

Abstract
Expression of leucine-rich alpha-2 glycoprotein (LRG), a member of the leucine-rich repeat family of proteins, was recently shown to be upregulated during neutrophil differentiation. Its precise role in granulopoiesis, however, remains unknown. In this paper, we show that the transcription factors PU.1 and C/EBPε that regulate the expression of multiple myeloid-specific genes also bind to the LRG promoter. We also demonstrate that LRG localizes to the same cytoplasmic compartment as myeloperoxidase and that G-CSF treatment of the 32Dcl3 myeloid cell line induces nuclear translocation of LRG. Stable transfection of LRG into 32Dcl3 cells resulted in accelerated G-CSF-mediated neutrophil differentiation and induction of CD11b expression. In contrast, constitutive expression of LRG in 32Dwt18 cells expressing a chimeric Epo/G-CSF receptor consisting of the Epo receptor extracellular domain fused to the G-CSF receptor transmembrane and cytoplasmic domains failed to induce accelerated neutrophil differentiation and CD11b expression in response to Epo stimulation. LRG-mediated accelerated differentiation and CD11b expression were found to correlate with an increased level of phospho-Stat3 but not with PU.1 or p27kip1 levels. Hence, similar to other genes involved in neutrophil differentiation, the expression of LRG also appears to be regulated by PU.1 and C/EBPε. Collectively, these findings suggest a role for LRG in modulating neutrophil differentiation and expression of CD11b via non-redundant G-CSF receptor signals.
Keywords: Granulopoiesis, Transcription Factors, G-CSF
INTRODUCTION
Leucine-rich α2-glycoprotein (LRG) was purified from human serum more than 25 years ago [1], establishing a novel family of proteins characterized by the presence of leucine-rich repeats in their amino acid sequences. Although several family members have been shown to be involved in signal transduction, protein-protein interaction or cell adhesion and development [2], a function for LRG has remained elusive. Consensus binding sites for several myeloid specific transcription factors are present within the putative promoter region of the gene for human LRG and its expression was previously shown by our laboratory to be upregulated during neutrophilic differentiation [3]. Notably, the gene for human LRG localizes to the same region on chromosome 19p13.3, to which the genes for multiple primary neutrophil granule enzymes also localize.
A specific role for LRG in granulopoiesis has not been elucidated. G-CSF has been shown to be the major regulator of granulopoiesis [4] and supports not only the survival, proliferation, and neutrophilic differentiation of myeloid progenitor cells but also activates certain functions of mature, terminally differentiated neutrophils [5-7]. The biological activities of G-CSF are mediated by specific receptors on the surface of responsive cells. G-CSF binds to the extracellular portion of the G-CSF receptor (G-CSFR) resulting in activation of a complex signaling cascade that includes the Jak and Stat kinases. Expression of the G-CSFR, like many other myeloid-specific genes including genes for several neutrophil granule proteins, has been shown to be regulated by PU.1 and the C/EBP family of transcription factors.
Studies in mice with knock-out or knock-in mutations in the G-CSFR gene suggest that the G-CSFR generates unique nonredundant signals required for neutrophil (PMN) production and marrow egress to maintain homeostatic levels of circulating PMN [7-10]. G-CSFR knock-out mice have chronic neutropenia with a uniform decrease in myeloid cells in the bone marrow. PMN from these mice also exhibit selective defects in activation [11,12]. These observations suggest that the G-CSFR is a critical regulator of both PMN differentiation and activation.
Previous work by our laboratory to further characterize the mechanisms by which G-CSF promotes neutrophil differentiation using representational difference analysis (RDA) resulted in the isolation of cDNA and genomic clones for human and murine LRG. To further characterize the physiological role of LRG in granulopoeisis, we stably transfected the murine LRG cDNA into 32Dcl3 cells endogenously expressing the wild-type murine G-CSFR and also into 32Dwt18 cells stably transfected with a chimeric Epo/G-CSF receptor, and examined the effects of constitutive expression of LRG on their proliferation, differentiation and activation. Previous investigators have utilized the 32Dwt18 cell line in place of 32Dcl3 cells to study myeloid maturation since 32Dwt18 cells transferred to Epo-containing media do not undergo the early massive cell death observed when 32Dcl3 cells are transferred to G-CSF containing media [13]. We report here significant differences in these cell lines and provide further evidence that unique signaling pathways are activated by the G-CSFR which involve LRG. Our data also suggest a role for the extracellular domain of the G-CSFR in signal transmission.
MATERIALS AND METHODS
Cell Culture and Stable Transfection
G-CSF responsive murine 32Dcl3 cells were kindly provided by Dr. Giovanni Rovera (The Wistar Institute, Philadelphia, PA), and were maintained in RPMI-1640 medium supplemented with 10% FBS and 10% WEHI 3B conditioned medium, as a source of IL-3. The 32Dwt18 cells were generously provided by Dr. Dan Link (Washington University, St Louis, MO), and were maintained in IMDM medium supplemented with 10% FBS and 10% WEHI 3B conditioned medium.
Murine LRG (mLRG) cDNA was cloned into the pcDNA 3.1D/V5-His-TOPO vector (Invitrogen, Carlsbad, CA) in frame with the V5 epitope tag and transfected into 32Dcl3 cells by electroporation (300V, 960μF, single pulse). mLRG cDNA in frame with the V5 epitope tag was also cloned into the pcDNA6/HisB vector (Invitrogen, Carlsbad, CA), and transfected into 32Dwt18 cells by electroporation as described above. Clones expressing mLRG were selected by growth in media containing either G418 (300μg/ml, for 32Dcl3 cells transfection) or Blasticidin (10μg/ml, for 32Dwt18 cells transfection) at 48 hours after transfection. Expression of mLRG mRNA in positive clones was confirmed by RT-PCR. To confirm expression of mLRG protein, western blotting was performed on whole cell lysates (WCL) from positive clones with the anti-V5 antibody (Invitrogen, Carlsbad, CA).
Differentiation Assay
Purified recombinant human (rh)G-CSF was generously provided by Amgen Inc. (Thousand Oaks, CA), and purified human erythropoietin (Procrit, epoitin alfa, Epo) by Ortho Biotech Inc. (Bridgewater, NJ). 32Dcl3 and 32Dwt18 cells were removed from IL-3 containing media, washed twice in PBS to remove residual IL-3, then transferred to G-CSF (10ng/ml, for 32Dcl3 cells) or Epo (0.1-1u/ml, for 32Dwt18 cells) containing media to induce neutrophilic granulocyte differentiation. At varying time points, aliquots were removed for analysis of differentiation. Differentiation was monitored by Wright-Giemsa staining and flow cytometric analysis.
