

Abstract
Cell adhesion molecules (CAMs) are known to be involved in a variety of developmental processes that play key roles in the establishment of synaptic connectivity during embryonic development, but recent evidence implicates the same molecules in synaptic plasticity of the adult. In the present study, we have used neural CAM (NCAM)-deficient mice, which have learning and behavioral deficits, to evaluate NCAM function in the hippocampal mossy fiber system. Morphological studies demonstrated that fasciculation and laminar growth of mossy fibers were strongly affected, leading to innervation of CA3 pyramidal cells at ectopic sites, whereas individual mossy fiber boutons appeared normal. Electrophysiological recordings performed in hippocampal slice preparations revealed that both basal synaptic transmission and two forms of short-term plasticity, i.e., paired-pulse facilitation and frequency facilitation, were normal in mice lacking all forms of NCAM. However, long-term potentiation of glutamatergic excitatory synapses after brief trains of repetitive stimulation was abolished. Taken together, these results strongly suggest that in the hippocampal mossy fiber system, NCAM is essential both for correct axonal growth and synaptogenesis and for long-term changes in synaptic strength.
Keywords: cell adhesion molecules/frequency facilitation/paired-pulse facilitation/long-term potentiation/excitatory synapses
Activity-dependent changes in synaptic strength play a key role in various behaviors ranging from cortical arousal to learning and memory (1–3). The mechanisms contributing to the different forms of plasticity are still the subject of intense debate. One of the hypotheses that has been proposed is that specific patterns of activity could lead to structural modifications of synapses (for reviews, see refs. 4 and 5). However, little is known about the molecular machinery that orchestrates the coordination of the structural changes in synaptic remodeling with functional modifications of synaptic strength during learning and memory. In line with the hypothesis of structural reorganization of synapses by activity is the finding that cell adhesion molecules (CAMs) contribute to synaptic plasticity. For example, reduced cell-surface expression of Fasciclin II (Fas II) at the Drosophila neuromuscular synapse and of apCAM in Aplysia, two homologues of vertebrate neural CAMs (NCAMs), are prerequisites for the induction of activity-dependent synaptic plasticity in these species (6, 7). In both cases, presynaptic mechanisms have been proposed to underlie these plastic changes. At the Drosophila neuromuscular synapse, Fas II is not required for initial activity-independent synapse formation, but is required for synapse stabilization, remodeling, and sprouting of additional synaptic contacts (8, 9). These findings suggest a role for this class of molecules during different forms of synaptic plasticity, triggered at a presynaptic level.
One of the most commonly used paradigms for the study of the different presynaptic forms of plasticity are the mossy fibers of the hippocampus (2). These axons of dentate gyrus granule cells form en passant synapses on the proximal portion of the apical dendrite of CA3 pyramidal neurons in a narrow band termed stratum lucidum. In contrast, associational and commissural fibers form synapses on the rest of the dendritic tree. The latter have properties similar to those of excitatory synapses found on dentate granule and CA1 pyramidal cells: they are associated with high concentrations of N-methyl-d-aspartate (NMDA) receptor-binding sites (10), and repetitive activation results in an NMDA-receptor-dependent long-term potentiation (LTP) of synaptic strength. In striking contrast, the mossy fiber synapses are associated with a low level of NMDA receptor binding, and high-frequency stimulation results in an NMDA receptor-independent form of LTP (11), which is triggered presynaptically (12).
Variations in hippocampal mossy fiber distribution have been shown to be correlated with spatial learning abilities and exploratory behavior in mice (13, 14). Consistent with these observations, we reported that mice deficient for all the different isoforms of the NCAM protein, which exhibit deficits in exploratory behavior and perform less well in the Morris water maze (15), show alterations in mossy fiber growth and fasciculation (16).
In the present study, we investigate further the role of NCAM in the formation, function, and plasticity of the mossy fiber system. Using immunohistochemistry and confocal microscopy we show that high levels of NCAM are associated specifically with mossy fiber terminals and that the laminated distribution of these terminals is perturbed in NCAM knockout mice. To address the consequences of NCAM deficiency on the modulation of synaptic transmission, we investigated mossy fiber plasticity in the NCAM mutants. Our results show that only long-term and not short-term plasticity requires the presence of these adhesion molecules.
