

Abstract
Background
Long QT syndrome type 2 (LQT2) is caused by mutations in the human ether-a-go-go-related gene (hERG). More than 30% of the LQT2 mutations result in premature termination codons (PTCs). Degradation of PTC-containing mRNA transcripts by nonsense-mediated mRNA decay (NMD) is increasingly recognized as a mechanism for reducing mRNA levels in a variety of human diseases. However, the role of NMD in LQT2 mutations has not been explored.
Methods and Results
We examined the expression of hERG mRNA in lymphocytes from patients carrying the R1014X mutation using a technique of allele-specific transcript quantification. The R1014X mutation led to a reduced level of mutant mRNA compared to that of the wild-type allele. The decrease in mutant mRNA was also observed in LQT2 nonsense mutations W1001X and R1014X using hERG minigenes expressed in HEK293 cells or neonatal rat ventricular myocytes. Treatment with the protein synthesis inhibitor cycloheximide or RNAi-mediated knockdown of the Upf1 protein resulted in the restoration of mutant mRNA to levels comparable to that of the wild-type minigene, suggesting that hERG nonsense mutations are subject to NMD.
Conclusions
These results indicate that LQT2 nonsense mutations cause a decrease in mutant mRNA levels by NMD rather than production of truncated proteins. Our findings suggest that the degradation of hERG mutant mRNA by NMD is an important mechanism in LQT2 patients with nonsense or frameshift mutations.
Keywords: arrhythmia, ion channels, myocytes, long QT syndrome
Long QT syndrome is a disease associated with delayed cardiac repolarization and prolonged QT intervals on the electrocardiogram, which can lead to ventricular arrhythmias and sudden death.1 The inherited long QT syndrome type 2 (LQT2) is caused by mutations in the human ether-a-go-go-related gene (hERG), which encodes the pore forming subunit of the rapidly activating delayed rectifier K+ channel (IKr) in the heart.2,3 More than 250 hERG mutations have been identified in patients with LQT2.4-7 The mechanisms of hERG channel dysfunction in LQT2 mutations have been studied extensively in the last 10 years.8-11 Most previous studies, however, have been focused on the analysis of mutant proteins and channel function. More than 30% of LQT2 mutations are nonsense or frameshift mutations that introduce premature termination codons (PTCs).4-7 These PTC mutations are generally assumed to result in truncated dysfunctional channel proteins, and several nonsense and frameshift mutations have been studied at the protein level.8,12-18 However, it is now becoming clear that nonsense and frameshift mutations bearing PTCs can destabilize mRNA transcripts via a mechanism known as nonsense-mediated mRNA decay (NMD) in many human diseases, resulting in decreased abundance of mutant mRNA transcripts, rather than in production of truncated proteins.19,20
NMD is an RNA surveillance mechanism that selectively degrades mRNA transcripts containing PTCs due to nonsense or frameshift mutations. The role of NMD as a disease-causing mechanism of PTC mutations is becoming increasingly evident.19,20 According to the proposed rule, NMD occurs when translation terminates more than 50-55 nt upstream of the 3′-most exon-exon junction.21,22 The molecular mechanisms of NMD have been studied extensively. These studies have shown that pre-mRNA splicing deposits the exon junction complex (EJC) about 20-40 nt upstream of the exon-exon junction in spliced mRNA. The EJC can recruit Upf proteins, which are required for NMD.22 Several Upf proteins (Upf1, Upf2 and Upf3a, b) have been identified.23 The Upf1 protein appears to play a key role in the distinction between proper and improper translation termination. Upf1 is a group I helicase that has RNA-dependent ATPase and ATP-dependent 5′ to 3′ helicase activities. Knockdown of Upf1 by RNA interference (RNAi) has been shown to inhibit NMD.24,25
The objective of this work was to determine whether NMD occurs in hERG mutations that contain PTCs. We investigated two nonsense mutations W1001X and R1014X in the C-terminal region of the hERG channel. The W1001X and R1014X mutations have previously been studied at the protein level using hERG cDNAs.14,15 It was found that both mutations produced truncated hERG channel proteins and reduced hERG current amplitude. The R1014X mutation also caused a dominant negative effect on the wild-type (WT) hERG current, which is expected to result in a severe clinical phenotype. However, the R1014X carriers have presented with a mild phenotype. In the present study, we demonstrate that rather than the production of truncated proteins, the primary defect of the W1001X and R1014X mutations is the degradation of mutant mRNA by NMD.
Methods
Subjects
The study was approved by the institutional review board and carried out upon receipt of informed consent. The participants were blood-related members of a large family previously identified as having the R1014X mutation.4 The pedigree included 25 blood-related family members in four generations (Fig. 1). Phenotyping was performed based on the history of LQTS-related cardiac events, the assessment of QT intervals and T-wave morphology, and pedigree analysis.26 Genotyping was conducted by sequencing of DNA samples collected from buccal swabs. Normal control subjects were unrelated individuals.
Figure 1.

The pedigree of the family with the R1014X mutation.
RNA and DNA preparations from blood samples
Total RNA was isolated from peripheral blood lymphocytes using the RiboPure-Blood kit (Ambion, Austin, TX). The isolated RNA was treated with RNase-free DNase to remove genomic DNA. Genomic DNA was isolated from lymphocytes or Epstein Barr virus-transformed lymphoblastoid cells using the DNeasy tissue kit (Qiagen, Valencia, CA).