Flow Cytometric Analysis
Cells (1× 106) were washed twice with cold HBSS solution supplemented with 1% BSA and 0.1% sodium azide, resuspended in 500μl of cold HBSS/BSA/Azide solution, and incubated with FITC conjugated rat anti-mouse CD11b or FITC conjugated rat anti-mouse CD13 antibody (Pharmingen, San Diego, CA) for 1hr at 4°C. After incubation, cells were washed twice with cold HBSS/BSA/sodium azide solution, and fixed in 500 μl of cold 1% paraformaldehyde solution. Cells were then analyzed by flow cytometry on FACS Calibur (Becton-Dickinson Immunocytometry System, Manassas VA) using Cell Quest software.
Immunofluorescence Staining and Confocal Microscopy
A total of 5×104 32Dcl3 cells per clone were spun onto non-charged slides, and fixed in 4% formaldehyde. The fixed cells were washed in PBS, and then permeabilized with 1% TX-100 in TBS for 30 min at room temperature. To minimize non-specific antibody binding, the slides were incubated with 1% BSA in TBS for 1hr at room temperature. To detect expression of the V5-tagged mLRG protein, a murine anti-V5 antibody (Invitrogen, Carlsbad, CA) was used as the primary antibody in conjunction with the TRITC-conjugated donkey anti-mouse F(ab)’2 secondary antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). To detect mouse myeloperoxidase, goat anti-human myeloperoxidase antibody (cross reactive with mouse, Santa Cruz Biotechnology Inc., Santa Cruz, CA) and donkey anti-goat F(ab)’2 antibody conjugated with Cy5 (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) were used. Hoescht (Molecular Probes, Eugene, OR) was used for nuclear staining. ProLong anti-fade mounting media (Molecular Probes, Eugene, OR) was placed on each slide after staining. Slides were examined under confocal microscopy using a Zeiss LSM510 multiphoton confocal microscope. The excitation/emission spectra for the fluorophores used were as follows: Hoescht 360/415 nm, TRITC 550/570 nm, Cy5 633/660 nm.
Subcellular Fractionation
Subcellular fractionation of 32Dwt18 transfectants was performed as described [14]. Briefly, cells were washed with PBS, resuspended in cold KRG buffer (130mM NaCl, 5mM KCl, 1.27mM MgSO4, 0.95mM CaCl2, 5mM Glucose, 10mM NaH2PO4/Na2HPO4, pH 7.4, 5mM PMSF), and incubated on ice for 5 min. Cells were then spun down, resuspended at 108cells/ml in disruption buffer (100 mM KCl, 3mM NaCl, 3.5mM MgCl2, 10mM Piperazine N,N’-bis2[ethan-sulfonic acid], pH 7.2, 1mM ATPNa2, 0.5mM PMSF) with one complete mini protease inhibitor cocktail tablet (Roche Diagnostic Corporation, Indianapolis, IN), and disrupted by nitrogen cavitation (pressurized under nitrogen for 5 min at 380 psi). The cavitate was collected dropwise into disruption buffer containing a final concentration of 0.5mM EGTA. Nuclei and intact cells were pelleted by centrifugation at 400g for 15 min. The postnuclear supernatant was applied onto a three-layer Percoll gradient solution (densities 1.050, 1.065, and 1.090 g/ml), and was centrifuged at 37,000g for 30 min at 4°C. After centrifugation, six fractions of 5ml each were collected. Percoll was removed by ultracentrifugation at 100,000g for 90 min. Biological materials above the Percoll pellet were collected, and were next subjected to Western blot analysis.
Western blot analysis
Whole cell lysates (WCL) from unstimulated and differentiated cells were prepared as described [15]. Protein concentrations were determined using a BCA kit (Rockford, IL). For each sample, 50μg of protein was separated on 10% SDS-PAGE and the proteins transferred to nitrocellulose membranes. The membranes were probed with the respective antibodies, and proteins visualized by enhanced chemiluminescence (ECL) (Amersham, Piscataway, NJ). Antibodies to pStat3 and p27kip1 were purchased from Cell Signaling Technology Inc. (Beverly, MA). Antibodies to Stat3, PU.1 and Raf-1 were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Antibody to V5 tag was purchased from Invitrogen Corporation (Carlsbad, CA). A rabbit polyclonal antibody to the p85 subunit of PI3 kinase was purchased from Upstate Biotechnology Inc. (Lake Placid, NY).
Chromatin Immunoprecipitation Assay (ChIP)
ChIP assays were performed following the protocol for the acetyl-histone H4 ChIP Assay Kit (Upstate Biotechnology, Lake Placid, NY), using buffers supplied in the kit. Briefly, 5×106 cells were cross-linked with 1% formaldehyde for 30 min at 37°C. Prior to cross-linking, an aliquot of the cells was removed for analysis of input chromatin DNA. After cross-linking, cells were washed twice in cold PBS, resuspended in lysis buffer, and incubated on ice for 10 min. The samples were then subjected to sonification to shear protein-cross-linked DNA into fragments of 1-3 kb, and diluted with IP buffer. After pre-clearing with a salmon sperm DNA/protein A agarose slurry, samples were then incubated with antibodies to either PU.1 (4μg), C/EBPα (8μg), C/EBPε (4μg), or with normal rabbit IgG (4μg) at 4°C overnight. All antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Samples containing no antibody were also included as controls. After sequential washing with low salt, high salt, LiCl and TE buffers, the protein-DNA complex was eluted with freshly made elution buffer (1% SDS, 0.1M NaHCO3). Cross-linking was then reversed by addition of 5M NaCl at 65°C for 4 hrs. After proteinease K treatment, DNA was isolated by phenol/chloroform extraction followed by ethanol precipitation. Equal volumes of DNA products were used as PCR templates. Primers (Forward: AATCCCCACCTCACCCTTAACTAG, Reverse: CTATGTCTGTCCTCAAGACCCCAG) were designed to specifically amplify the sequences within the mLRG promoter corresponding to consensus binding sites for C/EBPα, C/EBPε and PU.1 (nt. 794-990, 197 nt product).
RESULTS
PU.1 and C/EBPε bind to the LRG promoter
Within the putative promoter region of LRG, consensus binding sequences for PU.1 and C/EBP are present [3]. To determine whether these sequences are functional binding sites for these transcription factors, we performed chromatin immunoprecipitation (ChIP) assays on parental 32Dwt18 cells. As shown in Figure 1, a low basal level of binding of both PU.1 and C/EBPε to the amplified sequence within the putative LRG promoter could be detected (Figure 1, lanes 5 and 9) which increased significantly after three days of culture in Epo-containing media to induce differentiation (Figure 1, lanes 6 and 10). In contrast, no significant changes in binding of C/EBPα to the same region in the LRG promoter was observed during the same time period following transfer to Epo-containing media. As negative controls, reactions were included in which either no antibody (Figure 1, lanes 1 and 2) or normal rabbit IgG (Figure 1, lanes 3 and 4) was added. As an additional negative control, PCR reactions were also performed in the absence of input DNA (Figure 1, lane 11). As a positive control, isolated DNA (Input, Figure 1, lane 12) was subjected to PCR amplification prior to immunoprecipitation using the same primers.
Figure 1. Chromatin immunoprecipitation assay (ChIP) indicated functional binding sites for PU.1 and C/EBPε within the promoter region of mLRG gene.