MATERIALS AND METHODS
Generation of the NCAM-mutant mice has been described previously (15). All data were acquired from either wild-type or homozygous mutant mice on a C57BL/6J background (five backcrosses), and all recordings were made “blind” to mice genotype. For immunohistochemistry, 4- to 5-month-old animals were deeply anesthetized with Rompun/Imalgene and perfused with 50 ml 4% paraformaldehyde in PBS. Brains were postfixed in the same solution, and 50-μm-thick sagittal sections were cut on a standard vibratome. Staining for NCAM was performed on floating sections in DMEM containing 10% fetal calf serum by using polyclonal antibodies (17). Confocal analysis was performed by using a standard Leitz confocal microscope and the complementary software package. For three-dimensional reconstruction, 10 individual virtual sections were reconstructed. For electron microscopy, mice were superfused with 4% paraformaldehyde (PFA)/0.1% glutaraldehyde followed by 4% PFA containing sodium m-periodate and lysine. Brains were postfixed overnight in the second solution. Fifty-micron sections were cut on a vibratome and kept in osmium tetroxide for 1 h. After dehydration, the tissue was immersed in propylene oxide, followed by propylene oxide/epon (1:1) and, finally, pure epon for infiltration. Subsequently, slices were flat-embedded in epon between two plastic slides. Selected areas from the distal CA3 hippocampal region were cut from the plastic weaver and ultrathin-sectioned. After mounting on single-hole copper grids, the sections were stained with uranyl acetate and lead citrate and examined under a Zeiss electron microscope.
Hippocampal slices were prepared from adult mice (4–5 months old). After decapitation, the brain was quickly removed and submerged in artificial cerebrospinal solution (ACSF: 119 mM NaCl/2.5 mM KCl/2.5 mM CaCl2/1.2 mM MgSO4/26.2 mM NaHCO3/1 mM NaH2PO4/10 mM d-glucose, equilibrated with 95% O2 and 5% CO2) at 4°C for about 2 min. The brain was sectioned further into a block and glued to the stage of a Vibratome. Transverse hippocampal slices (400-μm thick) were allowed to recover for at least 1 h at 32°C and then at room temperature for the rest of the experiment. They were transferred one at a time to a submersion recording chamber for electrophysiological experiments, where they were superfused at room temperature (flow rate of about 2 ml/min) with ACSF.
Extracellular field recordings were made with glass electrodes containing 1 M NaCl (impedance ranging between 5 and 20 MΩ) by using a DAM-80 amplifier (WPI Instruments, Waltham, MA) and were located in the stratum lucidum to record evoked mossy fiber excitatory postsynaptic potentials (fEPSPs). To evoke synaptic responses, stimuli (100-μs duration) were delivered through fine bipolar tungsten electrodes placed in the cell-body layer of the granule cells. Field responses were filtered at 1 kHz, digitized at 4 kHz on a TL-1 interface (Axon Instruments), and collected on a Pentium IBM-compatible computer. On- and off-line data processing were carried out by using in-house software (acquis1, Gérard Sadoc, Centre National de la Recherche Scientifique-Agence Nationale pour la Valorisation de la Recherche). Average values are expressed as mean ± SEM as a percentage of the baseline. Student’s t test was used to determine whether there was a significant difference in the means between the results from wild-type and mutant mice.
Paired-pulse facilitation was defined as [(A2 − A1)/A1] × 100, where A1 and A2 are the amplitude of the fEPSPs evoked by the first and second pulse, respectively. For LTP experiments, baseline transmission was monitored at 0.05 Hz and groups of three potentials were averaged to yield one measurement per minute of the initial slope of the field EPSP. The initial values of EPSP were adjusted to about half-maximal. The LTP-inducing stimulus consisted of 1 train at 100 Hz, repeated four times, after at least 30 min of stable baseline in the presence of 100 μM d-2-amino-5-phosphonovaleric acid. The restricted anatomy of the mossy fiber input and the reversal of the waveform as the recording electrode is moved from stratum lucidum to stratum radiatum serve to define mossy fiber inputs clearly. Moreover, bath application of 10 μM (2S,3S,4S)-CCG/(2S,1′S,2′S)-2-(carboxycyclopropyl)-glycine (L-CCG1) was used to distinguish mossy fiber synaptic responses from non-mossy-fiber synaptic responses. At the end of all the experiments, 10 mM kynurenic acid (dissolved directly into ACSF solution with pH adjusted to 7.35 using NaOH) was applied to the bath to assess the fiber volley component of the response. If necessary, this component was subtracted from the synaptic potentials during off-line analysis.
RESULTS
Confocal analysis of sagittal sections stained for the presence of NCAM reflected the laminated organization of the hippocampal CA3 region (Fig. 1a). The strongest coloration was visible in the stratum lucidum, which corresponds to the termination field of the mossy fibers on the apical dendrites of CA3 pyramidal neurons. A lower degree of staining was found in stratum radiatum, whereas the cell body layer was almost negative over its entire length (Fig. 1 b–d). Coimmunostaining with the presynaptic marker synaptophysin confirmed the restriction of large boutons, typical of mossy fiber terminals, to stratum lucidum (Fig. 1 a–d). At a higher magnification, it became evident that synaptophysin and NCAM immunoreactivities were closely associated, with the boutons embedded in NCAM-positive structures (Fig. 1c).
