

Abstract
Closed head injury to the developing rat brain causes an acute excitotoxic lesion and axonal disruption at the impact site followed by a delayed pattern of apoptotic damage at various distant sites. Using an electromagnetic impact device to deliver a precisely controlled degree of mechanical deformation to the P7 infant rat skull, we studied the distribution of distant apoptotic lesions and the sequence and time course with which these lesions evolve following relatively mild closed head injury. The first major wave of apoptotic neurodegeneration occurred at 8 hrs post-impact in the retrosplenial cortex and pre- and para-subiculum. The next major wave occurred in the 16 to 24 hr interval and was localized to the anterior thalamic nuclei. A third wave was detected at 36 to 48 hrs in the mammillary nuclei. We propose that the first and second waves were triggered by injury to a specific fiber tract, the corpus callosum/cingulum bundle that conveys reciprocal connections between the anterior thalamic nuclei and retrosplenial/pre and parasubicular neurons. This fiber tract passes through a zone of maximum mechanical strain, as measured by tagged MRI. The third wave affecting mammillary neurons occurred because the principal synaptic targets of these neurons are the anterior thalamic neurons that were destroyed in the second wave of degeneration. Prevention of these apoptotic waves of brain damage is a realistic goal in view of the long delay between the impact event and onset of apoptotic degeneration.
Keywords: Brain Development, Apoptosis, Axonal Injury , Biomechanics, Caspase-3
1. INTRODUCTION
Traumatic brain injury (TBI) has been studied much more extensively in the adult than in the developing nervous system. A major focus of attention in adult studies has been on traumatic axonal injury (TAI), often caused by rotational acceleration of the skull, that can focally injure axonal processes, often with little or no discernible damage to neuronal cell bodies. Geddes et al. (2001a,b) studied 53 cases of fatal pediatric head injury and found that in infants >1 yr old a pattern of TAI similar to that associated with adult TBI was present, but in infants < 1 yr old, TAI was not a prominent finding. Thus, although significant mechanistic insights have been gained in recent years pertaining to TAI in the adult CNS (recently reviewed by Buki and Pavlishock, 2006), it is not clear how this information can be applied to an understanding of mechanisms underlying TBI in neonates.
Several authors have investigated mechanisms of neuronal cell death in adult TBI, and have postulated that apoptosis may play a role, based on positive TUNEL staining (Rink et al., 1995; Fox et al., 1998) or evidence for caspase-3 activation (Yakovlev et al., 1997; Clark et al., 2000). However, the relative contribution of apoptosis to total neuronal degeneration after adult TBI remains unclear. TUNEL staining is not a specific marker for apoptosis (Ishimaru et al., 1996), and in a study of adult TBI by Rink et al., (1995), when morphological criteria were applied, some populations of TUNEL-positive neurons were judged to be dying by apoptosis and others by necrosis.
Animal studies of TBI in the neonatal period are relatively scarce. Olney and colleagues (Ikonomidou et al., 1996; Bittigau et al., 1999; Pohl et al., 1999) have reported that in infant rats concussive head trauma can cause extensive cell death, consisting of neuronal degeneration directly subjacent to the impact site, and subsequent degeneration of other neurons in distant brain regions. Neuronal degeneration at the impact site occurred rapidly (within 2–4 hours) and displayed ultrastructural characteristics of excitotoxicity (Ikonomidou et al., 1996), a cell death picture that does not resemble apoptosis. In contrast, the distant neurodegenerative response was delayed (> 6 hrs post-impact) and was ultrastructurally consistent with apoptosis, as originally described by Kerr, Wyllie and colleagues (1972).
Mechanism(s) underlying the delayed apoptotic response in neonatal TBI are poorly understood. Immature neurons are more prone than mature neurons to undergo apoptosis. Indeed developing neurons are programmed to commit suicide if, due to adverse circumstances, they are unsuccessful in established normal synaptic connections. For example, by suppressing neuronal activity, certain drugs interfere with synaptogenesis, and this is a sufficient adverse circumstance to cause widespread neuroapoptosis in the developing brain (Olney et al., 2002). Similarly, brain trauma can disrupt synaptogenesis, and thereby trigger neuroapoptosis. Thus, following neonatal TBI, the initial excitotoxic wave of neurodegeneration deletes neurons that project to distant sites, and this may cause distant neurons to undergo apoptosis because they are deprived of synaptic inputs. Alternatively, death of neurons at the impact site may delete synaptic targets that distant neurons depend on for survival. In addition, physical strain may damage the distant neurons or their axons. Thus, either direct physical injury, or loss of synaptic inputs or synaptic targets, or both, are candidate mechanisms but their relative importance remains unclear.
To further study developmental TBI, we devised a system to deliver a precisely controlled impact to the flexible skull of the infant rat. This system uses an electromagnetic actuator mounted on a stereotaxic instrument to impact the exposed skull at a specified angle, velocity and depth. The device is intended to produce injury similar to that achieved with a weight-drop device (Ikonomidou et al., 1996; Bittigau et al., 1999), but with improved precision and flexibility.
We also have begun studying mechanical strain associated with TBI in infant rats, using methods described in a recent publication (Bayly et al., 2006). This method entails measurement of maximum principal strain using tagged magnetic resonance imaging (MRI) of the brain during controlled indentation of the skull at the same location and depth used in the current study. Measurement of strain fields by this approach provides both a qualitative picture and quantitative assessment of the mechanical insult to the brain.