Allele-specific quantification of RNA transcripts and genomic DNA
The relative abundance of RNA transcripts from WT and R1014X alleles was determined by a modified “hot-stop” PCR method.27,28 In this assay, the regular RT-PCR was carried out using the primers in exon 13 (E13-F, forward 5′-GCCTTCTCAGGAGTGTCCAA-3′) and exon 14 (E14-R, reverse 5′-GAAAGCGAGTCCAAGGTGAG-3′). After 35 cycles, [32P]-dCTP was added and subjected to a single cycle of PCR.28 With hot-stop PCR only homoduplexes incorporated 32P-labels, and any heteroduplexes formed during previous cycles were unlabeled. Thus, hot-stop PCR will prevent the detection of WT/mutant heteroduplexes, which are resistant to restriction enzyme digestion. Because hot-stop PCR analysis yields a relative measure of transcripts from two alleles, normalization to a reference housekeeping gene is unnecessary. The hERG genomic DNA was analyzed by hot-stop PCR with the same forward primer as used in RT-PCR and a reverse primer in intron 13 (I13-R, 5′-CTCCGCGCTAGAGGTGTG-3′). For analysis of allelic variation in hERG mRNA expression in normal subjects, the ratio of a common polymorphism 1692A/G was determined by hot-stop PCR using the primers in exon 6 (E6-F, forward 5′-ATCAACTTCCGCACCCCTA-3′) and exon 7 (E7-R, reverse 5′-TGTGTGGCTGCTCCATGT-3′). The labeled PCR products were treated with TaqI or NheI restriction enzyme and analyzed by 5% polyacrylamide gel electrophoresis and autoradiography. For quantitative analysis, the intensity of each band was quantified using Scion Image software (Scion Corp., Frederick, MD). The ratio of two alleles was calculated and a correction factor according to the respective GC content of each digested product was applied to the ratio.28
Construction of minigenes
Human genomic DNA was used as a template for PCR amplification of fragments spanning from hERG exons 12 to 15. The PCR products were cloned into pCRII vector using the TA cloning kit (Invitrogen, Carlsbad, CA), and verified by DNA sequencing. The minigenes were then subcloned into a mammalian expression vector pcDNA5/FRT (Invitrogen). The N-terminus of the minigene was tagged by Myc epitope, which is in-frame with the hERG translation sequence. The W1001X and R1014X mutations in the minigenes were generated using the pAlter in vitro site-directed mutagenesis system (Promega, Madison, WI) and verified by DNA sequencing.
Stable expression of mingene constructs in HEK293 cells
The minigenes in pcDNA5/FRT vector were stably transfected into HEK293 cells by using the Flp-In method (Invitrogen). In this approach, an FRT site sequence is integrated into the genome of HEK293 cells and is recombined by Flp recombinase with the FRT site of the pcDNA5/FRT vector. The pcDNA5/FRT vector carries the hygromycin resistance gene, which is used for the selection of stable cell lines.
RNase protection assay of mRNA transcripts from minigene transfected cells
RNA isolation and RNase protection assay (RPA) were performed as previously described.29 Briefly, cytosolic RNA was isolated from HEK293 cells or neonatal rat ventricular myocytes expressing hERG minigenes using the RNAeasy kit (Qiagen). The antisense RNA riboprobes were transcribed in vitro in the presence of biotin-16-UTP (Roche, Indianapolis, IN). Thirty μg of RNA were analyzed with the riboprobes using the RPAIII and BrightStart BioDetect kits (Ambion). Yeast RNA was used as a control for the complete digestion of the probes by RNase. The expression level of the hygromycin resistance gene from the pcDNA5/FRT vector or the E2 gene from adenovirus was used as a loading control for normalization. The intensity of each band was quantified using Scion Image software.
RNA interference
Two plasmids, pSUPERpuro-hUpf1/I and pSUPERpuro-hUpf1/II (kindly provided by Dr. Oliver Mühlemann), were used to inhibit expression of Upf1 as described by Paillusson et al.25 These plasmids contain short hairpin RNAs targeting two sequences of hUpf1 (5′-GAGAATCGCCTACTTCACT-3′ for pSUPERpuro-hUpf1/I and 5′-GATGCAGTTCCGCTCCATT-3′ for pSUPERpuro-hUpf1/II). The HEK293 cells stably expressing WT or R1014X minigenes were transfected with a mixture of 1 μg pSUPERpuro-hUpf1/I and 1 μg pSUPERpuro-hUpf1/II, or 2 μg pSUPERpuro with scrambled sequence of hUpf1/I using LipofectAmine 2000 (Invitrogen). At 24 h post-transfection, puromycin was added to the final concentration of 1.5 μg/ml for 48 h to eliminate the untransfected cells. Before analysis, the cells were cultured without puromycin for at least 24 h to avoid potential ffects of this translation inhibitor on NMD. The knockdown of the Upf1 protein was analyzed by Western blot as described.9,25
Construction and use of recombinant adenovirus
The AdEasy vector kit was used to generate WT and R1014X minigene recombinant adenoviruses (Stratagene, La Jolla, CA). First, the WT and R1014X minigenes were subcloned into pShuttle-CMV vector and recombined with the pAdEasy plasmid in Escherichia coli strain BJ5183. The pAdEasy/minigene plasmids were transfected into HEK293 cells. After 2 days, the transfected cells were cultured in growth medium containing 1.25% Seaplaque-agarose to promote the formation of recombinant viral plaques. Approximately two to three weeks later, individual plaques were picked, amplified in HEK293 cells, and purified over a discontinuous CsCl gradient.