Figure 1.
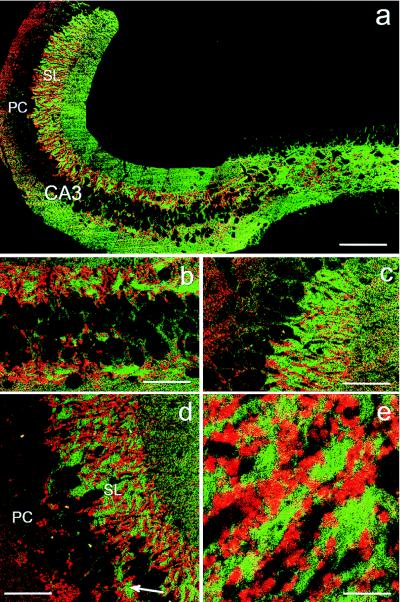
Expression of NCAM and synaptophysin in the hippocampus. (a) Reconstruction of confocal images of a sagittal brain section stained for the presence of NCAM (green) and the synaptic vesicle marker synaptophysin (red). Strongest NCAM immunoreactivity is visible in the hilus and the stratum lucidum, thus, in regions of mossy fiber terminals. (b–d) Higher magnification of a showing the region closest to the hilus (b), at the border to CA2 (c) and at the midpoint of CA3 (d). In all situations mossy fiber terminals are linked to high expression of NCAM protein. Note that the few giant synapses formed within stratum pyramidale also are associated with NCAM expression (arrow in d). Smaller synapses, also expressing synaptophysin and representing other types of terminals, are present in regions of the hippocampal formation expressing lower amounts of NCAM, as, for example, stratum radiatum (upper right part of d). (e) Higher magnification of d showing the closely associated and partially overlapping distribution of NCAM and synaptophysin immunoreactivities in stratum lucidum. CA3, hippocampal CA3 region; DG, dentate gyrus; PC, pyramidal cell layer or stratum pyramidale; SL, stratum lucidum. [Bars = 200 μm (a); 50 μm (b–d); and 10 μm (e).]
In a former study, we showed altered axonal growth and fasciculation in the mossy fiber system of NCAM-deficient mice (16). We used immunohistochemistry and confocal microscopy to further investigate these morphological alterations. Immunostaining for the axonal marker neurofilament and reconstitution of serial sections of the CA3 region revealed that in adult wild-type mice, the axons are oriented strictly parallel to the direction of the CA3 pyramidal cell layer (Fig. 2a). In addition, they appear to be organized in fascicles that are located between the neurons as well as in the supra- and infrapyramidal bundles (Fig. 2a). In the mutant, this configuration of the mossy fibers is lost entirely (Fig. 2b). Although the fibers still travel along the CA3 region, a parallel organization is no longer visible. In contrast, many of the remaining small bundles or individual mossy fibers travel through the CA3 region perpendicular to their normal direction.
Figure 2.
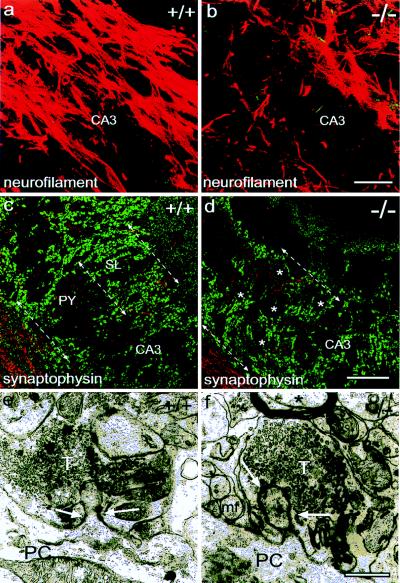
Consequences of NCAM deficiency on mossy fiber axons and terminals. (a and b) Neurofilament staining and confocal analysis reveals the parallel and fasciculated organization of the mossy fibers following the direction of the axis of the CA3 region (a). In the mutant (b), this arrangement is lost. (c and d) Staining for the presynaptic marker synaptophysin (green) demonstrates the laminated organization of the wild-type CA3 region (c). In the mutants, the ectopic formation of mossy fiber terminals within the stratum pyramidale, where the pyramidal cell bodies (asterisks) are located, leads to the loss of a defined stratum lucidum (d). In these experiments, the neurofilament staining of the axons (red) has been attenuated to better reveal the boutons. (e and f) Ultrastructural analysis of mossy fiber terminals revealed no obvious alterations in the NCAM-deficient situation. Note the presence of individual mossy fiber axons close to the vesicle-filled bouton in the mutant (arrows in f). CA3, hippocampal CA3 region; DG, dentate gyrus; PC, pyramidal cell; sl, stratum lucidum; mf, mossy fiber axon; T, mossy fiber terminal. [Bars = 40 μm (a–d); 0.5 μm (e–f).]