The present study was undertaken to further characterize the delayed apoptotic response to impact trauma in the infant (P7) rat brain. It is important to develop a better understanding of this phenomenon because TBI is a leading cause of death and permanent disability in human infants and young children, and our prior observations suggest that the number of neurons deleted from the developing brain post-trauma by the delayed apoptosis mechanism, may be much greater than the number deleted by the excitotoxic mechanism at the local impact site. Moreover, the delay between trauma and onset of apoptotic degeneration may be long enough for therapeutic intervention. Therefore, a major aim of the present study was to characterize in greater detail the time course and spatial pattern of apoptotic neurodegeneration triggered by concussive trauma in the postnatal day 7 (P7) rat brain. Another aim was to compare the temporospatial pattern of neurodegeneration with the pattern of mechanical strain, as measured by tagged MRI. To identify apoptotic neurons and study their pattern of distribution, we relied most heavily on activated caspase-3 immunohistochemistry, a method we have found very useful for studying evolving patterns of neuroapoptosis in the developing rodent brain (Olney et al., 2002a,b; Young et al., 2004). To confirm the apoptotic nature of the cell death process, neurons undergoing delayed degeneration were examined by electron microscopy.
2. RESULTS
2.1 Gross anatomical observations
In the acute post-impact interval, there were no signs of cavitation or gross distortion of the cortical mantle at the site of impact, and there was no evidence of hemorrhage at the cortical surface. However, there was evidence of small hemorrhagic foci in the subcortical white matter, especially in a zone where the corpus callosum and cingulum are joined together.
2.2 Temporospatial progression of neurodegeneration
Zero to four hours
Caspase-3 IHC during the 0 to 4 hr interval, revealed no evidence of caspase-3 activation or of any other pathological changes in any brain region. Of particular note, was the total absence of caspase-3 activation in the vicinity subjacent to the impact site (Fig. 1A), a region where a fulminant neurodegenerative reaction was occurring during this post-impact interval. In thin plastic sections evaluated by light microscopy, this acute reaction was evident in a circumscribed tissue zone immediately subjacent to the impact site, and was characterized by massive edematous swelling of dendrites and cell bodies of neurons, with edematous swelling and dilatation of intracellular organelles (both mitochondria and endoplasmic reticulum) within the affected neuronal cell bodies. The degenerating neurons reached a stage of advanced cell death within 4 hours after the impact. This acute reaction, which meets all criteria for an excitotoxic cell death process, is illustrated by both light and electron microscopy in Fig. 1 and described in further detail in the legend of that figure.
Fig. 1.

Panel A is a 50 µm thick AC-3 stained vibratome section from a P7 rat brain 4 hrs post-impact. An acute excitotoxic neurodegenerative reaction is occurring at the site of impact within the region demarcated by the dashed semi-circular line, but this is not detected by the AC-3 stain because the excitotoxic lesion induced by head trauma does not entail caspase-3 activation. Panels B and C are 1 µm thin plastic sections from the same brain region demarcated by the dashed line in A depicting the appearance of normal neurons in a control brain (B) and abnormal appearance of neurons undergoing acute excitotoxic degeneration in an experimental brain 4 hrs post-impact (C). Panel D is an electron micrograph showing the ultrastructural appearance of a neuron (from the scene in C) undergoing excitotoxic cell death. These cells display all the morphological changes characteristic of an excitotoxic process. In the initial stages, dendrites and cell bodies undergo extreme edematous swelling which is accompanied by dilatation of mitochondria and endoplasmic reticulum, disaggregation of polyribosomes and dissolution of the formed structural elements in the cytoplasm. Nuclear changes occur slightly later and begin by the formation of small aggregates of clumped chromatin which give the nucleus a floccular appearance. These small clumps migrate to the perimeter of the nucleus in a clockface pattern, then coalesce into progressively larger clumps that consolidate into a large irregular dense mass at the center of the nucleus (nuclear pyknosis). The nuclear membrane remains intact but the plasma membrane becomes ruptured and loses its integrity early in the degenerative process. Compare this description with that given for neuroapoptosis in the legend of Fig. 7.
The only evidence of a tissue reaction in other regions during this acute period were small hemorrhagic foci in the corpus callosum, typically at or near its junction with the cingulum. Light micrographs of the corpus callosum/cingulum bundle (CCCB) at 4 h (Fig. 2A and B) show evidence of tissue disruption, including interstitial edema and apparent stretch injury evidenced by disarray and apparent discontinuity of some of the axonal processes.
Fig. 2.

Thin plastic sections from the corpus callosum (CC) and cingulum bundle (CB) region of the infant rat brain 4 hours (A and B) or 16 hours (C and D) after TBI. The typical findings at 4 hours are a mild degree of interstitial edema giving portions of the CCCB track a rarefied appearance, and focal accumulations of red blood cells (arrows) due apparently to rupture of small blood vessels. The boxed region in A, shown at higher magnification in B, exhibits axonal fibers in disarray, surrounded by edema fluid and accompanied by phagocytic cells that are just beginning to respond to the pathological reaction. At 16 hrs, cystic spaces have formed at the site of tissue injury. The boxed regions in C, shown at higher magnification in D, reveals many phagocytic cells lining the cystic spaces and occupying the surrounding tissue area where they are ingesting red blood cells (dark round masses) and other less dense debris.