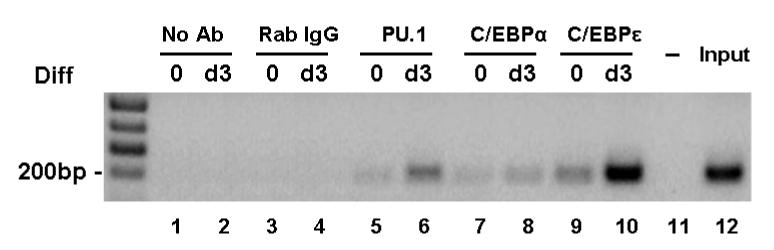
32Dwt18 cells were cross-linked with 1% formaldehyde, washed and resuspended in lysis buffer. Samples were sonicated to shear the protein-DNA complexes and immunoprecipitated with antibodies for PU.1, C/EBPα and C/EBPε. The complexes were washed, eluted, and the cross-linkage was reversed. Isolated DNA was then subjected to PCR using primer sets specific for the consensus binding sites of PU.1, C/EBPα and C/EBPε with the putative promoter region of mLRG gene. Normal rabbit IgG was used as a negative control. No-antibody control and input DNA control were also included.
Subcellular localization of LRG
Since the gene for human LRG maps to chromosome 19p13.3, which is the same region to which the genes for multiple other neutrophil granule proteins also localize [3], we were interested in determining whether LRG is targeted to myeloid granules. For these experiments, we examined the intracellular localization of constitutively expressed LRG containing a V5-epitope tag and endogenously expressed myeloperoxidase (MPO) in 32Dcl3 transfectants by immunofluorescence staining and multiphoton confocal microscopy. 32Dcl3 cells endogenously express MPO and we have previously shown that expression of LRG in these cells can be induced by G-CSF treatment after 16 h. In LRG-transfected cells, LRG (shown as green, Figure 2A) and MPO (shown as red, Figure 2B) were detected by anti-V5 and anti-MPO primary antibodies, which were bound to secondary antibodies conjugated with TRITC and Cy5, respectively. Nuclei were stained with Hoescht (shown as blue, Figure 2C). LRG was found to localize to the cytoplasmic compartment in a diffuse granular pattern that overlapped with the pattern observed for MPO (Figure 2D). Cells transfected with vector alone only displayed MPO staining, although both antibodies were added (Figure 2E). When stained with secondary antibodies only,LRG-transfected cells displayed only nuclear staining with a clear background (Figure 2F).
Figure 2. LRG exhibited the same cellular localization as that of myeloperoxidase (MPO).
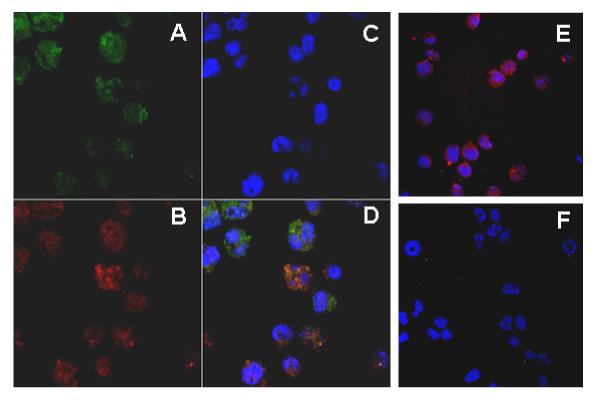
32Dcl3 cells stably transfected with V5 epitope-tagged mLRG or vector alone were cytospun onto slides and fixed with 4% formaldehyde. LRG and MPO were detected by mouse monoclonal anti-V5 and goat anti-human MPO primary antibodies, which were then detected using TRITC-conjugated donkey anti-mouse IgG and Cy5-conjugated donkey anti-goat IgG secondary antibodies, respectively. Nuclei were stained with Hoescht. (A) Cellular localization of LRG in LRG-transfected cells. (B) Cellular localization of MPO in LRG transfectants. (C) Nuclear staining with Hoescht in LRG transfectants. (D) Merged figure ofA, B and C. (E) Cells transfected with vector alone only stained positively for MPO. (F) LRG transfectants stained with secondary antibodies only.
To further confirm that LRG indeed localizes to a granular compartment within the cytoplasm of myeloid cells that also corresponds to the same compartment to which MPO is targeted, namely, the primary neutrophil granules, we analyzed different subcellular fractions from our 32Dwt18 transfectants. Using standard methods employing nitrogen cavitation and Percoll density gradient centrifugation, we isolated six continuous subcellular fractions from these transfectants for analysis by Western blot with antibodies to MPO and V5, the latter to recognize V5-tagged LRG. Both LRG and MPO were found to localize to the same fractions (Figure 3A, B). Notably, when these cells were cultured in Epo-containing media for seven days to induce differentiation and maturation, LRG was found to localize more to the lower density fractions (Figure 3B, upper panel). Cells transfected with vector alone were used as a control, showing the localization of MPO only (Figure 3C). Thus, our preliminary data suggest that LRG localizes to the subcellular fractions corresponding to the primary neutrophil granules and secretory vesicles. These findings, along with results from confocal imaging studies, suggest that LRG like MPO is targeted to the primary (azurophilic) granule compartment within the cytoplasm of myeloid cells.
Figure 3. Analysis of subcellular fractions from 32Dwt18 transfectants.
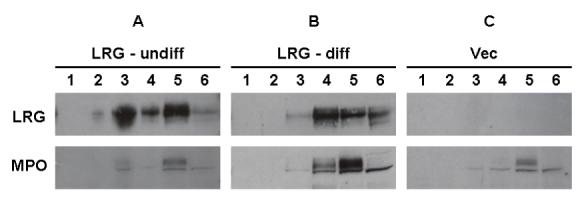
32Dwt18 cells stably transfected with V5-tagged murine LRG (undifferentiated, A; differentiated, B) or with empty vector only (C) were resuspended in disruption buffer, and disrupted by nitrogen cavitation. The nuclei and intact cells were pelleted by centrifugation, and the resultant postnuclear supernatants were loaded onto Percoll gradients. After centrifugation, six continuous fractions were collected. After ultracentrifugation to remove Percoll, the biological materials were collected, and were then subjected toSDS-PAGE analysis. Samples were immunoblotted with an antibody to the V5 epitope (upper panels) or murine MPO (lower panels).
Constitutive expression of LRG accelerates neutrophilic differentiation in 32Dcl3 cells
To characterize the physiologic role of LRG in neutrophilic differentiation, we stably transfected the murine LRG cDNA tagged with a V5 epitope into the murine IL-3 dependent 32Dcl3 cell line. This cell line was established from normal murine diploid bone marrow cells. These cells express wild-type G-CSFR on the cell surface, and undergo terminal differentiation into mature neutrophils in response to G-CSF [16]. 32Dcl3 cells do not constitutively express LRG, but upon neutrophilic differentiation in response to G-CSF, LRG expression is induced [3]. Positive clones from transfection were verified by RT-PCR, western blotting and immunoprecipitation (data not shown). Using an anti-V5 antibody, the LRG protein appeared as a single band of approximately 50kDa on SDS-PAGE. Three single clones with the highest LRG expression levels were used for the differentiation assay. A mixed pool of three single clones transfected with empty vector only was also used as control.