The distribution of mossy fiber terminals was examined by synaptophysin immunohistochemistry. In wild-type animals (Fig. 2c, see also Fig. 1), the typical giant boutons representing mossy fiber synapses were organized in the well described laminated pattern (18) and permitted a clear distinction between pyramidal cell layer and stratum lucidum (arrows in Fig. 2c). The most obvious alteration in NCAM-deficient mice at this level of resolution was the loss of lamination. The mossy fiber terminals appear to be distributed throughout the CA3 region, and many of them are present between the pyramidal cell bodies (Fig. 2d), which is in accordance with the altered Timm’s staining already described in these mutants (16). The arrangement of the boutons, surrounding the contours of pyramidal cells (asterisks in Fig. 2d), suggests that terminals are formed mainly on the cell bodies and not on the proximal part of their dendrites as in wild-type mice. Nevertheless, ultrastructural analysis revealed no fundamental difference in the structure or size of the mossy fiber boutons (Fig. 2 e and f). Both mutant and wild-type terminals represent multilobulated complexes providing intense contact between pre- and postsynaptic membranes and a variety of synaptic contacts. Note the presence of nonmyelinized mossy fibers adjacent to the mutant terminals, which is a result of the ectopic position of the boutons. This is never seen in wild-type animals where fasciculated axons are spatially separated from terminals (see also ref. 16).
To investigate the electrophysiological properties of mossy fiber synapses in NCAM-deficient mice, we first asked whether the basic properties of the mutant mossy fiber synapses were altered. Testing the functional integrity of the mossy fiber pathway is not trivial as, even in the absence of functional mossy fibers, activation of neighboring associational/commissural fibers still would be expected to elicit a response. Thus, to distinguish mossy fiber synaptic responses from other synaptic responses, we took advantage of the fact that in the mouse, mossy fiber terminals but not associational/commissural fibers possess inhibitory presynaptic metabotropic glutamate receptors (mGluRs) of the group 2 subtype (19, 20). Thus, application of the group 2-selective agonist L-CCG1 (10 μM) did not affect the response to associational/commissural stimulation, but blocked the response evoked by a stimulating electrode placed in the dentate granule cell layer (data not shown, n = 7). The magnitude of the inhibition induced by L-CCG1 was the same in wild-type and knock out mice (Fig. 3B). This shows that mossy fibers terminals are present and functional in NCAM-deficient mice, as predicted from their normal ultrastructural appearance. In all subsequent experiments, L-CCG1 was applied to assess the purity of the mossy fiber response and inputs were used only if the inhibition was at least 80%. Although L-CCG1 did not distinguish basal synaptic transmission between the two animal groups, it is noteworthy that, for the same stimulation intensity, field synaptic responses were reduced systematically in NCAM-deficient mice (data not shown). Consistent with the reduced Timm’s staining, this observation suggests a lower number of mossy fiber terminals in the mutant CA3 region, as described previously (16).
Figure 3.

Mossy fiber synapses have functional presynaptic group 2 metabotropic glutamate receptors in NCAM-deficient mice. (A) Bath application of 10 μM L-CCG1 (horizontal bar) during 2 min abolished the mossy fiber responses in NCAM-mutant mice. (B) Summary graphs showing that inhibitory effects of L-CCG1 measured at the end of the application were similar in mutant mice (Upper, solid circles, three animals, five slices) and wild-type mice (Lower, open circles; four animals, six slices), suggesting a normal targeting of metabotropic glutamate receptors in NCAM knockout mice.
Mossy fiber and associational/commissural synaptic responses also differ from each other in their sensitivity to different forms of short-term synaptic plasticity. A synapse activated twice at short intervals shows an increase of the response to the second stimulus because of augmented transmitter release, a process referred to as paired-pulse facilitation (PPF). This facilitation, which is stronger in mossy fiber synapses than in other synapses, was measured in knockout mice. No significant change in duration or magnitude of PPF was observed in the mutants compared with wild-type animals (Fig. 4A and B). Another characteristic of mossy fiber synapses is their sensitivity to repetitive stimulation. This process, which also represents a form of short-term plasticity, occurs as a consequence of a change in the frequency of stimulation and results in a reversible increase in the mossy fiber responses. This phenomenon, referred to as frequency facilitation, also was unaffected in the knockout mice (Fig. 4 C and D). For both groups of animals, changing the frequency of stimulation from 0.05 to 0.2 Hz resulted in a more than 2-fold reversible increase in the size of the mossy fiber responses. Changing the frequency from 0.05 to 0.33 Hz gave a 4-fold increase. Since these two forms of plasticity depend on the probability of transmitter release, we concluded from this first set of experiments that in NCAM-deficient mice, the probability of glutamate release is not altered markedly. Together this suggests that basic synaptic transmission parameters are unaffected in these animals.