Four to eight hours
The 4 to 8 hr interval was essentially a quiescent interval during which no new signs of neurodegeneration became evident by any method of evaluation. In the cingulum region where small hemorrhagic foci were evident, numerous foamy cellular profiles appeared on the scene. These were activated microglia performing a phagocytic function at this scene of apparent injury to axonal pathways.
Eight to twelve hours
The first signs of delayed degeneration appeared at about 8 hours and were detected as AC-3 positivity in neuronal profiles in a deep cortical zone at the medial edge of the impact site (Fig. 3A). These profiles were not in the core region of the excitotoxic lesion, but rather were in a penumbral zone at its ventromedial edge. Anatomically, these neurons were in a border zone between the agranular retrosplenial (RS) cortex and occipital cortex and were primarily in layers V and VI. In terms of rostro-caudal distribution they were not limited to the zone of impact, but rather began at this level then extended caudally away from it all the way to the caudal pole of the forebrain.
Fig. 3.

All panels are AC-3 stained sections from the P7 rat brain showing the first wave of caspase-3 activation that occurs at distant sites on a delayed basis following brain impact. The earliest AC-3 response occurs at 8–10 hrs and selectively affects the retrosplenial cortex (A) and parasubiculum (B). Initially, a relatively small number of neurons are involved, but in the ensuing 6 hours many more neurons in these same locations become involved. For example, panel C shows the extensive involvement of many neurons in the pre and parasubicular region 16 hrs following impact, compared to the relatively small patch of neurons involved at 8–10 hrs (panel B).
In addition, during the 8 to 12 hr interval, a dense pattern of caspase-3 positivity became progressively more evident in neurons of the pre and para-subicular cortices (Fig. 3B). These profiles were distributed across several of the middle layers in a continuous pattern from the impact zone to the caudal-most extent of these brain regions.
In addition to these heavily affected neuronal populations, there were scattered individual AC-3 positive profiles beginning to appear in a radial pattern in all directions (mediolateral, rostrocaudal and ventral) from the impact zone. These profiles were relatively sparsely distributed but were clearly more frequent on the side of the brain ipsilateral to the impact.
There also was a laminar display of AC-3 positivity in layer VI neurons of the granular RSC at a level slightly rostral to the impact zone. Initially, this was confined to neurons in a ventral location relatively close to the cingulum bundle.
Twelve to sixteen hours
The 12 to 16 hr interval was devoted to a consolidation and extension of the patterns described above. Some new neurons were added in the same distribution pattern (Fig. 3C) but no new zones of dense involvement became evident.
At 16 h, the pathological reaction in the region of the cingulum bundle was still evident and consisted of small cystic spaces lined with phagocytes and surrounded by tissue debris, apparently from degenerating axonal processes (Figs. 2C and D).
Sixteen to twenty four hour interval
At approximately 16 hrs, neurons in several nuclei of the anterior thalamic group became AC-3 positive, and the numbers of these positive profiles steadily increased in the 16 to 24 hr interval (Figs. 4A and B). The most frequently involved anterior thalamic nuclei during this interval were the laterodorsal and anterodorsal nuclei, although a discrete cluster of neurons at the junction of anteroventral and anteromedial nuclei also became AC-3 positive.
Fig. 4.

Panel A illustrates, at 24 hrs post-impact, the vestiges of the first wave of caspase-3 activation in the retrosplenial (RS) cortex, and the second wave that has evolved in the anterior thalamus in the 16–24 hr period. The anterior thalamic nuclear complex consists of the laterodorsal (LD), anterodorsal (AD), anteroventral (AV) and anteromedial (AM) thalamic nuclei. At 16 hrs (not shown), only a small number of thalamic neurons are AC-3 positive, but by 24 hrs (A), large numbers have turned positive, especially in LD and AD. Note that the hippocampus (HC), which lies between the point of impact (arrow head) and the distant neurodegeneration in the anterior thalamus is essentially devoid of AC-3 positivity. Although the anterior thalamic lesion is primarily confined to the ipsilateral side, there is a modest increase in the number of AC-3 positive neurons in the contralateral AD as well. The neurons shown at higher magnification in panel B are from the ipsilateral LD at 20 hrs. It is clear that these neurons are in a relatively early stage of degeneration, because in later stages the dendritic processes become more fragmented, the cell bodies more shrunken and condensed, and finally at about 36 hrs, the staining assumes a faded, smudged appearance (due to loss of immunoreactivity and/or leakage of the AC-3 molecule into the neuropil as the neurons disintegrate.
At 16 hrs, neurons in the RS cortex and presubiculum began to show a more dense multilaminar involvement, apparently reflecting the death of intrinsic neurons within these brain regions, as a secondary response to the earlier death of primary projection neurons.
In the 16 to 24 hr interval, there was a tendency for individual neurons distributed sparsely in a scattered radial pattern in relation to the impact zone to become AC-3 positive.