Following initial transfer from IL-3 to G-CSF containing media, massive cell death was repeatedly observed in five independent experiments. Cell viability decreased by more than 85% within 24 hours. Surviving cells were observed to slowly recover, and consequently undergo terminal differentiation into mature neutrophils. Notably, LRG transfectants displayed higher viability compared to cells transfected with vector alone despite the massive cell death (data not shown), suggesting that constitutive expression of LRG increased the survival of cells after initial transfer to G-CSF containing media.
Constitutive expression of LRG also significantly accelerated the differentiation of 32Dcl3 cells into mature neutrophils. Morphological analysis of Wright-Giemsa stained cells demonstrated that by day 7 of culture with G-CSF, more than 50% of LRG transfectants displayed segmented nuclei consistent with the nuclear morphology of mature neutrophils (Figure 4A, 4B). In contrast, only about 5-10% of cells transfected with vector alone exhibited segmented nuclei (Figure 4A, 4B). To confirm neutrophil maturation, we examined the surface expression level of CD11b (mac-1), a marker for neutrophil differentiation, by flow cytometry. Compared to untransfected parental 32Dcl3 cells, there was a clear-cut log shift to the right in LRG-transfected cells (Figure 4C), with accelerated expression of CD11b deteced in these cells. Taken together, these results indicate that constitutive expression of LRG accelerates neutrophil differentiation in 32Dcl3 cells.
Figure 4. Accelerated neutrophilic differentiation in LRG transfected 32Dcl3 cells in response to G-CSF treatment.
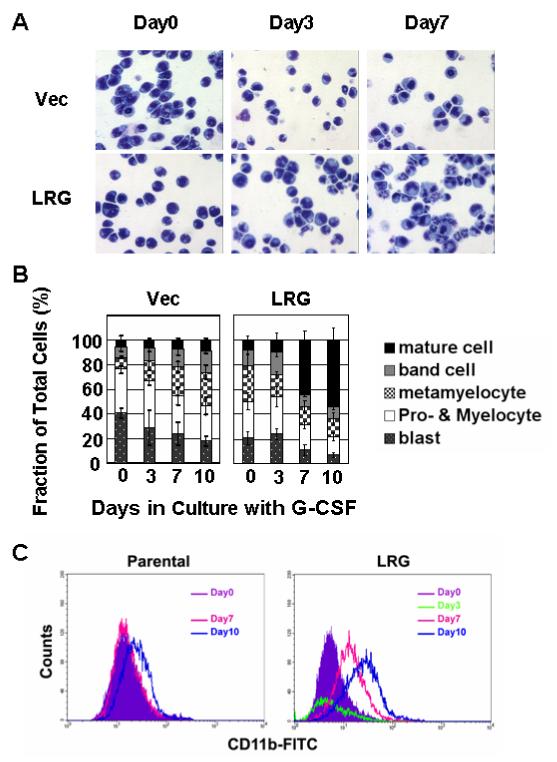
(A) Cells stably transfected with vector alone or mLRG cDNA were washed out of IL-3 and transferred to G-CSF containing media. At indicated time points, aliquots of cell were cytospun onto slides. Neutrophilic differentiation was monitored by Wright-Giemsa staining. (B) Bar graphs indicate the fraction of the total cell population of cells transfected with vector alone and LRG clone at each stage of differentiation at the indicated time points. (C) Aliquots of cell were stained with FITC conjugated CD11b antibody and subjected to FACS analysis.
Constitutive expression of LRG fails to accelerate neutrophilic differentiation in 32Dwt18 cells
We were interested in clarifying the mechanisms of LRG-accelerated neutrophil differentiation. However, the massive cell death observed after initial transfer of 32Dcl3 cells to G-CSF containing media made this cell line difficult to use for lengthy studies. To overcome this, we stably transfected the murine LRG cDNA tagged with a V5 epitope into anther 32D subline, the 32Dwt18 cell line, and used this subline as an alternative model system for our studies. These cells are stably transfected with a chimeric form of the G-CSFR, in which the extracellular domain has been replaced by the extracellular domain of the erythropoietin (Epo) receptor, while the transmembrane and cytoplasmic domains of the G-CSFR remain intact. Similar to 32Dcl3 cells, these cells also do not constitutively express LRG. In response to Epo, 32Dwt18 cells differentiate along the granulocytic lineage into morphologically normal neutrophils, but without the massive cell death observed in 32Dcl3 cells [13]. Positive clones from transfection were verified by RT-PCR and western blotting (data not shown). Three single clones with the highest LRG expression levels were used for the differentiation assay. A mixed pool of three single clones transfected with the empty vector alone was used as a control.
Initially, we used a high concentration of Epo (1u/ml) as previously used to study the CCAAT displacement protein (CDP/cut) in differentiating 32Dwt18 cells [13]. Massive cell death was not observed after transfer of the cells from IL-3 to Epo containing media. However, at this concentration, we did not observe accelerated neutrophil differentiation in LRG-transfected clones. Due to concerns that any accelerated differentiation might be masked by strong Epo signaling at the high concentration, we reduced the Epo concentration to as low as 0.1u/ml. At the lower concentration of Epo, we observed decreased viability of the cells similar to that observed with 32Dcl3 cells following their transfer to G-CSF containing media. Notably, the LRG-transfected clones displayed a higher viability than cells transfected with empty vector alone (data not shown). However, even at the lower concentration of Epo, there was no evidence of accelerated differentiation in LRG transfectants either by morphology (Figure 5A, 5B), or by CD11b expression (Figure 5C). These results suggest different signaling pathways are activated in response to G-CSF and Epo in 32Dcl3 and 32Dwt18 cells, respectively, during neutrophilic differentiation.
Figure 5. Lack of accelerated neutrophilic differentiation in LRG-transfected 32Dwt18 cells in response to Epo treatment.