Primary culture of neonatal rat ventricular myocytes
Neonatal rat ventricular myocytes were prepared as described.30 Briefly, one to three-day-old Sprague-Dawley rat pups were killed under ether anesthesia by decapitation, and hearts were removed through a sternotomy. The ventricles were trimmed free of atria, fat and connective tissues. Myocytes were dissociated by several 20-minute cycles of collagenase/pancreatin treatment and serum neutralization. Myocytes were cultured in DMEM with 17% Media 199, 10% horse serum, 5% fetal bovine serum, penicillin (100 units/ml) and streptomycin (100 μg/ml). After one day in culture, myocytes were infected with the recombinant adenoviruses.
Statistical analysis
Data are presented as mean±SD for QTc intervals or mean±SEM for PCR and RPA analyses. Statistical comparison of QTc intervals between R1014X mutation carriers and non-carriers was performed with a family-based analysis approach using the software package PedGenie, a Monte Carlo simulation-based program.31 ANOVA with the Bonferroni correction for multiple pair-wise comparisons between mutation/treatment groups was used for statistical analysis of RPA data. P<0.05 was considered statistically significant.
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
Patient description
A total of 22 family members were tested for the presence of the R1014X mutation. Nine family members were identified as R1014X mutation carriers (Fig. 1). The ECG data were available for seven of the mutation carriers, all of which showed a prolonged QTc interval and typical LQT2 ECG pattern with the subtle bifid T waves. The mean initial QTc interval in the mutation carriers was 461±7 ms (n=7) vs. 420±13 ms (n=8) in non-carriers, P<0.001. Four mutation carriers had exercise tests, with Ex QTc-max of 510±10 ms. In this family 89% (8/9) of the R1014X mutation carriers were asymptomatic. The only one with a history of cardiac events is the 71-year-old proband. From age 32 to 42, she had multiple syncopal episodes and one cardiac arrest that were associated with the presence of hypokalemia (serum K+ = 2.7 mEq/L) caused by a dietary supplement containing potassium-wasting diuretics or taking QT-prolonging antihistamines. Since then she has remained asymptomatic by stopping the potassium-wasting diet, avoiding QT-prolonging drugs, and taking beta-blockers.
Analysis of mRNA isolated from blood samples
The R1014X mutation causes premature termination of the hERG channel protein. This mutation has previously been studied at the protein level.15 However, it has been known that nonsense and frameshift mutations that contain PTCs can lead to the degradation of mRNA transcripts by NMD in many diseases.19,20 In order to determine the underlying pathogenic mechanism of the R1014X mutation, it is important to study this mutation at the mRNA level. Because the affected heart tissue from the mutation carriers was not available for this study, we analyzed hERG mRNA transcripts isolated from lymphocytes of the patients carrying the R1014X mutation. To distinguish between WT and R1014X alleles, we performed allele-specific quantification analysis by using the hot-stop PCR assay. The WT allele contains a TaqI restriction site, which is destroyed by the R1014X mutation. Following reverse transcription of mRNA, cDNA was amplified by hot-stop PCR. After digestion of the PCR products with TaqI, the WT allele should yield two fragments of 287 and 72 bp, and the R1014X allele should give a fragment of 359 bp. As shown in Fig. 2A, cDNA from a normal subject showed a single band at 287 bp, corresponding to the WT alleles, whereas in cDNA from the proband, in addition to WT 287 bp band, a weak 359 bp band from the R1014X mutant allele was observed. Quantitative analysis of the samples from three patients carrying the R1014X mutation revealed that the level of the R1014X mutant was reduced to 23±1% of the WT level, suggesting that the mRNA derived from the R1014X mutant allele is decreased. As a control, we also analyzed genomic DNA from these three patients and showed that the ratio of R1014X to WT alleles was 1.03±0.03, very close to the expected ratio of 1 (Fig. 2A).
Figure 2.
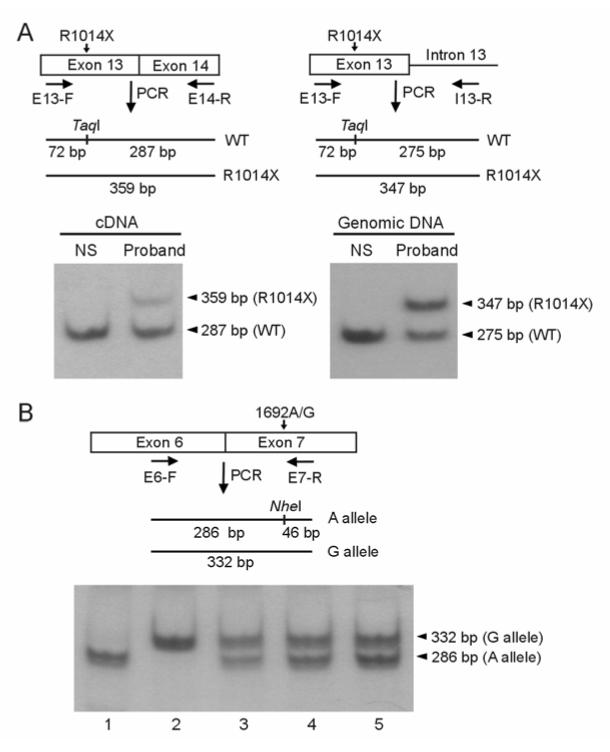
Hot-stop PCR analysis of mRNA and genomic DNA isolated from lymphocytes. A: Analysis of WT and R1014X mutant alleles in a normal subject (NS) and the proband. Schematic diagrams are shown for hot-stop PCR analysis of cDNA (left panel) and genomic DNA (right panel). In cDNA analysis a forward primer in exon 13 (E13-F) and a reverse primer in exon 14 (E14-R) were used, and in genomic DNA analysis the same forward primer and a reverse primer in intron 13 (I13-R) were used. The position of TaqI and the size of the fragments from WT and mutant PCR products are shown. After digestion with TaqI, the 32P labeled hot-stop PCR products were analyzed by polyacrylamide gel electrophoresis and autoradiography. In cDNA analysis (left panel) the bands from WT and R1014X alleles are 287 bp and 359 bp, respectively and in genomic DNA analysis (right panel) the bands from WT and R1014X alleles are 275 bp and 347 bp, respectively (the 72 bp band ran off the gel). Similar results were obtained in two additional R1014X carriers and three to five independent experiments were performed for each patient. B: Analysis of allelic variation of hERG expression by analyzing 1692A/G polymorphism in five normal subjects. A schematic diagram is shown for the position of NheI and the size of the fragments from A and G alleles. The 32P labeled hot-stop PCR products were digested with NheI. The 286 bp and 332 bp bands represent 1692A and 1692G alleles, respectively (the 46 bp band ran off the gel). Results shown are representative of two independent experiments.