Twenty four to forty eight hours
In the 24 to 36 hr interval, AC-3 positivity tended to disappear or become blurred and less distinct in the various regions where cell-specific staining had previously been prominent. During this interval, a few new regions showed light to moderate involvement, including pyramidal neurons in the frontal neocortex and neurons in the nucleus reuniens of the thalamus. In both of these locations the involvement was confined to the side ipsilateral to the impact site.
In the 36 to 48 hr interval, neurons in the ipsilateral medial and lateral mammillary nuclei began showing fresh AC-3 positivity (Fig. 5). This represents a third major wave of distant delayed apoptotic neurodegeneration.
Fig. 5.

This figure illustrates at 48 hrs post-impact the third wave of caspase-3 activation, which begins at 36 hrs, becomes more prominent by 48 hrs, and is selectively localized to the mammillary nuclei (MN) on the side of the brain ipsilateral to the impact site.
Ipsilateral versus contralateral
The pattern of AC-3 staining was largely confined to the side of the brain ipsilateral to the impact site, with the exception that a mild increase in AC-3 staining of anterior thalamic neurons on the contralateral side was evident in some animals (Fig. 4A).
Summary of the temporospatial progression of neurodegeneration
In Fig. 6 the time course and pattern of spatial distribution of apoptotic neuronal degeneration, indicated by AC-3, following a mild concussive blow to the skull of the P7 rat are summarized schematically across the time period from 4 to 48 hrs post-impact. These spatial and temporal patterns were highly consistent, although the density of neurodegeneration showed some variability.
Fig. 6.
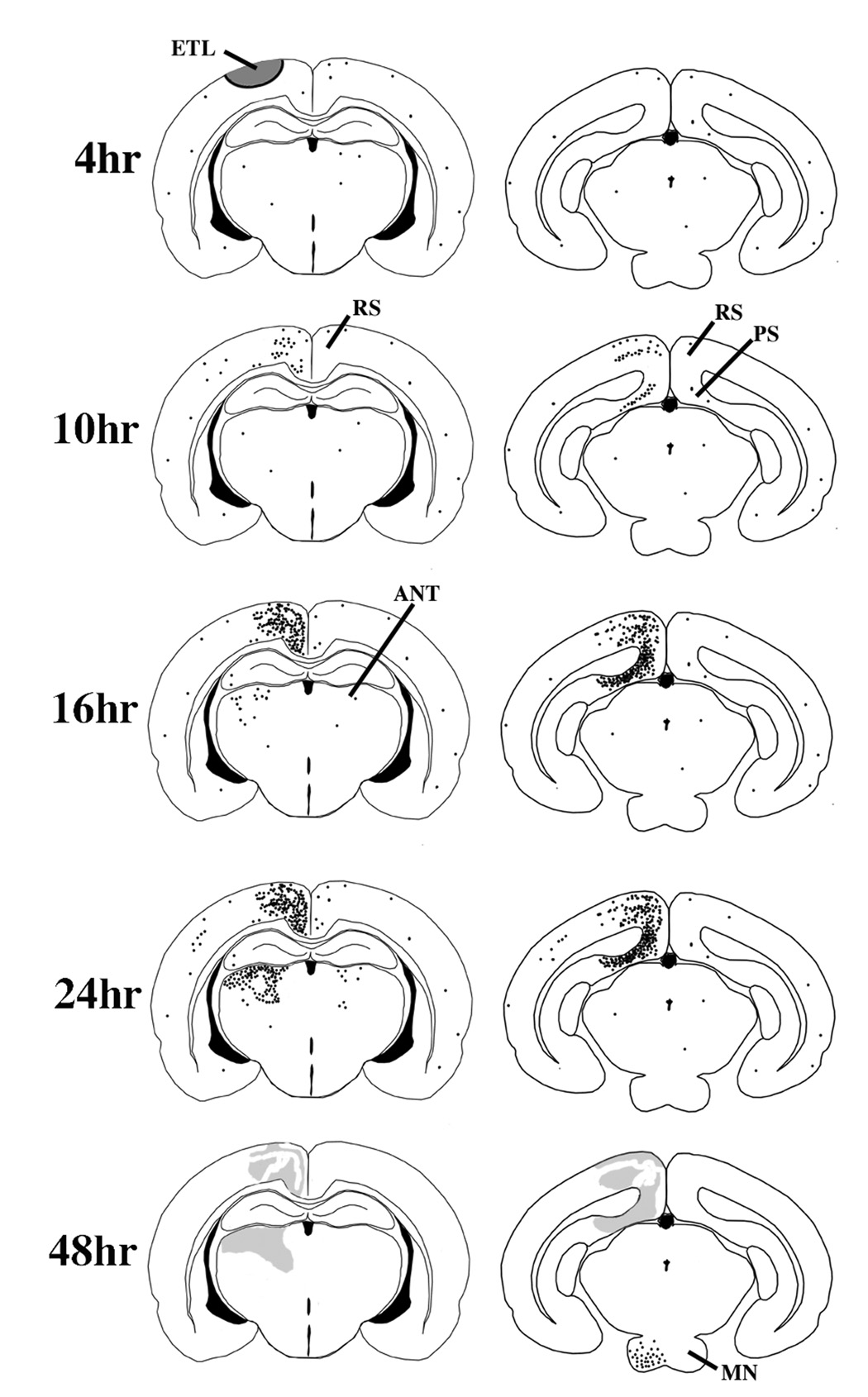
This schematic summarizes the nature, time course and localization of pathological changes in the brain following relatively mild concussive impact to the head of a P7 rat. At 4 hrs post-impact, AC-3 staining does not reveal any pathological reaction, but other histological procedures demonstrate an acute excitotoxic lesion (ETL) that rapidly kills neurons in the semicircular zone shaded in grey. At 8 – 10 hrs AC-3 staining begins to reveal a wave of neuroapoptosis affecting the ipsilateral retrosplenial (RS) and pre/para-subicular (PS) cortices. By 16 hrs, the RS and PS cortical involvement has progressed substantially, and a new wave of neuroapoptosis begins to appear in the ipsilateral anterior thalmic nuclei (ANT). By 24 hrs the ANT lesion has progressed to its full extent and there is mild increased staining in the contralateral ANT and RS and also in scattered neurons in the ipsilateral cortical mantle. In the 36–48 hr interval a third wave of neuroapoptosis appears in the ipsilateral mammillary nuclei (MN) and the apoptotic lesions in other brain regions are no longer visible, except as vague smudged areas in AC-3 stained sections.