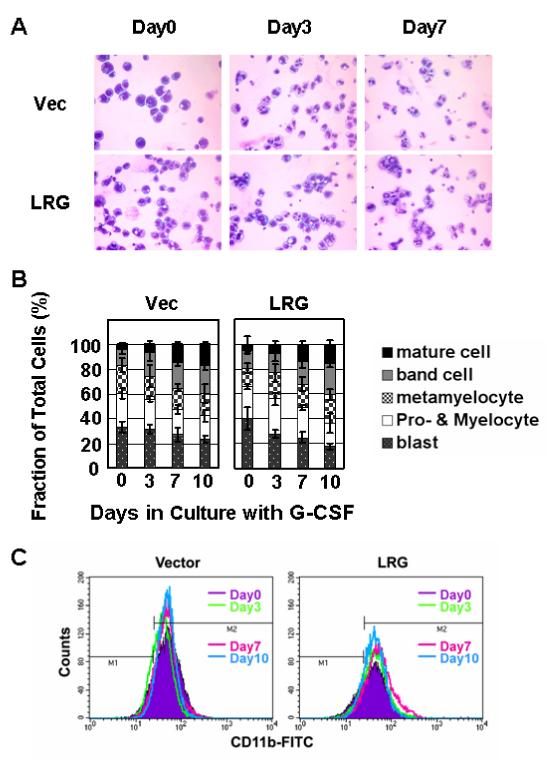
(A) Cells stably transfected with vector alone or mLRG cDNA were washed out of IL-3 and transferred to Epo containing media. At the indicated time points, aliquots of cells were cytospun onto slides and neutrophilic differentiation monitored by Wright-Giemsa staining. (B) Bar graphs indicate the fraction of the total cell population transfected with vector alone or LRG at various stages of neutrophilc differentiation at each time point. (C) Aliquots of cells were stained with FITC-conjugated CD11b antibody and analyzed for CD11b expression by FACS analysis.
Differing expression patterns of phospho-Stat3 but not PU.1 or p27kip1 in 32Dcl3 and 32Dwt18 cells
To clarify the discrepant effects of constitutive expression of LRG on neutrophilic differentiation of 32Dcl3 and 32Dwt18 cells, we examined several proteins reported to be involved in cytokine-induced signal transduction, cell cycle regulation and gene transcription.
We first investigated activation of Stat3, which has been reported to be required for G-CSF-induced neutrophilic differentiation [17]. In 32Dcl3 cells, we detected a rather weak signal for phospho-Stat3 in cells transfected with vector alone and cultured for 24 hours in G-CSF. In contrast, we detected a strong signal for phospho-Stat3 whichh was apparent as early as 8 hours in LRG transfectants. Notably, the expression level of Stat3 remained the same throughout this time (Figure 6A), suggesting that Stat3 may be involved in LRG-accelerated neutrophilic differentiation of 32Dcl3 cells. In 32Dwt18 cells, we detected similar patterns of Stat3 and phospho-Stat3 in both vector transfected cells and LRG transfectants cultured in Epo-containing media (Figure 6B). This may explain why we did not observe accelerated neutrophilic differentiation of these cells following stable transfection with LRG. We also examined the 32Dwt18 cells in G-CSF containing media. To our surprise, we did not detect any phospho-Stat3 signals even as late as 24 hours following G-CSF treatment (Figure 6C). In addition, the expression level of Stat3 significantly decreased at 24 hours in G-CSF in both LRG-expressing cells and cells transfected with vector alone (Figure 6C). This suggests that the endogenous full-length G-CSFR may not be functional in 32Dwt18 cells.
Figure 6. Stat3 is activated at an early stage in LRG transfected 32Dcl3 cells but not in LRG transfected 32D wt18 cells.
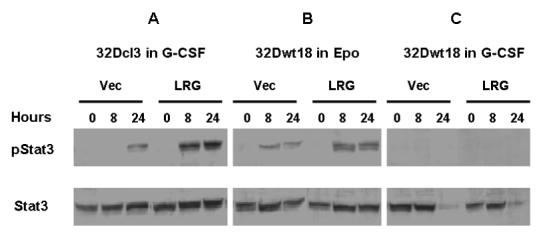
Cells were washed out of IL-3 containing media and transferred to media containing G-CSF or Epo as indicated. Whole cell lysates were made from cells harvested at the indicated time points and used for immunoblotting with Stat3 and phospho-Stat3. (A) Vector or LRG transfected 32Dcl3 cells in G-CSF containing media. (B) Vector or LRG transfected 32Dwt18 cells in Epo containing media. (C) Vector or LRG transfected 32Dwt18 cells in G-CSF containing media.
We next investigated the expression levels of the cyclin-dependent kinase inhibitor p27kip1 and the transcription factor PU.1. In 32Dcl3 cells, we detected decreased expression of p27kip1 in cells transfected with vector alone following 24 hours of G-CSF treatment, but not in LRG-transfected cells (Figure 7A). The expression of PU.1 was undetectable in 32Dcl3 cells transfected with vector alone after 24 hours culture in G-CSF, whereas it was still detectable in LRG transfectants at 24 hours, although decreased from basal levels (Figure 7A). The same expression patterns for both PU.1 and p27kip1 were detected in 32Dwt18 cells as those observed in 32Dcl3 cells (Figure 7B). Taken together, these results suggest that Stat3, but not p27kip1 or PU.1, is involved in LRG-accelerated neutrophil differentiation in 32Dcl3 cells.
Figure 7. p27kip1 and PU.1 display similar patterns of expression in LRG-transfected 32Dcl3 and 32Dwt18 cells.
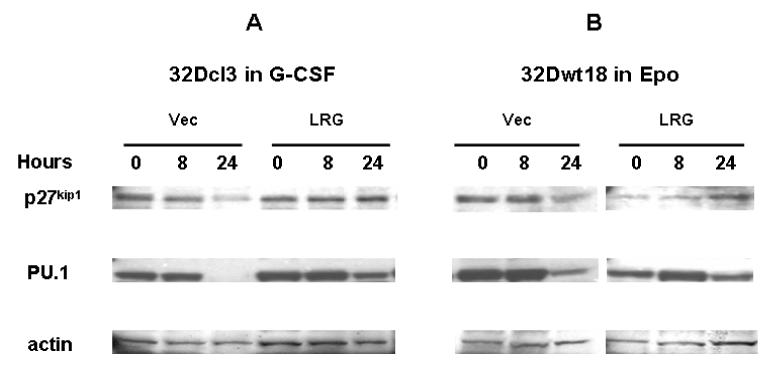
Cells were washed out of IL-3 containing media and transferred to media containing G-CSF or Epo as indicated. Whole cell lysates were made from cells harvested at the indicated time points and used for immunoblotting with p27kip1 antibody (top panels). The membrane was stripped and reblotted with PU.1 (middle panels) and actin antibodies (bottom panels). (A) Vector or LRG transfected 32Dcl3 cells in G-CSF containing media. (B) Vector or LRG transfected 32Dwt18 cells in Epo containing media.