The allele-specific quantification analysis depends on the assumption that there is no significant allelic variation in hERG mRNA expression. To rule out possible allelic variation in hERG expression in the general population, we examined the allele-specific expression of hERG mRNA in normal subjects. We analyzed three normal subjects that are heterozygous for a common polymorphism, 1692A/G. To distinguish between 1692A and 1692G alleles, the relative levels of mRNA transcripts from 1692A and 1692G alleles were measured by the hot-stop PCR assay. The 1692A allele contains an NheI restriction site, which is absent in the 1692G allele. Thus, digestion with NheI should allow us to determine the relative ratio of the two WT alleles. After digestion of the PCR products with NheI, the 1692A allele should be cut into two fragments of 286 bp and 46 bp, and the 1692G alleles should remain uncut (332 bp). As shown in Fig. 2B, in subjects 3, 4, and 5 (lanes 3 to 5) there are two bands of 268 bp and 332 bp, suggesting that they are heterozygous for the 1692A/G polymorphism. In these three normal subjects, the average ratio of 1692G to 1692A was 0.97±0.07. This result suggests that there is no significant allelic variation in hERG mRNA expression in normal subjects. Subjects 1 and 2 (lanes 1 and 2) are homozygous for 1692A and 1692G, respectively.
Minigene analysis of the R1014X and W1001X mutations
To study whether the decrease in the abundance of mRNA levels in the R1014X mutation is due to NMD, we constructed minigenes containing the hERG genomic sequence spanning from exon 12 to exon 15 and expressed the minigenes in HEK293 cells. In the minigene experiments two LQT2 nonsense mutations R1014X and W1001X were analyzed by RPA. Figure 3A shows the structure of the minigene and the mRNAs after splicing. The R1014X and W1001X mutations lead to a PTC in exon 13, which is expected to trigger NMD. As shown in Fig. 3B, the mRNA level of the R1014X minigene was significantly lower than that of the WT minigene. Because degradation of mRNA by NMD is dependent on protein synthesis, we examined whether inhibition of protein synthesis by cycloheximide (CHX) abrogates NMD of the mutant mRNA, as has been shown for other PTC-containing transcripts.32 The cells expressing WT and R1014X minigenes were treated with CHX for 3 hours before RNA isolation. Treatment with CHX had no effect on the level of WT mRNA, but significantly increased the level of R1014X mutant mRNA, suggesting that the mutant mRNA is degraded by NMD. Similar results were observed in the W1001X minigene (Fig. 3C), suggesting that the degradation of PTC-containing mRNAs by NMD may represent a common mechanism in LQT2 patients with nonsense mutations.
Figure 3.
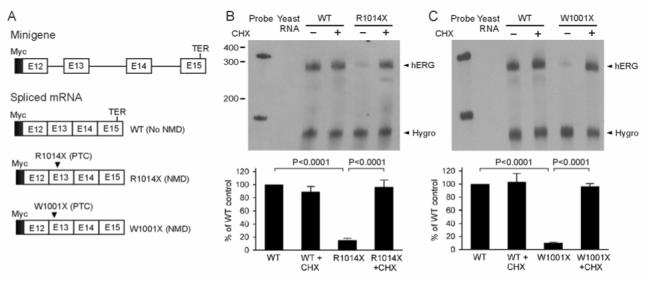
Analysis of the R1014X and W1001X mutations using minigene constructs. A: The structure of the Myc-tagged minigene and spliced mRNAs. The positions of WT termination codon (TER) and mutation-induced PTCs are indicated. B and C: Analysis of mRNA by RNase protection assay (RPA). HEK293 cells were stably transfected with WT, R1014X (B) or W1001X (C) minigenes, and the expressed mRNA was analyzed by RPA. Cells expressing WT and mutant minigenes were treated (+) or not treated (-) with 100 μg/ml of CHX for three hours before RNA isolation. The level of hygromycin resistance gene transcripts (Hygro) served as a loading control. The quantitative data after normalization using protected hygromycin resistance gene mRNA are plotted as percentage of WT control from four (B) or three (C) independent experiments. P-values are Bonferroni-corrected.