2.3 Electron Microscopy observations
Electron microscopic evaluation confirmed the excitotoxic nature of the acute neurodegenerative reaction at the impact site (Fig. 1) and the apoptotic nature (Fig. 7) of the delayed reaction at distant sites. The distinctly different appearance of cells degenerating by these two different modes of cell death is clear in Fig. 2 and Fig. 7. The accompanying figure legends describe the detailed morphological features that distinguish these two forms of cell death.
Fig. 7.

Light and electron microscopic appearance of apoptotic neurons following head trauma in the P7 rat brain. Panel A illustrates neurons showing AC-3 positivity in the early stages of apoptotic cell death while they still retain a relatively normal morphological profile. Panel B depicts the appearance of apoptotic neurons in thin plastic sections at stages when they have formed multiple dense spherical balls of clumped nuclear chromatin, which is a hallmark sign of neuroapoptosis. Panels C and D are electron micrographs illustrating a relatively early stage (C) and a later stage (D) of apoptotic cell death. In the early stage (C), the cytoplasmic components remain relatively intact except for deteriorative changes in mitochondria, and nuclear chromatin is beginning to aggregate into dense masses that assume a geometrically spherical shape. In addition, the nuclear membrane is beginning to become discontinuous, which allows intermingling of the cytoplasmic and nuclear constituents. At a later stage (D), multiple nuclear chromatin balls become evident, the nucleolus disaggregates into worm-like structures (not visible at this magnification, but see Dikranian et al., 2001), the nuclear membrane becomes more fragmented, mitochondria are reduced to vacuous debris, and the entire cell becomes shrunken and condensed. Compare these features of neuroapoptosis with the excitotoxic cell death process illustrated in Fig.2.
2.4 Comparison of strain patterns with patterns of neurodegeneration
The histological results of the current study can be presented in the context of the mechanical strain field typical of such impacts. Figs. 8A and B depict typical strain fields measured by tagged MRI during dynamic impact of the P7 rat skull to the same depth (Bayly et al., 2006). These measured strain patterns are superimposed on line drawings of the P7 brain to illustrate which brain regions sustained the greatest degree of strain. During impact, maximum strain values greater than 0.20 (i.e., a local spherical element of tissue is stretched by more than 20%) occur over a period of 1–2 ms, i.e. at strain rates of 100/200 strain/sec. This strain level is well above thresholds for axonal injury proposed on the basis of studies of optic nerve stretch (Bain and Meaney, 2000; Bain et al., 2001). By comparison, strains of 0.02–0.05 (2–5%) occur at strain rates of about 2–3 strain/sec during head accelerations typical of normal human athletic activity (Bayly et al., 2005).
Fig. 8.