Figure 4.

Two forms of short-term synaptic plasticity are normal at mossy fiber synapses from NCAM knockout mice. (A and B) PPF for different interstimulus intervals was compared in normal and mutant mice. (A) Sample traces are averages of five consecutive mossy fiber responses recorded from hippocampal slices from NCAM-deficient (KO) or wild-type (WT) mice. The fiber volley was subtracted from both responses. (B) Summary graphs showing a similar duration and magnitude (mean ± SEM) of PPF between the two animal groups (n = 6 for each animal genotype). (C and D) Frequency facilitation of mossy fiber responses. The graph summarizes the results of six experiments performed by changing the frequency of stimulation for 2 min (horizontal bars) from 0.05 to 0.2 Hz (C) or from 0.05 to 0.33 Hz (D). There are no significant differences (P > 0.5) in PPF or frequency facilitation between wild-type and mutant mice.
Lastly, we investigated whether long-term plasticity would be altered in the absence of NCAM. Mossy fiber pathway LTP was induced by high-frequency trains (100 Hz for 1 sec given four times with a 20-sec interval between trains) in the presence of the NMDA receptor antagonist d-2-amino-5-phosphonopentanoic acid (100 μM) to prevent any contamination of the responses by the NMDA receptor-dependent LTP, which occurs at the associational/commissural synapses onto CA3 pyramidal cells. Hippocampal slices (n = 8) from four wild-type mice showed a robust mossy fiber LTP, which lasted for the entire time period analyzed (a mean of 174 ± 8% measured 60 min after the tetanus). In contrast, LTP was essentially absent in four NCAM-deficient mice when measured 1 h after induction (a mean of 96 ± 9% measured after 60 min; 8 slices) (Fig. 5). The potentiation measured immediately after the tetanus, however, did not seem to be significantly altered in the knockout mice.
Figure 5.

Mossy fiber long-term potentiation is reduced in NCAM-deficient mice. (A) Superimposed sample field potential recordings before and 55–60 min after tetanization in wild-type (WT) and knockout (KO) mice. (B) Mossy fiber field potentials are plotted against time. After baseline responses were stable for at least 30 min, a tetanus was given to induce long-term potentiation. This summary graph (mean ± SEM) shows that the magnitude of LTP was reduced in NCAM-deficient animals (solid circles; four animals and eight slices) when compared with normal animals (open circles; four animals and eight slices, P < 0.001 with Student’s unpaired t test).
DISCUSSION
In previous work, we have shown that in the absence of NCAM, the organization of the mossy fiber pathway is perturbed, probably because of the failure of the fiber bundles to fasciculate correctly (16). Similar findings have been reported for mice lacking the major NCAM isoform (21). In the present study, we show that not only the pathway taken by the mossy fibers but also the orderly distribution of their terminals are perturbed in the mutant hippocampus. Instead of being restricted to the proximal dendrites in the stratum lucidum, many of the typical giant mossy fiber boutons, which are otherwise of normal size and shape, appear in the pyramidal cell layer. We then asked how the function of mossy fiber synapses in the CA3 region of the hippocampus would be affected by the lack of all NCAM isoforms. The results show that basic synaptic parameters, including two forms of short-term plasticity described previously (22), were unaltered. In contrast, long-term plasticity of these synapses was severely affected, as mossy fiber LTP was essentially absent.
Mossy fibers, which arise from dentate granule cells and which synapse on the proximal dendrites of CA3 pyramidal cells, exhibit a form of LTP that is fundamentally different from NMDA-receptor-dependent LTP (for a review, see ref. 2). This LTP is independent of NMDA receptors, thought to be initiated presynaptically by calcium entry into the presynaptic terminal, and thought to result from an increase of the probability of neurotransmitter release. Therefore, our observation has to be distinguished from the reduction of LTP reported in the CA1 region of NCAM-deficient mice (23), a form of NMDA-receptor-mediated LTP that cannot be seen in our experiments because of the presence of D-2-amino-5-phosphonopentanoic acid. Furthermore, these authors used young, not adult mice.