Effect of suppression of Upf1 on NMD of the R1014X mutation
Recently, the Upf1 protein has been identified as a key factor for NMD. Reducing Upf1 expression by RNAi has been used as a functional assay to assess the NMD-sensitivity of PTC-containing mRNA transcripts.24,25 To study the role of Upf1 in the reduced mRNA level of the R1014X mutation, we used the RNAi method to knock down Upf1 protein expression. In these experiments, HEK293 cells stably expressing the WT and R1014X minigenes were transfected with pSUPERpuro-hUpf1/I and pSUPERpuro-hUpf1/II.25 The Upf1 knockdown in the transfected cells was confirmed by Western blot analysis using anti-Upf1 antibody (a gift from Dr. Jens Lykke-Andersen) (Fig. 4A).23 Detection of tubulin with anti-tubulin antibody served as a loading control. In the RPA analysis of hERG minigene mRNA transcripts, the level of R1014X mutant mRNA was significantly increased in Upf1-siRNA-transfected cells (Fig. 4B). These results suggest that mRNA transcripts of the R1014X mutation undergo NMD.
Figure 4.
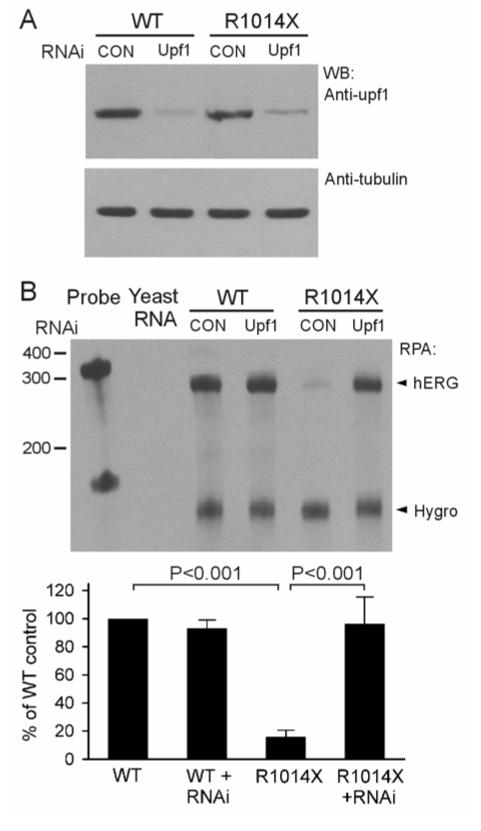
Effect of suppression of Upf1 by RNAi on NMD of the R1014X mutation. HEK293 cells stably expressing the WT and R1014X minigenes were transfected with pSUPERpuro-hUpf1/I and pSUPERpuro-hUpf1/II (Upf1) or pSUPERpuro-scrambled (CON) constructs. A: Western blot analysis of Upf1 protein. B: Analysis of mRNA by RPA. The quantitative data after normalization using protected hygromycin resistant gene mRNA are plotted as percentage of WT control from four independent experiments. P-values are Bonferroni-corrected.
Analysis of NMD in neonatal rat myocytes using R1014X adenovirus minigene
The above experiments indicate that mRNA transcripts of the R1014X mutation are subject to NMD in lymphocytes and HEK293 cells. The noncardiac cells may behave differently from cardiac cells in the degradation of mutant mRNA by NMD. Therefore, it is important to evaluate whether the defects observed in noncardiac systems are present in cardiac myocytes. To test whether NMD of the R1014X mutation occurs in cardiac myocytes, we infected neonatal rat ventricular myocytes with WT or R1014X minigene adenovirus and performed RPA analysis. As shown in Fig. 5, the mRNA level of the R1014X mutant was significantly lower than that of WT. Treatment with CHX had no effect on the level of WT mRNA, but significantly increased the level of R1014X mutant mRNA, suggesting that the R1014X mutant mRNA is degraded by NMD in cardiac myocytes. No protected bands in the control lane indicate that the riboprobe is specific for exogenous hERG transcripts.
Figure 5.
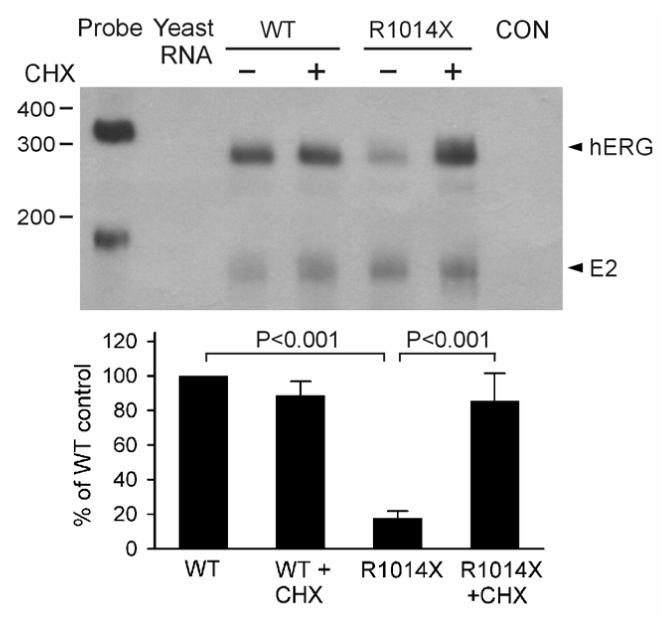
Analysis of NMD in neonatal rat ventricular myocytes using R1014X minigene adenovirus constructs. Myocytes infected by WT and R1014X mutant minigene adenoviruses were treated (+) or not treated (-) with 100 μg/ml of CHX for three hours, and expressed mRNA analyzed by RPA. The mRNA from uninfected myocytes was used as a control (CON). The level of E2 transcripts (E2) from adenovirus served as a loading control. The quantitative data after normalization using protected E2 mRNA are plotted as percentage of WT control from four independent experiments. P-values are Bonferroni-corrected.