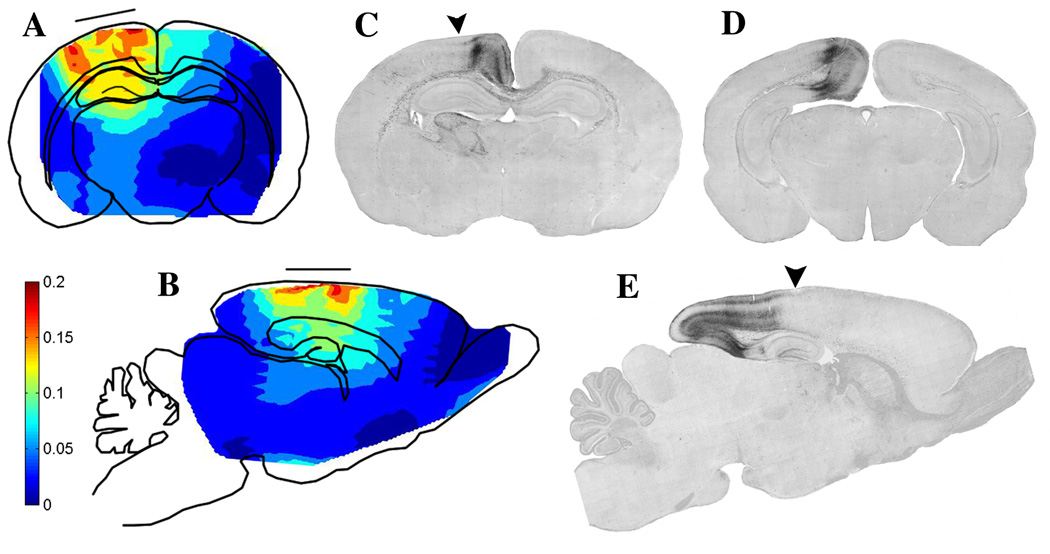
Panels A and B are schematic depictions of coronal (A) and sagittal (B) sections of the P7 rat brain illustrating the pattern of strain magnitude following a focal impact force causing parasagittal indentation of the flexible infant rat skull. The positioning of the impacter tip (solid horizontal line) is shown to allow comparison with the resultant pattern of strain (tissue deformation). A more detailed account of the method of strain measurement is described in Bayly et al. (2006). Colors represent degrees of deformation: red = 0.20 (20%) strain; deep blue = 0 strain; intermediate levels indicated by the color bar. Panels C, D, E are AC-3 stained sections from the P8 rat brain 24 hrs post impact. The arrows in C and E indicate the direction of impact and the center of the impact site. Panels C and D are coronal sections cut in a rostrocaudal plane slightly caudal to the impact site (C), or at a much more caudal level (D). These sections show that the delayed pathological reaction at the cerebrocortical level is primarily concentrated in a tissue zone medial to the point of impact. Panel E is a sagittal section revealing that this cerebrocortical delayed pathological reaction begins at the point of impact, and extends in a caudal (but not rostral) direction to the caudal pole of the brain. It encompasses continuous rows of neurons in the retrosplenial and pre and para-subicular cortices, all lying in a medio-caudal sector of the brain.
Notably, the strain field is quite symmetric with respect to the midline of the impacter, both in the medial-lateral and rostral-caudal directions. Strain concentrations occur under the edge of the impacter tip. Areas of highest strain (>0.20, or 20% deformation) occur in the parietal cortex and cingulate cortex. The intersection of the corpus callosum and cingulum bundle occurs directly within the mediodorsal area of high strain. Strain drops off smoothly with distance from the impacter; the hippocampus receives significantly less strain than the overlying cortex, and the deeper structures (basal ganglia and thalamus) are deformed only slightly. No evidence of contrecoup or distant strain concentrations is seen; this is attributable to the relatively focal nature of the insult, aided by the flexibility of the skull combined with firm, distributed, support of the animal’s head.
Strain fields can be related to specific regions of neurodegeneration. In some areas dense neuropathology directly reflects high strain. Early excitotoxic cell death occurs in cortical areas exposed to high strain. Injury to fibers in the corpus callosum/cingulum bundle coincides with large deformation in these areas. Early apoptotic degeneration in the posterior cingulate cortex also occurs in, or near to, areas of high strain. However, significant degeneration also is seen in areas that do not undergo large deformations; the anterior thalamic and mamillary nuclei that exhibit delayed apoptosis are not subjected to high strain. Conversely, some areas, particularly rostral and lateral areas of the cortex, receive high strain but do not exhibit significant apoptotic degeneration. Generally, except for the anterior thalamic nuclei and mammillary nuclei the distribution of dense neuropathology is all in a dorsomediocaudal sector, whereas the strain pattern emanates more uniformly in a radial pattern from the impact site.
3. DISCUSSION
In this study we have characterized the temporospatial progression of neuropathological changes over a 48 hr period following head trauma in the P7 rat brain and studied the potential relationship between the pattern of neuropathological changes and the MRI-measured spatial pattern of mechanical deformation induced in the brain by the traumatic event.
The TBI model employed in this study is one that causes a rapid but relatively mild deformation of the cortical surface of the brain during a critical stage in neonatal development. The tissue reaction to this insult consists of two major components, an acute reaction at the impact site and a more delayed reaction at distant sites. Nerve cell bodies at the impact site react immediately in a manner that displays all of the characteristics of an excitotoxic reaction presumably triggered by excessive activation of glutamate excitatory transmitter receptors. This excitotoxic reaction kills these neurons very rapidly, with end-stage cell death being reached within 2 to 4 hrs. This component of the tissue reaction entails direct tensile injury to nerve cell bodies and appears to be the way developing nerve cell bodies respond to rapid deformation.
The other component to the acute tissue reaction is a fiber tract component. Developing axonal fibers are fragile and they course through the brain in bundles that are anchored at certain points, which determines how far they can stretch without tearing or exceeding some injury threshold. Any fiber tract passing through a portion of the impact zone where strains are high will be subjected to stretch injury and potential severance, either abruptly or on a more slowly evolving basis. Axonal processes that are severed will be prevented from establishing synaptic connections between the parent neuron and neurons at distant sites. Loss of synaptic connections in the developing brain can cause nerve cells to commit suicide.
We propose that the distant, delayed pattern of apoptotic neurodegeneration is either a direct or indirect response to the local tissue injuries. The most conspicuous early component of the distant response was apoptotic degeneration of neurons in brain regions dorsomedial and caudal to the point of impact. This suggests that a fiber tract vital for the survival of neurons in this brain sector was severely injured. Nerve cells in this brain sector, including those in the retrosplenial and pre- and para-subicular cortices, receive many of their inputs and deliver many of their outputs through a single fiber pathway, the CCCB. This pathway passes through a portion of the impact zone where maximal strains were measured, and we did detect signs of injury to axonal fibers in this pathway. Therefore, we propose that neurons in this dorsomediocaudal sector committed suicide because a high percentage of both their incoming and outgoing communication channels were injured or destroyed. Neurons in the developing brain are programmed to commit suicide under such circumstances.
