mice. Brain Res 880: 102-1088. [DOI] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR (2003) p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci 23: 4017-4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesnikov Y, Jain S, Wilson R, Pasternak GW (1996) Peripheral kappa 1-opioid receptor-mediated analgesia in mice. Eur J Pharmacol 310: 141-143. [DOI] [PubMed] [Google Scholar]
- Laughlin TM, Vanderah TW, Lashbrook J, Nichols ML, Ossipov M, Porreca F, Wilcox GL (1997) Spinally administered dynorphin A produces long-lasting allodynia: involvement of NMDA but not opioid receptors. Pain 72: 253-260. [DOI] [PubMed] [Google Scholar]
- Li H, Wu L, Li YQ (2003) Opioid peptides modulate the response of neurons of the superficial laminae of the rat spinal dorsal horn to GABA. Biochem Biophys Res Commun 307: 730-736. [DOI] [PubMed] [Google Scholar]
- Ma W, Quirion R (2002) Partial sciatic nerve ligation induces increase in the phosphorylation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) in astrocytes in the lumbar spinal dorsal horn and the gracile nucleus. Pain 99: 175-184. [DOI] [PubMed] [Google Scholar]
- Malan TP, Ossipov MH, Gardell LR, Ibrahim M, Bian D, Lai J, Porreca F (2000) Extraterritorial neuropathic pain correlates with multisegmental elevation of spinal dynorphin in nerve-injured rats. Pain 86: 185-194. [DOI] [PubMed] [Google Scholar]
- Mayer DJ, Mao J, Holt J, Price DD (1999) Cellular mechanisms of neuropathic pain, morphine tolerance, and their interactions. Proc Natl Acad Sci USA 96: 7731-7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Marton-Popovici M, Chavkin C (2003a) κ opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci 23: 5674-5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Xu M, Mackie K, Chavkin C (2003b) Phosphorylation of a carboxyl-terminal serine within the kappa-opioid receptor produces desensitization and internalization. J Biol Chem 278: 34631-34640. [DOI] [PubMed] [Google Scholar]
- McLaughlin JP, Myers LC, Zarek PE, Caron MG, Lefkowitz RJ, Czyzyk TA, Pintar JE, Chavkin C (2004) Prolonged kappa-opioid receptor phosphorylation mediated by G-protein receptor kinase underlies sustained analgesic tolerance. J Biol Chem 279: 1810-1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa T, Furuya Y, Kaneko T, Yamatsu K (1991) Spinal kappa receptor-mediated analgesia of E-2078, a systemically active dynorphin analog, in mice. J Pharmacol Exp Ther 256: 76-81. [PubMed] [Google Scholar]
- Obara I, Mika J, Schafer MK, Przewlocka B (2003) Antagonists of the kappa-opioid receptor enhance allodynia in rats and mice after sciatic nerve ligation. Br J Pharmacol 140: 538-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, Malan Jr TP, Porreca F (2000) Spinal and supraspinal mechanisms of neuropathic pain. Ann NY Acad Sci 909: 12-24. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, Vanderah TW, Porreca F (2003) Induction of pain facilitation by sustained opioid exposure: relationship to opioid antinociceptive tolerance. Life Sci 73: 783-800. [DOI] [PubMed] [Google Scholar]
- Peppel K, Boekhoff I, McDonald P, Breer H, Caron MG, Lefkowitz RJ (1997) G protein-coupled receptor kinase 3 (GRK3) gene disruption leads to loss of odorant receptor desensitization. J Biol Chem 272: 25425-25428. [DOI] [PubMed] [Google Scholar]
- Przewlocki R, Przewlocka B (2001) Opioids in chronic pain. Eur J Pharmacol 429: 79-91. [DOI] [PubMed] [Google Scholar]
- Randic M, Cheng G, Kojic L (1995) κ-opioid receptor agonists modulate excitatory transmission in substantia gelatinosa neurons of the rat spinal cord. J Neurosci 15: 6809-6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley RC, Zhao ZQ, Duggan AW (1996) Spinal release of immunoreactive dynorphin A(1-8) with the development of peripheral inflammation in the rat. Brain Res 710: 131-142. [DOI] [PubMed] [Google Scholar]
- Seltzer Z, Dubner R, Shir Y (1990) A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain 43: 205-218. [DOI] [PubMed] [Google Scholar]
- Sharifi N, Diehl N, Yaswen L, Brennan MB, Hochgeschwender U (2001) Generation of dynorphin knockout mice. Brain Res Mol Brain Res 86: 70-75. [DOI] [PubMed] [Google Scholar]
- Sivilotti L, Woolf CJ (1994) The contribution of GABAA and glycine receptors to central sensitization: disinhibition and touch-evoked allodynia in the spinal cord. J Neurophysiol 72: 169-179. [DOI] [PubMed] [Google Scholar]
- Somers DL, Clemente FR (2002) Dorsal horn synaptosomal content of aspartate, glutamate, glycine and GABA are differentially altered following chronic constriction injury to the rat sciatic nerve. Neurosci Lett 323: 171-174. [DOI] [PubMed] [Google Scholar]
- Stiene-Martin A, Zhou R, Hauser KF (1998) Regional, developmental, and cell cycle-dependent differences in mu, delta, and kappa-opioid receptor expression among cultured mouse astrocytes. Glia 22: 249-259. [PMC free article] [PubMed] [Google Scholar]
- Todd AJ, Sullivan AC (1990) Light microscope study of the coexistence of GABA-like and glycine-like immunoreactivities in the spinal cord of the rat. J Comp Neurol 296: 496-505. [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Gardell LR, Burgess SE, Ibrahim M, Dogrul A, Zhong CM, Zhang ET, Malan Jr TP, Ossipov MH, Lai J, Porreca F (2000) Dynorphin promotes abnormal pain and spinal opioid antinociceptive tolerance. J Neurosci 20: 7074-7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderah TW, Ossipov MH, Lai J, Malan Jr TP, Porreca F (2001) Mechanisms of opioid-induced pain and antinociceptive tolerance: descending facilitation and spinal dynorphin. Pain 92: 5-9. [DOI] [PubMed] [Google Scholar]
- Wagner R, DeLeo JA, Coombs DW, Willenbring S, Fromm C (1993) Spinal dynorphin immunoreactivity increases bilaterally in a neuropathic pain model. Brain Res 629: 323-326. [DOI] [PubMed] [Google Scholar]
- Walker JM, Moises HC, Coy DH, Baldrighi G, Akil H (1982) Nonopiate effects of dynorphin and des-Tyr-dynorphin. Science 218: 1136-1138. [DOI] [PubMed] [Google Scholar]
- Wang Z, Gardell LR, Ossipov MH, Vanderah TW, Brennan MB, Hochgeschwender U, Hruby VJ, Malan Jr TP, Lai J, Porreca F (2001) Pronociceptive actions of dynorphin maintain chronic neuropathic pain. J Neurosci 21: 1779-1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Maier SF (2003) GLIA: a novel drug discovery target for clinical pain. Nat Rev Drug Discov 2: 973-985. [DOI] [PubMed] [Google Scholar]
- Yaksh TL (1989) Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: effects of modulatory receptor systems and excitatory amino acid antagonists. Pain 37: 111-123. [DOI] [PubMed] [Google Scholar]
- Zimmermann M (1983) Ethical guidelines for investigations of experimental pain in conscious animals. Pain 16: 109-110. [DOI] [PubMed] [Google Scholar]
- Zimmermann M (2001) Pathobiology of neuropathic pain. Eur J Pharmacol 429: 23-37. [DOI] [PubMed] [Google Scholar]