Our present data do not permit us to determine whether the failure of the mutant animals to exhibit mossy fiber LTP is a consequence of the altered topology of the terminals or whether it reflects a functional change of the mature synapses. However, the observations that the postsynaptic cell plays little or no role in triggering mossy fiber LTP and that the mossy fiber boutons from the mutants seem morphologically normal argue in favor of a role of NCAM in synapse function. Moreover, a great variety of studies have strengthened the notion that CAMs in general, and NCAM in particular, are essential for various forms of synaptic plasticity (reviewed in refs. 24–26). For example, antibodies against NCAM were found to interfere with LTP expressed in the CA1 region of hippocampal slices without affecting basal synaptic transmission (27, 28). In both chicks and rats, intraventricular injection of antibodies against NCAM has been shown to block memory formation in a passive avoidance task, a form of associative learning thought to involve activity-dependent synaptic plasticity. Moreover, learning of the avoidance task was found to be associated with increased NCAM labeling at active synaptic zones in the chicken striatum (ref. 29; for a review of the earlier literature see ref. 30).
Genetic analysis of Fas II function, the Drosophila homolog of NCAM, in the neuromuscular system provides perhaps the best evidence to date for the implication of a CAM in axonal fasciculation, pathfinding, synaptic growth and stability during development, as well as synaptic plasticity once the synapses have formed. Work on defined motoneuron pathways first showed that Fas II controls fasciculation and sorting during axon pathfinding (31). Thus, during development, Fas II is essential for orderly axonal growth as is NCAM in the mossy fiber system. Later in development, Fas II controls growth and stabilization of the neuromuscular synapse and is required pre- and postsynaptically to carry out these functions (8, 32). Finally, the levels of Fas II expressed at the synapse play a critical role in controlling structural and, in conjunction with activator of transcription factor (CREB activator), functional plasticity (7, 9). As is the case for mossy fiber LTP, the CREB- and Fas II-dependent changes in synaptic strength at the Drosophila neuromuscular synapse seem to be due to an increase in glutamate release. This raises the possibility that in vertebrates as well, modulation of the cyclic AMP system, which is thought to mediate LTP presynaptically at mossy fiber terminals (33), functions together with changes in NCAM at the cell surface to increase synaptic strength.
There is one major difference between Drosophila and our system: an increase in synaptic strength at the Drosophila neuromuscular synapse requires a decrease in Fas II levels, whereas we see loss of long-term plasticity in the absence of NCAM. Similarly, training for long-term memory in the gill-withdrawal reflex paradigm of Aplysia leads to down-regulation of the NCAM homolog, apCAM, at the neuronal cell surface (6, 34). In contrast, in vertebrates, mobilization of polysialylated NCAM to the cell surface was found to require electrical activity (23, 35) and enhancement of glutamatergic transmission resulted in increased NCAM promoter activity (36). These contrasting results may be taken as evidence for fundamental differences in the functioning of vertebrate and invertebrate synapses. However, studying the effect of complete removal of apCAM or Fas II has not been possible in Aplysia, because of the lack of genetic techniques, and in Drosophila, since a 90% reduction in Fas II levels already results in synapse elimination. Conceivably, optimal levels of NCAM or Fas II are required to trigger synaptic plasticity.
The studies on Fas II also suggest an explanation for the altered topology of mossy fiber terminals observed in the NCAM null mutants. In the Drosophila system, preventing the normal decrease in extrasynaptic Fas II on the target cells results in the formation of ectopic synapses (32). Similarly, we found the highest levels of NCAM in the stratum lucidum, where mossy fibers normally synapse, and greatly reduced levels in the stratum pyramidale. This laminated expression of NCAM is, of course, abolished in the mutants, and this, together with the misrouting of the fibers due to lack of fasciculation, may contribute to the absence of a clear preference for one or the other of the two layers.
In NCAM mutants, both the disorganized growth of mossy fibers (21) and the lack of LTP induction in the CA1 region of the hippocampus (23) have been attributed to lack of the polysialic acid (PSA) moiety of NCAM, since these alterations can be partly (mossy fiber growth) or fully (LTP in the CA1 region) reproduced by enzymatic removal of PSA. There is, however, an important difference between the mossy fiber system and the synapses formed by Schaffer collaterals on CA1 pyramidal cell terminals: whereas the latter synapses, which are thought to be the site of LTP in the CA1 region, express PSA both pre- and postsynaptically at the stage studied (23), adult mossy fiber synaptic boutons and their targets are devoid of PSA (21). Furthermore, only a subset of dentate granule neurons and their axons, probably the newly generated ones, express PSA in adult mice (37). In contrast, we found that NCAM protein colocalizes with the giant boutons in the stratum lucidum, suggesting that here the protein itself may function in synaptic plasticity. Conceivably then, although in-growing mossy fibers, which are added continuously during adult life, may need PSA to fasciculate correctly, this carbohydrate is not required for synaptic function at the mossy fiber terminal.