DISCUSSION
We previously identified DNA-binding motifs for the PU.1 and C/EBP transcription factors within the putative LRG promoter region. Similar to the regulatory mechanisms that modulate the expression of other neutrophil granule proteins, the PU.1 and C/EBP transcription factors could also bind to the promoter region of LRG to regulate its expression. To test this possibility, we performed chromatin immunoprecipitation (ChIP) analysis with untreated and Epo-treated 32Dwt18 cells using specific antibodies and primers to amplify the regions corresponding to the PU.1 binding site or the C/EBP binding site within the putative LRG promoter. Induction of 32Dwt18 cells towards neutrophil maturation by Epo treatment appears to change the dynamics of protein binding to both the LRG PU.1 and C/EBP binding sites. Binding of PU.1 and C/EBPε to their respective LRG binding sites dramatically increases within the initial three days of Epo-induced neutrophil maturation, while the binding pattern for C/EBPα did not vary significantly. PU.1 has been shown to activate transcription of primary neutrophil granule enzymes such as neutrophil elastase [18]. C/EBPα is the predominant C/EBP family member in immature myeloid cells and appears to play an important role in regulation of early granulopoiesis [19-21]. C/EBPε is expressed in late stage granulocytes, and C/EBPε null mice have increased numbers of immature myeloid cells in their bone marrow and hyposegmented dysfunctional neutrophils [20-22]. Our results indicate that PU.1 and C/EBPε bind to the putative LRG promoter region that we previously identified, and binding of these transcription factors appears to modulate the expression of LRG during neutrophil maturation. Up-regulation of LRG expression seems to be an event that occurs at a later stage during granulocytic differentiation and maturation.
The gene for human LRG localizes to the same chromosome region to which genes for multiple primary neutrophil granule enzymes also localize, suggesting that LRG may be another primary granule protein. Lack of commercially available antibodies to LRG has hindered the determination of its cellular localization. We have stably transfected a V5-tagged LRG cDNA into either 32Dcl3 or 32Dwt18 cells, so that specific antibody to the V5 epitope tag could be utilized. Immunofluorescence staining and confocal microscopy results displayed similar diffuse granular distribution patterns within the cytoplasmic compartment for both LRG and MPO, suggesting that LRG localizes to primary neutrophil granules. Subcellular fractionation experiments further confirmed the co-localization of both LRG and MPO within the primary neutrophil granules. Notably, LRG in differentiated cells tended to move to fractions of lighter density, indicating its progression in post-translational modification along the secretion pathways, which is possibly related to its functions in neutrophil maturation. Heparin-binding protein, another primary neutrophil granule protein that belongs to the family of serine proteases, was also recently shown to localize to secretory vesicles in addition to primary granules in mature human neutrophils but not in HL-60 cells [23]. It is plausible that LRG, similar to HBP, has dual localization. In undifferentiated 32D cells, LRG was produced and stored in primary granules. When its expression is upregulated during differentiation and maturation, however, LRG could also be targeted to other compartments, such as the secretory vesicles. This dual localization may imply that LRG has multiple functions in granulopoiesis.
The most import issue in LRG characterization is to determine its physiological roles. Our previous work showed that the expression of LRG is induced during neutrophilic granulocyte differentiation in response to G-CSF, leading us to the hypothesis that LRG may be involved in granulopoiesis. Constitutive expression of LRG significantly accelerated neutrophilic differentiation in 32Dcl3 cells, demonstrated by both nuclear morphologic changes and cell surface expression levels of CD11b. Proliferation of LRG transfectants was decreased compared to cells transfected with empty vector alone, consistent with an expected decrease in proliferation as cells undergo differentiation and also consistent with our observation that LRG transfectants undergo accelerated neutrophilic differentiation (data not shown).
The mechanisms of this accelerated differentiation are still under investigation. By immunoblot analysis, LRG transfected 32Dcl3 cells displayed a stronger phospho-Stat3 signal at an earlier time point in response to G-CSF treatment. Jak kinases and Stat proteins have been shown to be activated in hematopoietic cells by the majority of hematopoietic cytokines [24]. Over-expression of Jak3 in 32Dcl3 cells was recently reported to accelerate neutrophil differentiation in response to G-CSF [25]. This effect was mediated in part through Stats and cell cycle regulatory proteins. It is possible that accelerated differentiation induced by over-expression of LRG is mediated by similar signaling pathways involving Stat3. Whether LRG facilitates the phosphorylation and action of Stat3 or LRG directly interacts with other genes involved in granulocytic differentiation after it translocates to nucleus upon G-CSF stimulation remains to be determined. On the other hand, our results suggested that PU.1 or the cyclin-dependent kinase inhibitor p27kip1 might not be involved in the accelerated differentiation of 32Dcl3 cells, since we did not detect any difference in the expression patterns of either p27kip1 or PU.1 between 32Dcl3 and 32Dwt18 cells (in which constitutive expression of LRG does not accelerate neutrophilic differentiation). This is in contrast with previous reports on Stat3 mediated activation of p27kip1 [26] and PU.1 [27]. One explanation for this is that Stat3 may be necessary, but not sufficient for the activation of p27kip1 and PU.1.
Previous studies suggest that LRG interacts with transforming growth factor beta (TGF-β) receptor and modulates the activity of TGF-β [28]. Notably, TGF-β has been shown to modulate hematopoietic cell proliferation, with both inhibitory and stimulatory activities. TGF-β was reported to increase granulocyte/macrophage-colony stimulating factor (GM-CSF)-driven granulocyte differentiation, which was postulated to occur via increased surface expression of the GM-CSF receptor induced by TGF-β [29]. Recently, Saito et al. reported isolation of LRG from high endothelial venule cells and proposed a role for LRG in adhesive interactions between lymphocytes and the endothelium [30]. They also reported that LRG immobilized on plastic wells could bind to TGF-β in vitro. Therefore, LRG may aid in the control of granulopoiesis by helping to modulate the surface expression of different receptor types, including TGF-β receptor, GM-CSFR, and possibly the G-CSFR. Another recent publication provided evidence that specific isoforms of LRG were detected in the proliferative endometrium of women undergoing assisted reproduction, suggesting that LRG has a role in implantation and/or decidualization [31]. As G-CSFR is highly expressed in endometrium, the interaction between LRG and G-CSFR may also play an important part in the preparation of the uterus for embryonic growth factor activity.
In these experiments, we used two model systems: 32Dcl3 and 32Dwt18 cells. It would be expected that the signaling pathways mediated by the same cytoplasmic domain of the G-CSFR should lead to the same cellular processes in the two cell model systems. However, the effects of constitutive expression of LRG in the two systems are quite different. Our data indicate that the full length G-CSFR plays a critical role in neutrophil activation, in addition to neutrophilic differentiation. In vivo studies by Link’s group [32] demonstrated that in homozygous mutant mice expressing a G-CSF/Epo receptor in which the extracellular and transmembrane domains of G-CSFR are fused to the cytoplasmic domain of the Epo receptor, morphologically mature neutrophils were produced, but their mobilization was greatly impaired. Surface adhesion molecules are important factors involved in these biological activities. Interestingly, we showed that CD11b, the major β2 integrin expressed on neutrophils, is only induced by the endogenous full-length G-CSFR; and its expression is accelerated by the constitutive expression of LRG. Thus, LRG may interfere with the metabolism of adhesion molecules to facilitate functional maturation of neutrophils in response to G-CSF.