Discussion
The present results demonstrate that the W1001X and R1014X mutations lead to a reduction of mutant mRNA transcripts by NMD. Our findings provide the first evidence that PTC-containing mRNA transcripts in LQT2 are subject to NMD. NMD is an evolutionarily conserved mRNA surveillance pathway that detects and eliminates PTC-containing mRNA transcripts, thereby preventing the synthesis of truncated and potentially harmful proteins.33 NMD occurs when translation terminates more than 50-55 nt upstream of the 3′-most exon-exon junction.21,22 According to this rule, more than 90 LQT2 nonsense and frameshift mutations are potential targets for NMD. Several LQT2 nonsense and frameshift mutations have been studied at the functional and protein levels using cDNA constructs.8, 12-18 All previous studies, however, have been carried out under the assumption that these nonsense and frameshift mutations lead to the production of truncated proteins. Because NMD requires introns, the absence of introns in cDNA constructs would preclude the degradation of PTC-containing transcripts by NMD. As a result, NMD effects could not be observed when cDNAs were used in these studies. In the present study we used minigene constructs that contain the hERG genomic DNA with both exons and introns and showed that the W1001X and R1014X mutations cause a marked decrease in mutant mRNA transcripts. Inhibition of protein synthesis by CHX or knockdown of Upf1 by RNAi results in the restoration of mutant mRNA to levels comparable to the WT minigene. These results strongly suggest that the degradation of mutant mRNA by NMD is an important mechanism in LQT2 mutations carrying PTCs.
Previous studies have shown that different LQT2 mutations cause hERG channel dysfunction by different mechanisms. This led to a proposed classification of LQT2 mutations according to their underlying mechanisms.11 The classification scheme shown in figure 6 illustrates mechanisms underlying LQT2 mutations. Class 1 mutations cause abnormal protein synthesis by defective transcription or translation. Class 2 mutations lead to defective protein trafficking. Class 3 mutations result in abnormal gating and/or kinetics. Class 4 mutations result in altered or absent channel selectivity or permeability.11 In the present study, we show that LQT2 nonsence mutations cause a decrease in mutant mRNAs by NMD, thereby altering the amount of mRNA available for subsequent hERG protein generation. We propose that the degradation of PTC-containing mRNA transcripts by NMD represents a new class of LQT2 pathogenic mechanism (class 5).
Figure 6.
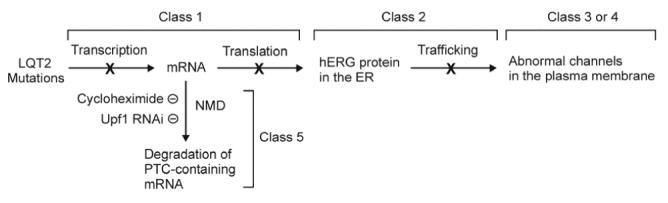
Classification scheme for LQT2 mutations.
The mutations that undergo NMD will result in the degradation of mutant mRNAs before they produce large quantities of truncated proteins. By eliminating abnormal mRNA transcripts carrying PTCs, NMD prevents the production of truncated proteins that could act in a dominant-negative manner, leading to deleterious effects on the cells. One of the physiological roles of NMD is to protect against severe disease phenotypes by converting the dominant-negative effect to haploinsufficiency.32 NMD as a modifier of phenotypic severity has been reported in many human diseases.19,20,32,34 For example, in Marfan syndrome, an autosomal dominant connective tissue disorder caused by mutations in the fibrillin 1 gene, nonsense mutations that result in reduced levels of mutant mRNA are associated with a mild phenotype. In contrast, patients with nonsense alleles that escape NMD develop a severe phenotype due to the dominant negative effect.19,34
Most R1014X mutation carriers in this family have presented with a mild LQT2 phenotype. In contrast to patients with pore-region mutations, who usually present with a longer QT interval and more frequent cardiac events,35 the QTc interval in the R1014X mutation carriers is only mildly prolonged (461±7ms) and only the proband experienced arrhythmia-related cardiac events that were always associated with hypokalemia or taking QT-prolonging drugs. We have previously shown that the R1014X mutation causes hERG channel dysfunction by defective trafficking of the mutant protein.15 In addition, the truncated mutant protein exhibits a dominant negative effect on the WT hERG. This implied that a severe phenotype would be expected in the R1014X mutation carriers. However, our present study reveals that the R1014X mutant mRNA transcripts are markedly decreased by NMD, and as a result, the dominant negative effect caused by the production of truncated proteins would be minimized. Therefore, haploinsufficiency rather than a dominant negative effect is probably the underlying mechanism for the R1014X mutation, which is consistent with the observed clinical presentation of this family. It is interesting to note that the W1001X mutation carriers also present a mild LQT2 phenotype.35 Moss et al reported that LQT2 patients with mutations in the pore region of hERG have a significantly higher risk of arrhythmia-related cardiac events than patients with non-pore mutations.35 Although the difference may be explained by in vitro electrophysiological effects of reported hERG mutations, with pore mutations having a greater negative effect on hERG current than non-pore mutations,35 it is also possible that NMD may play a role. It is noted that only 6% of LQT2 mutations in the pore region are nonsense or frameshift mutations, whereas more than 40% of the mutations in non-pore regions are nonsense or frameshift mutations. Clearly, further genotype-phenotype correlation studies are required to test whether NMD contributes to the observed differences in clinical presentations of pore and non-pore LQT2 mutations.