In summary, we have found that a lack of all isoforms of NCAM results in a selective functional deficit at mossy fiber synapses: LTP is abolished whereas short-term changes in synaptic strength are preserved. It remains to be seen to what extent these changes are a consequence of the altered distribution of mossy fiber terminals or of altered functioning of the mature synapses. The generation of mice in which the inactivation of NCAM is inducible will be required to definitively resolve this point. However, a variety of studies, both in vertebrates and invertebrates, point to an involvement of NCAM and NCAM-like molecules in structural and functional plasticity of mature synapses (for reviews see refs. 25, 26, and 30). Therefore, by modulating the strength of adhesive interactions at the synapse or by triggering growth-conductive second-messenger systems, we propose that modulations in the concentration of adhesion molecules play essential roles in the structural remodeling of synapses that are thought to underlie learning and memory. Alternatively, intracellular transduction mechanisms triggered by homo- or heterophilic interactions between CAMs may directly modulate the mechanisms leading to enhanced synaptic activity. Whatever the case, our findings provide genetic evidence for the role of NCAM in activity-induced synaptic plasticity and support the results of earlier studies suggesting an important role for CAMs in learning and memory.
Acknowledgments
We thank Profs. P. E. Castillo and R. A. Nicoll for helpful comments on the manuscript. This work was supported by institutional support from the Centre National de la Recherche Scientifique and the Institut Universitaire de France and a grant from the European Community Biomed program to H.C. (BIO4-CT96-0730).
ABBREVIATIONS
- CAMs
cell adhesion molecules
- NCAMs
neural CAMs
- PSA
polysialic acid
- PPF
paired-pulse facilitation
- NMDA
N-methyl-d-aspartate
- LTP
long-term potentiation
- EPSP
excitatory postsynaptic potential
- Fas II
fasciclin II
References
- 1. Bliss T V P, Collingridge G L. Nature (London) 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 2.Nicoll R A, Malenka R C. Nature (London) 1995;377:115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- 3.Castro-Alamancos M A, Connors B W. Science. 1996;272:274–277. doi: 10.1126/science.272.5259.274. [DOI] [PubMed] [Google Scholar]
- 4.Geinisman Y, de Toledo-Morrell L, Morrell F, Heller R E, Rossi M, Parshall R F. Hippocampus. 1993;3:435–445. doi: 10.1002/hipo.450030405. [DOI] [PubMed] [Google Scholar]
- 5.Edwards F. Physiol Rev. 1995;75:759–787. doi: 10.1152/physrev.1995.75.4.759. [DOI] [PubMed] [Google Scholar]
- 6.Mayford M, Barzilai A, Keller F, Schacher S, Kandel E R. Science. 1992;256:638–644. doi: 10.1126/science.1585176. [DOI] [PubMed] [Google Scholar]
- 7.Davis G W, Schuster C M, Goodman C S. Neuron. 1996;17:669–679. doi: 10.1016/s0896-6273(00)80199-3. [DOI] [PubMed] [Google Scholar]
- 8.Schuster C M, Davis G, Fetter R, Goodman C S. Neuron. 1996;17:641–654. doi: 10.1016/s0896-6273(00)80197-x. [DOI] [PubMed] [Google Scholar]
- 9.Schuster C M, Davis G W, Fetter R D, Goodman C S. Neuron. 1996;17:655–667. doi: 10.1016/s0896-6273(00)80198-1. [DOI] [PubMed] [Google Scholar]
- 10.Monaghan D T, Cotman C W. J Neurosci. 1985;5:2909–2919. doi: 10.1523/JNEUROSCI.05-11-02909.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris E W, Cotman C W. Neurosci Lett. 1986;70:132–137. doi: 10.1016/0304-3940(86)90451-9. [DOI] [PubMed] [Google Scholar]
- 12.Castillo P E, Weisskopf M G, Nicoll R A. Neuron. 1994;12:261–269. doi: 10.1016/0896-6273(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 13.Schwegler H, Crusio W E, Lipp H P, Heimrich B. Behav Genet. 1988;18:153–165. doi: 10.1007/BF01067837. [DOI] [PubMed] [Google Scholar]