The second most conspicuous but more delayed component of distant apoptosis was the apoptotic degeneration of anterior thalamic neurons beginning at about 16 hrs post-impact. These thalamic neurons are reciprocally connected to neurons in the retrosplenial and pre- and para-subicular cortices via a single fiber pathway, the CCCB (Shibata, 1993; Price, 1995). Thus, the initial injury to this fiber pathway deprived these thalamic neurons of a high percentage of both their incoming and outgoing communication channels. Why did they respond later than the retrosplenial and pre- and para-subicular cortical neurons? Probably because they are located much farther away from the impact site, so that more time is needed for the distress signal to travel via the connecting fibers.
The very late apoptotic response of neurons in the mammillary nuclei at 36 to 48 hr post-impact, may occur because these neurons are unidirectionally connected via the mammillothalamic tract to neurons in the anterior thalamic nuclei. The mammillary neurons take longer to respond, presumably because they are even farther removed from the primary site of tissue injury and because they were responding not directly to the fiber tract injury at the impact site, but to the loss of thalamic neurons, a loss that did not occur until the thalamic neurons degenerated in the 16 to 24 hr post-impact interval. Mammillary neurons are thought to be a major source of excitatory drive for anterior thalamic neurons, whereas other inputs to the anterior thalamus are thought to serve a modulatory function.
Individual neurons widely scattered outside the impact zone also undergo sporadic apoptosis. This may be explained by a synaptic relationship between these neurons and neurons at the impact site, or with neurons that died by delayed apoptosis at various distant sites. We propose that the primary factor that determines whether a developing neuron will undergo delayed apoptosis in response to brain trauma is whether it has sustained loss of a substantial percentage of its communication channels. The extent to which the loss entails both incoming and outgoing channels may also be important. Most of the major neuronal populations that underwent distant apoptotic degeneration in the present situation (retrosplenial, pre- and para-subicular, and anterior thalamic neurons) had lost a high percentage of both their incoming and outgoing communication channels. The mammillary neurons lost a high percentage of their synaptic targets and perhaps also lost feed-back communications that they would normally receive via a circuit that has been referred to as an extended hippocampal circuit that sustained significant damage. This circuit is discussed further in the following paragraph.
Important behavioral consequences might ensue from a TBI event of the type described here. In the learning and memory literature a great deal of attention has been focused on the hippocampus, a region that was not substantially damaged in this model. However, important orientation and spatial learning functions may rely on an intact extended hippocampal circuit which communicates extensively with the hippocampus, and includes the retrosplenial cortex, subicular complex, anterior thalamic nuclei and the mammillary nuclei (Mitchell et al., 2002; Aggleton et al., 1995, 1996, 1999; van Groen et al., 2002; Krazem et al., 1995; Sziklas and Petrides, 1993; Wozniak et al., 2004). Remarkably, our findings document that all components of this extrahippocampal circuit on the ipsilateral side of the brain are selectively and irreversibly damaged by a relatively mild blow to the skull overlying the parietal cortex. Head injury of this degree and type is not uncommon in a human pediatric setting. Therefore, we propose that the current model is useful for studying the long-term consequences of developmental TBI and for learning how to prevent or mitigate any ensuing disability.
Ironically, although Geddes et al., (2001a, b) found that head injury in human neonates <1 yr old differs from head injury in older children or adults, in not involving a prominent display of TAI, it appears from our observations that axonal injury may, nevertheless, be an extremely important, but subtle and inconspicuous, component of neonatal head injury. The most conspicuous neuropathological consequence of impact injury in our model were waves of apoptotic degeneration of nerve cell bodies in several major brain regions located at a distance from one another and at a distance from the trauma impact site. Preceding these conspicuous but delayed neuropathological events was a much less obvious tissue disturbance in the CCCB region which we believe may be causally related to all of the subsequent neuroapoptotic degenerative events in several distant regions of the brain. The conclusion that the developing brain responds differently to trauma than the mature brain (Koskiniemi et al., 1995; Geddes et al., 2001a, b) is valid, we believe, but the difference is complex and relates not only to the presence or absence of TAI, but to the propensity of developing neurons to commit suicide when their synaptogenesis mission is thwarted. TAI in the traumatized adult brain is a fascinating phenomenon for the very reason that it can occur in the absence of and independent of any apparent injury to the parent cell bodies. In our neonatal model, subtle injury to the CCCB axonal pathway was preceded (or accompanied) by excitotoxic cell death of nerve cell bodies at the impact site directly overlying this axonal pathway, and it was followed by subsequent apoptotic cell death of many nerve cell bodies at distant sites. We believe that injury to the CCCB pathway was directly or indirectly responsible for the delayed neuroapoptosis at several distant sites, but we do not know the mechanism that caused injury to the CCCB axons. In recent years, considerable progress has been made in understanding the mechanistic basis for TAI in the adult brain (see the review by Buki and Povlishock, 2006). Povlishock and colleagues (e.g., Buki and Povlishock, 2006; Stone et al., 2004; Reeves et al., 2005) have demonstrated that excitotoxicity-like mechanisms may initiate a process whereby increased axolemmal permeability to CA++ triggers a chain reaction, including mitochondrial injury which is followed by an apoptosis-like sequence of changes resulting in caspase-induced proteolytic digestion of cytoskeletal proteins, axonal failure and disconnection. We suspect that in future studies the mechanisms underlying CCCB axonal degeneration in our neonatal TBI model can also be deciphered by methods similar to those employed by these authors.