In the conventional view, the role of the extracellular domains of cytokine receptors is simply to bind to the respective cytokine and mediate receptor dimerization. It is the cytoplasmic domains that play critical roles in signal transduction, as they bear the docking sites for cytoplasmic tyrosine kinases. A recent study on the Δ319 G-CSFR mutation identified in a patient with SCN has provided additional insights into G-CSFR signaling [33]. This mutation deletes part of the extracellular portion and the entire transmembrane and cytoplasmic domains. Expression of this mutant in Ba/F3 cells, either alone or in combination with the wild-type G-CSFR, reproduces the dominant-negative phenotype observed in the patient. Proper targeting of the receptor to the cell membrane is disrupted, and the response to G-CSF is also abrogated. Similar defects in receptor processing and assembly have been reported with truncation mutants of the Epo receptor as well, indicating that sequences in the extracellular domain are critical for correct expression and sorting of both mature receptor complexes to the plasma membrane [34,35]. Here, we present further evidence that the extracellular domain of the G-CSFR is indeed indispensable for the unique signaling pathways mediated by the full length G-CSFR. We thus propose the model shown in Figure 8. In response to G-CSF, the full-length wild-type G-CSFR undergoes unique conformational changes in the cytoplasmic domain, activating the associated tyrosine kinases. This results in the recruitment and activation of Stat proteins, including Stat3, and possibly other unknown molecules. The activated Stats then translocate to the nucleus and activate the transcription of target genes. The cell undergoes terminal differentiation, and CD11b expression is induced. On the other hand, in response to Epo, the chimeric Epo/G-CSFR may undergo different conformational changes within the cytoplasmic domain. This may lead to the activation of different Stat molecules or other factors; and in the end, different target genes. LRG may be an important factor involved in the non-redundant signaling pathway mediated by the full-length G-CSFR, and its constitutive expression accelerates neutrophilic differentiation in 32Dcl3 cells. Additional studies to more precisely define these signaling pathways should help to further clarify the functions of LRG in granuolopoiesis.
Figure 8. Schematic diagram for LRG function in the unique signaling pathway mediated by the full-length G-CSF receptor.
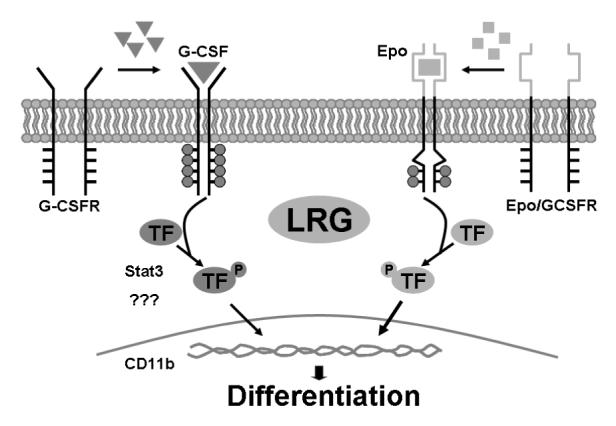
Refer to text for detailed description.
ACKNOWLEDGEMENTS
This work was supported by Grants CA75226, CA82859, and CA16058 from the National Cancer Institute, and Grant DK068639 from the National Institute of Diabetes and Digestive and Kidney Diseases.
REFERENCES
- 1.Haupt H, Baudner S. Z. Physiol. Chem. Vol. 358. Hoppe-Seyler’s; German: 1977. Isolation and characterization of an unknown, leucine-rich 3.1-S-alpha2-glycoprotein from human serum (author’s transl) pp. 639–646. [PubMed] [Google Scholar]
- 2.Buchanan SG, Gay NJ. Structural and functional diversity in the leucine-rich repeat family of proteins (Review) Prog. Biophys. Mol. Biol. 1996;65:1–44. doi: 10.1016/s0079-6107(96)00003-x. [DOI] [PubMed] [Google Scholar]
- 3.O’Donnell LC, Druhan LJ, Avalos BR. Molecular characterization and expression analysis of leucine-rich α2-glycoprotein, a novel marker of granulocytic differentiation. J. Leukoc. Biol. 2002;72:478–485. [PubMed] [Google Scholar]
- 4.Demetri GD, Griffin JD. Granulocyte colony-stimulating factor and its receptor (Review) Blood. 1991;78:2791–2808. [PubMed] [Google Scholar]
- 5.Williams GT, Smith CA, Spooncer E, Dexter TM, Taylor DR. Haematopoietic colony stimulating factors promote cell survival by surpporting apoptosis. Nature. 1990;343:76–79. doi: 10.1038/343076a0. [DOI] [PubMed] [Google Scholar]
- 6.Begley CG, Metcalf D, Nicola NA. Binding characteristics and proliferative action of purified granulocyte-colony stimulating factor (G-CSF) on normal and leukemic human promyelocytes. Exp. Hematol. 1988;16:71–79. [PubMed] [Google Scholar]
- 7.Avalos BR, Gasson JC, Hedvat C, Quan SG, Baldwin GC, Weisbart RH, Williams RE, Golde DW, Dipersio JF. Human granulocyte colony-stimulating factor: Biological activities and receptor characterization on hematopoietic cells and small lung cancer cell lines. Blood. 1990;75:851–857. [PubMed] [Google Scholar]
- 8.Avalos BR. Molecular analysis of the granulocyte colony-stimulating factor receptor. Blood. 1996;88:761–777. [PubMed] [Google Scholar]
- 9.Champagne B, Tremblay P, Cantin A, St. Pierre Y. Proteolytic cleavage of ICAM-1 by human neutrophil elastase. J. Immunol. 1998;161:6398–6405. [PubMed] [Google Scholar]
- 10.Nair KS, Zingde SM. Adhesion of neutrophils to fibronectin: role of the CD66 antigens. Cell. Immunol. 2001;208:96–106. doi: 10.1006/cimm.2001.1772. [DOI] [PubMed] [Google Scholar]
- 11.Le-Barillec K, Si-Tahar M, Balloy V, Chignard M. Proteolysis of monocyte CD14 by human leukocyte elastase inhibits lipopolysaccharide-mediated cell activation. J. Clin. Invest. 1999;103:1039–1046. doi: 10.1172/JCI5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li FQ, Horwitz M. Characterization of mutant neutrophil elastase in severe congenital neutropenia. J. Biol. Chem. 2001;276:14230–14241. doi: 10.1074/jbc.M010279200. [DOI] [PubMed] [Google Scholar]
- 13.Khanna-Gupta A, Zibello T, Kolla S, Neufeld EJ, Berliner N. CCAAT displacement protein (CDP/cut) recognizes a silencer element within the lactoferrin gene promoter. Blood. 1997;90:2784–2795. [PubMed] [Google Scholar]
- 14.Kjeldsen L, Sengeløv H, Borregaard N. Subcellular fractionation of human neutrophils on Percoll density gradients. J. Immuno. Methods. 1999;232:131–143. doi: 10.1016/s0022-1759(99)00171-4. [DOI] [PubMed] [Google Scholar]