- 14.van Daal J H, Herbergs P J, Crusio W E, Schwegler H, Jenks B G, Lemmens W A, van Abeelen J H. Behav Brain Res. 1991;43:57–64. doi: 10.1016/s0166-4328(05)80052-x. [DOI] [PubMed] [Google Scholar]
- 15.Cremer H, Lange R, Cristoph C, Plomann M, Vopper G, Roes J, Brown R, Baldwin S, Kraemer P, Scheff S, Barthels D, Rajewsky K, Wille W. Nature (London) 1994;367:455–459. doi: 10.1038/367455a0. [DOI] [PubMed] [Google Scholar]
- 16.Cremer H, Chazal G, Goridis C, Represa A. Mol Cell Neurosci. 1997;8:323–335. doi: 10.1006/mcne.1996.0588. [DOI] [PubMed] [Google Scholar]
- 17.Gennarini G, Hirsch M J, He H T, Hirn M, Finne J, Goridis C. J Neurosci. 1984;6:1983–1990. doi: 10.1523/JNEUROSCI.06-07-01983.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amaral D G, Witter M P. Neuroscience. 1989;31:571–591. doi: 10.1016/0306-4522(89)90424-7. [DOI] [PubMed] [Google Scholar]
- 19.Kamiya H, Shinozaki H, Yamamoto C. J Physiol (London) 1996;493:447–455. doi: 10.1113/jphysiol.1996.sp021395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castillo P E, Janz R, Südhof T C, Tzounopoulos T, Malenka R C, Nicoll R A. Nature (London) 1997;388:590–598. doi: 10.1038/41574. [DOI] [PubMed] [Google Scholar]
- 21.Seki T, Rutishauser U. J Neurosci. 1998;18:3757–3766. doi: 10.1523/JNEUROSCI.18-10-03757.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salin P, Scanziani M, Malenka R C, Nicoll R A. Proc Natl Acad Sci USA. 1996;93:13304–13309. doi: 10.1073/pnas.93.23.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muller D, Wang C, Skibo G, Toni N, Cremer H, Calaora V, Rougon G, Kiss J S. Neuron. 1996;17:413–422. doi: 10.1016/s0896-6273(00)80174-9. [DOI] [PubMed] [Google Scholar]
- 24.Fields R D, Itoh K. Trends Neurosci. 1996;19:473–480. doi: 10.1016/S0166-2236(96)30013-1. [DOI] [PubMed] [Google Scholar]
- 25.Martin K C, Kandel E R. Neuron. 1997;17:567–570. doi: 10.1016/s0896-6273(00)80188-9. [DOI] [PubMed] [Google Scholar]
- 26.Schachner M. Curr Biol. 1997;9:627–634. doi: 10.1016/s0955-0674(97)80115-9. [DOI] [PubMed] [Google Scholar]
- 27.Lüthi A, Laurent J-P, Figurov A, Muller D, Schachner M. Nature (London) 1994;372:777–779. doi: 10.1038/372777a0. [DOI] [PubMed] [Google Scholar]
- 28.Ronn L C, Bock E, Linnemann D, Jahnsen H. Brain Res. 1995;677:145–151. doi: 10.1016/0006-8993(95)00147-i. [DOI] [PubMed] [Google Scholar]
- 29.Skibo G G, Davies H A, Rusakov D A, Stewart M G, Schachner M. Neuroscience. 1998;82:1–5. doi: 10.1016/s0306-4522(97)00382-5. [DOI] [PubMed] [Google Scholar]
- 30.Rose S P. Trends Neurosci. 1995;18:502–506. doi: 10.1016/0166-2236(95)92774-k. [DOI] [PubMed] [Google Scholar]
- 31.Lin D M, Fetter R D, Kopczynsky C, Grenningloh G, Goodman C S. Neuron. 1994;13:1055–1069. doi: 10.1016/0896-6273(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 32.Davis G W, Schuster C M, Goodman C S. Neuron. 1997;19:561–573. doi: 10.1016/s0896-6273(00)80372-4. [DOI] [PubMed] [Google Scholar]
- 33.Weisskopf M G, Castillo P E, Zalutsky R A, Nicoll R A. Nature (London) 1994;376:256–259. [Google Scholar]
- 34.Bailey C H, Chen M, Keller F, Kandel E R. Science. 1992;256:645–649. doi: 10.1126/science.1585177. [DOI] [PubMed] [Google Scholar]
- 35.Kiss J Z, Wang C, Olive S, Rougon G, Lang J, Baetens D, Harry D, Pralong W F. EMBO J. 1994;13:5284–5292. doi: 10.1002/j.1460-2075.1994.tb06862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holst B D, Vanderklish P W, Krushel L A, Zhou W, Langdon R B, McWhirter J R, Edelman G M, Crossin K L. Proc Natl Acad Sci USA. 1998;95:2597–2602. doi: 10.1073/pnas.95.5.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seki T, Arai Y. Neurosci Res. 1993;17:265–290. doi: 10.1016/0168-0102(93)90111-3. [DOI] [PubMed] [Google Scholar]