There are potential limitations to this study. Our present experiments analyzed endogenously expressed mRNA from patients carrying the R1014X mutation, but the RNA was isolated from lymphocytes rather than the affected heart tissue. Although we have shown that the R1014X mutant minigene expressed in neonatal rat ventricular myocytes leads to reduced mRNA levels by NMD, further studies are required to determine whether the endogenous PTC-containing mRNA in human heart tissue is subject to NMD. Verification of our findings in human heart would strengthen the conclusion that hERG mutations that contain PTCs can lead to degradation of the mutant mRNA by NMD.
In summary, our findings that nonsense mutations in hERG lead to a reduced level of mutant mRNA by NMD add to our understanding of the disease-causing mechanisms of hERG mutations in LQT2. Thus, in studies of hERG nonsense and frameshift mutations, it is important to first analyze the abundance of mRNA to determine whether these PTC mutations are targeted by NMD. Obviously, this important point had been overlooked in previous studies, which analyzed hERG PTC mutations only at the protein and functional levels. Since PTC mutations account for more than 30% of LQT2 mutations, the RNA surveillance imposed by NMD is of fundamental importance in the pathogenesis of LQT2.
Acknowledgments
We thank Dr. Kent Thornburg for helpful comments on the manuscript.
Funding Sources:
This study was supported in part by NIH grant HL68854 (Dr. Zhou), Deseret Foundation grant DF400 (Dr. Vincent), and NIH grant 1UL1RRO24140-01 from the National Center for Research Resources.
Footnotes
Disclosure:
None
References
- 1.Schwartz PJ, Periti M, Malliani A. Fundamentals of clinical cardiology: the long QT syndrome. Am Heart J. 1975;89:378–390. doi: 10.1016/0002-8703(75)90089-7. [DOI] [PubMed] [Google Scholar]
- 2.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 3.Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 4.Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 5.Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, Bottelli G, Cerrone M, Leonardi S. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA. 2005;294:2975–2980. doi: 10.1001/jama.294.23.2975. [DOI] [PubMed] [Google Scholar]
- 6.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 7.Millat G, Chevalier P, Restier-Miron L, Da Costa A, Bouvagnet P, Kugener B, Fayol L, Gonzalez Armengod C, Oddou B, Chanavat V, Froidefond E, Perraudin R, Rousson R, Rodriguez-Lafrasse C. Spectrum of pathogenic mutations and associated polymorphisms in a cohort of 44 unrelated patients with long QT syndrome. Clin Genet. 2006;70:214–227. doi: 10.1111/j.1399-0004.2006.00671.x. [DOI] [PubMed] [Google Scholar]
- 8.Sanguinetti MC, Curran ME, Spector PS, Keating MT. Spectrum of HERG K+-channel dysfunction in an inherited cardiac arrhythmia. Proc Natl Acad Sci USA. 1996;93:2208–2212. doi: 10.1073/pnas.93.5.2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Z, Gong Q, Epstein ML, January CT. HERG channel dysfunction in human long QT syndrome: Intracellular transport and functional defects. J Biol Chem. 1998;273:21061–21066. doi: 10.1074/jbc.273.33.21061. [DOI] [PubMed] [Google Scholar]
- 10.Thomas D, Kiehn J, Katus HA, Karle CA. Defective protein trafficking in hERG-associated hereditary long QT syndrome (LQT2): molecular mechanisms and restoration of intracellular protein processing. Cardiovasc Res. 2003;60:235–2. doi: 10.1016/j.cardiores.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Delisle BP, Anson BD, Rajamani S, January CT. Biology of cardiac arrhythmias: ion channel protein trafficking. Circ Res. 2004;94:1418–1428. doi: 10.1161/01.RES.0000128561.28701.ea. [DOI] [PubMed] [Google Scholar]
- 12.Li X, Xu J, Li M. The human delta1261 mutation of the HERG potassium channel results in a truncated protein that contains a subunit interaction domain and decreases the channel expression. J Biol Chem. 1997;272:705–708. doi: 10.1074/jbc.272.2.705. [DOI] [PubMed] [Google Scholar]
- 13.Paulussen A, Yang P, Pangalos M, Verhasselt P, Marrannes R, Verfaille C, Vandenberk I, Crabbe R, Konings F, Luyten W, Armstrong M. Analysis of the human KCNH2 (HERG) gene: identification and characterization of a novel mutation Y667X associated with long QT syndrome and a non-pathological 9 bp insertion. Hum Mut. 2000;15:483. doi: 10.1002/(SICI)1098-1004(200005)15:5<483::AID-HUMU18>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 14.Kupershmidt S, Yang T, Chanthaphaychith S, Wang Z, Towbin JA, Roden DM. Defective human Ether-a-go-go-related gene trafficking linked to an endoplasmic reticulum retention signal in the C terminus. J Biol Chem. 2002;277:27442–27448. doi: 10.1074/jbc.M112375200. [DOI] [PubMed] [Google Scholar]