- 15.Shih CM, Wu JS, Ko WC, Wang LF, Wei YH, Liang HF, Chen YC, Chen CT. Mitochondria-mediated caspase-independent apoptosis induced by cadmium in normal human lung cells. J. Cell. Biochem. 2003;89:335–347. doi: 10.1002/jcb.10488. [DOI] [PubMed] [Google Scholar]
- 16.Valtieri M, Tweardy DJ, Caracciolo D, Johnson K, Mavilio F, Altmann S, Santoli D, Rovera G. Cytokine-dependent granulocyte differentiation: Regulation of proliferative and differentiative responses in a murine progenitor cell line. J. Immunol. 1987;138:3829–3835. [PubMed] [Google Scholar]
- 17.Shimozaki K, Nakajima K, Hirano T, Nagata S. Involvement of STAT3 in the granulocyte colony-stimulating factor-induced differentiation of myeloid cells. J. Biol. Chem. 1997;272:25184–25189. doi: 10.1074/jbc.272.40.25184. [DOI] [PubMed] [Google Scholar]
- 18.Oelgeschlager M, Nuchprayoon I, Luscher B, Friedman AD. C/EBP, c-Myb, and PU.1 cooperate to regulate the neutrophil elastase promoter. Mol. Cell. Biol. 1996;16:4717–4725. doi: 10.1128/mcb.16.9.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamanaka R, Lekstrom-Himes J, Barlow C, Wynshaw-Boris A, Xanthopoulos KG. CCAAT/enhancer binding proteins are critical components of the transcriptional regulation of hematopoiesis (Review) Int. J. Mol. Med. 1998;1:213–221. doi: 10.3892/ijmm.1.1.213. [DOI] [PubMed] [Google Scholar]
- 20.Wang W, Wang X, Ward AC, Touw IP, Friedman AD. C/EBPα and G-CSF receptor signals cooperate to induce the myeloperoxidase and neutrophil elastase genes. Leukemia. 2001;15:779–786. doi: 10.1038/sj.leu.2402094. [DOI] [PubMed] [Google Scholar]
- 21.Zhang DE, Zhang P, Wang ND, Hetherington CJ, Darlington GJ, Tenen DG. Absence of granulocyte coloby-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc. Natl. Acad. Sci. USA. 1997;94:569–574. doi: 10.1073/pnas.94.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamanaka R, Barlow C, Lekstrom-Himes J, Castilla LH, Liu PP, Eckhaus M, Decker T, Wynshaw-Boris A, Xanthopoulos KG. Impaired granulopoiesis, myelodysplasia, and early lethality in CCAAT/enhancer binding protein epsilon-deficient mice. Proc. Natl. Acad. Sci. USA. 1997;94:13187–13192. doi: 10.1073/pnas.94.24.13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tapper H, Karlsson A, Morgelin M, Flodgaard H, Herwald H. Secretion of heparin-binding protein from human neutrophils is determined by its localization in azurophilic granules and secretory vesicles. Blood. 2002;99:1785–1793. doi: 10.1182/blood.v99.5.1785. [DOI] [PubMed] [Google Scholar]
- 24.Rane SG, Reddy EP. JAKs, STATs and Src kinases in hematopoiesis. Oncogene. 2002;21:3334–3358. doi: 10.1038/sj.onc.1205398. [DOI] [PubMed] [Google Scholar]
- 25.Rane SG, Mangan JK, Amanullah A, Wong BC, Vora RK, Leibermann DA, Hoffman B, Grana X, Reddy EP. Activation of the Jak3 pathway is associated with granulocytic differentiation of myeloid precursor cells. Blood. 2002;100:2753–2762. doi: 10.1182/blood.V100.8.2753. [DOI] [PubMed] [Google Scholar]
- 26.de Koning JP, Soede-Bobok AA, Ward AC, Schelen AM, Antonissen C, van Leeuwen D, Lowenberg B, Touw IP. STAT3-mediated differentiation and survival and of myeloid cells in response to granulocyte colony-stimulating factor: role for the cyclin-dependent kinase inhibitor p27(Kip1) Oncogene. 2000;19:3290–3298. doi: 10.1038/sj.onc.1203627. [DOI] [PubMed] [Google Scholar]
- 27.Panopoulos AD, Bartos D, Zhang L, Watowich SS. Control of myeloid-specific integrin alpha Mbeta 2 (CD11b/CD18) expression by cytokines is regulated by Stat3-dependent activation of PU.1. J. Biol. Chem. 2002;277:19001–19007. doi: 10.1074/jbc.M112271200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun D, Kar S, Carr BI. Differntially expressed genes in TGF-β1 sensitive and resistant human hepatoma cells. Cancer. Letters. 1995;89:73–79. doi: 10.1016/0304-3835(95)90160-4. [DOI] [PubMed] [Google Scholar]
- 29.Fortunel NO, Hatzfeld A, Hatzfeld JA. Transforming growth factor-beta: pleiotropic role in the regulation of hematopoiesis. Blood. 2000;96:2022–2036. [PubMed] [Google Scholar]
- 30.Saito K, Tanaka T, Kanda H, Ebisuno Y, Izawa D, Kawamoto S, Okubo K, Miyasaka M. Gene expression profiling of mucosal addressin cells adhesion molecule-1+ high endothelial venule cells (HEV) and identification of a leucine-rich HEV glycoprotein as a HEV marker. J. Immunol. 2002;168:1050–1059. doi: 10.4049/jimmunol.168.3.1050. [DOI] [PubMed] [Google Scholar]
- 31.Gillott DJ, Al-Rumaih HM, Leung KY, Eldib A, Grudzinskas JG. Specific isoforms of leucine-rich alpha2-glycoprotein detected in the proliferative endometrium of women undergoing assisted reproduction are associated with spontaneous pregnancy. Fertil. Steril. 2007 Jun 19; doi: 10.1016/j.fertnstert.2007.01.094. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 32.Semerad CL, Poursine-Laurent J, Liu F, Link DC. A role for G-CSF receptor signaling in the regulation of hematopoietic cell function but not lineage commitment or differentiation. Immunity. 1999;11:153–161. doi: 10.1016/s1074-7613(00)80090-4. [DOI] [PubMed] [Google Scholar]
- 33.Druhan LJ, Ai J, Massullo P, Kindwall-Keller T, Ranalli MA, Avalos BR. Novel mechanism of G-CSF refractoriness in patients with severe congenital neutropenia. Blood. 2005;105:584–591. doi: 10.1182/blood-2004-07-2613. [DOI] [PubMed] [Google Scholar]
- 34.Miura O, Ihle JN. Dimer- and oligomerization of the erythropoietin receptor by disulfide bond formation and significance of the region near the WSXWS motif in intracellular transport. Arch. Biochem. Biophys. 1993;306:200–208. doi: 10.1006/abbi.1993.1501. [DOI] [PubMed] [Google Scholar]
- 35.Yoshimura A, Zimmers T, Neumann D, Longmore G, Yoshimura Y, Lodish HF. Mutations in the Trp-Ser-X-Trp-Ser motif of the erythropoietin receptor abolish processing, ligand binding, and activation of the receptor. J. Biol. Chem. 1992;267:11619–11625. [PubMed] [Google Scholar]