- 15.Gong Q, Keeney DR, Robinson JC, Zhou Z. Defective assembly and trafficking of mutant HERG channels with C-terminal truncations in long QT syndrome. J Mol Cell Cardiol. 2004;37:1225–1233. doi: 10.1016/j.yjmcc.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Teng S, Ma L, Dong Y, Lin C, Ye J, Bahring R, Vardanyan V, Yang Y, Lin Z, Pongs O, Hui R. Clinical and electrophysiological characterization of a novel mutation R863X in HERG C-terminus associated with long QT syndrome. J Mol Med. 2004;82:189–196. doi: 10.1007/s00109-003-0504-1. [DOI] [PubMed] [Google Scholar]
- 17.Paulussen AD, Raes A, Jongbloed RJ, Gilissen RA, Wilde AA, Snyders DJ, Smeets HJ, Aerssens J. HERG mutation predicts short QT based on channel kinetics but causes long QT by heterotetrameric trafficking deficiency. Cardiovasc Res. 2005;67:467–475. doi: 10.1016/j.cardiores.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 18.Choe CU, Schulze-Bahr E, Neu A, Xu J, Zhu ZI, Sauter K, Bahring R, Priori S, Guicheney P, Monnig G, Neapolitano C, Heidemann J, Clancy CE, Pongs O, Isbrandt D. C-terminal HERG (LQT2) mutations disrupt IKr channel regulation through 14-3-3ε. Hum Mol Genet. 2006;15:2888–2902. doi: 10.1093/hmg/ddl230. [DOI] [PubMed] [Google Scholar]
- 19.Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- 20.Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE. Nonsense-mediated decay approaches the clinic. Nat Genet. 2004;36:801–808. doi: 10.1038/ng1403. [DOI] [PubMed] [Google Scholar]
- 21.Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci. 1998;23:198–199. doi: 10.1016/s0968-0004(98)01208-0. [DOI] [PubMed] [Google Scholar]
- 22.Maquat LE. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol. 2004;5:89–99. doi: 10.1038/nrm1310. [DOI] [PubMed] [Google Scholar]
- 23.Lykke-Andersen J, Shu MD, Steitz JA. Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell. 2000;103:1121–1131. doi: 10.1016/s0092-8674(00)00214-2. [DOI] [PubMed] [Google Scholar]
- 24.Mendell JT, ap Rhys CM, Dietz HC. Related Separable roles for rent1/hUpf1 in altered splicing and decay of nonsense transcripts. Science. 2002;298:419–422. doi: 10.1126/science.1074428. [DOI] [PubMed] [Google Scholar]
- 25.Paillusson A, Hirschi N, Vallan C, Azzalin CM, Muhlemann O. A GFP-based reporter system to monitor nonsense-mediated mRNA decay. Nucleic Acids Res. 2005;33:e54. doi: 10.1093/nar/gni052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L, Timothy KW, Vincent GM, Lehmann MH, Fox J, Giuli LC, Shen J, Splawski I, Priori S, Compton SJ, Yanowitz F, Benhorin J, Moss AJ, Schwartz PJ, Robinson J, Wang Q, Zareba W, Keating M, Towbin JA, Napolitano C, Medina A. Spectrum of ST-T wave patterns and repolarization parameters in congenital long QT syndrome: ECG findings identify genotype. Circulation. 2000;102:2849–2855. doi: 10.1161/01.cir.102.23.2849. [DOI] [PubMed] [Google Scholar]
- 27.Uejima H, Lee MP, Cui H, Feinberg AP. Hot-stop PCR: a simple and general assay for linear quantitation of allele ratios. Na. Genet. 2000;25:375–376. doi: 10.1038/78040. [DOI] [PubMed] [Google Scholar]
- 28.Kurreeman FA, Schonkeren JJ, Heijmans BT, Toes RE, Huizinga TW. Transcription of the IL10 gene reveals allele-specific regulation at the mRNA level. Hum Mol Genet. 2004;13:1755–1762. doi: 10.1093/hmg/ddh187. [DOI] [PubMed] [Google Scholar]
- 29.Gong Q, Keeney DR, Molinari M, Zhou Z. Degradation of trafficking-defective long QT syndrome type II mutant channels by the ubiquitin-proteasome pathway. J Biol Chem. 2005;280:19419–19425. doi: 10.1074/jbc.M502327200. [DOI] [PubMed] [Google Scholar]
- 30.Kapiloff MS, Schillace RV, Westphal AM, Scott JD. mAKAP: an A-kinase anchoring protein targeted to the nuclear membrane of differentiated myocytes. J Cell Sci. 1999;112:2725–2736. doi: 10.1242/jcs.112.16.2725. [DOI] [PubMed] [Google Scholar]
- 31.Allen-Brady K, Wong J, Camp NJ. PedGenie: an analysis approach for genetic association testing in extended pedigrees and genealogies of arbitrary size. BMC Bioinformatics. 2006;7:209–220. doi: 10.1186/1471-2105-7-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inoue K, Khajavi M, Ohyama T, Hirabayashi S, Wilson J, Reggin JD, Mancias P, Butler IJ, Wilkinson MF, Wegner M, Lupski JR. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat Genet. 2004;36:361–369. doi: 10.1038/ng1322. [DOI] [PubMed] [Google Scholar]
- 33.Conti E, Izaurralde E. Nonsense-mediated mRNA decay: molecular insights and mechanistic variations across species. Curr Opin Cell Biol. 2005;17:316–325. doi: 10.1016/j.ceb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 34.Dietz HC, McIntosh I, Sakai LY, Corson GM, Chalberg SC, Pyeritz RE, Francomano CA. Four novel FBN1 mutations: significance for mutant transcript level and EGF-like domain calcium binding in the pathogenesis of Marfan syndrome. Genomics. 1993;17:468–475. doi: 10.1006/geno.1993.1349. [DOI] [PubMed] [Google Scholar]
- 35.Moss AJ, Zareba W, Kaufman ES, Gartman E, Peterson DR, Benhorin J, Towbin JA, Keating MT, Priori SG, Schwartz PJ, Vincent GM, Robinson JL, Andrews ML, Feng C, Hall WJ, Medina A, Zhang L, Wang Z. Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation. 2002;105:794–799. doi: 10.1161/hc0702.105124. [DOI] [PubMed] [Google Scholar]