If our interpretations are correct, an exceedingly important aspect of developmental TBI is relatively inconspicuous injury sustained by fiber tracts. In the present model, acute injury to CCCB axons appears responsible for the delayed apoptotic death of many more neurons than were killed acutely at the impact site. Our findings do not clarify to what extent the CCCB axons were severed or only stretched beyond some functional limit. In either case, the plasticity potential of the developing brain may be sufficient to allow repair or regrowth of the injured fibers, but this cannot happen if cell bodies connected to these fibers respond by committing suicide. Therefore, therapeutic strategies for traumatic injury to the developing brain should perhaps be aimed both at suppressing axonal injury and at preventing stressed neurons from committing suicide. The latter goal will probably require establishing and maintaining a favorable (pro-survival) balance between anti- and pro-apoptosis proteins in neurons whose communication channels have been injured. Fortunately, there is a substantial delay between the initial injury and onset of apoptosis, so the goal of rescuing these neurons is not unrealistic.
4. EXPERIMENTAL PROCEDURES
4. 1 Animals
Postnatal day 7 (P7) Sprague Dawley rat pups were used in all experiments. Each experimental animal was briefly anesthetized and subjected to an identical TBI procedure (see Injury Device and Procedure), then sacrificed at a specified time post-trauma (2, 4, 6, 8, 12, 16, 20, 24, 30, 36, or 48 hours; n = 4 to 6 animals at each time point), and their brains examined histologically as described below. Sham control animals (n = 1 to 2 at each time point) underwent anesthesia and scalp incision, and were placed in the stereotaxic holder, but did not undergo trauma. Animals underwent an impact procedure (below) designed to be mechanically similar to the controlled indentations performed inside an MR scanner in a previous study of mechanical strain in this model (Bayly et al., 2006; procedures described in detail in that reference). All procedures were approved by the Washington University Animal Studies Committee and performed in accordance with the Animal Welfare Act and the NIH Guide for the Care and Use of Laboratory Animals.
4.2 Injury Device and Procedures
An electromagnetic impact device based on a moving coil actuator (BEI Kimco, Vista, CA) was mounted on the arm of a stereotaxic instrument (Benchmark, MyNeuroLab, St. Louis, MO). Isoflurane (5% in air, via nosecone) was used to induce anesthesia. The animal was placed in a molded plastic head support; anesthesia was maintained with 2% isoflurane in air. A midline scalp incision was made and the skin was reflected to expose the skull. The actuator was energized in its downward position and the 3mm-diameter impact tip was positioned stereotaxically 3 mm anterior to lambda, 2 mm lateral to midline, and touching the skull. The actuator was retracted, and depth and speed of impact were set. Impact depth (2 mm) was controlled by advancing the position of the stereotaxic arm relative to its baseline position when contact with the skull was established. Speed (2.7 m/s) was specified by computer control of actuator current. Current was delivered by a servo-amplifier (BE12A8, Advanced Motion Controls, Camarillo, CA), under the control of a laptop PC (Gateway Computers, Irvine, CA). Communication between the computer and servo-amplifier was provided by a multi-function I/O card (6062E, National Instruments, Austin, TX) using Matlab software (The Mathworks, Natick, MA). Impact was induced by delivering the specified current pulse to the actuator. Anesthesia was terminated immediately after impact. The entire procedure required approximately 10 minutes.
4.3 Histological Methods
At specified times post-trauma a group of animals was deeply anesthetized with pentobarbital (160 mg/kg i.p.) and perfused through the left cardiac ventricle and ascending aorta with fixative, the composition of which is described below for each histological procedure. Several histological methods were employed; activated caspase-3 (AC-3) immunohistochemistry and electron microscopy were relied upon most heavily as they were found to be the most useful for clarifying the nature of the cell death process, and AC-3 staining was also useful for mapping the spatial pattern of distribution of dying cells and the time of onset of apoptotic cell death in each of the affected brain regions. DeOlmos cupric silver staining was also used, both to confirm that the cells were dying and as an alternate method for mapping the pattern of cell death throughout the brain.
Immunohistochemistry
Rat pups were anesthetized and perfused transcardially with a fixative composed of 4% paraformaldehyde in phosphate buffer, pH 7.4). Brains were postfixed for 2–24 hrs in the same fixative and kept in 0.1% PBS/Azide solution. Coronal, sagittal and horizontal vibratome sections, 50 µm thick, were washed in 0.01M PBS, quenched for 10 min in a solution of methanol containing 3% hydrogen peroxide, and incubated for 1 hr in blocking solution (2% BSA/0.2% milk/0.1 % triton X-100 in PBS) before incubation with primary antisera. Cells undergoing apoptotic cell death were detected with polyclonal antiserum to activated (cleaved) Caspase-3 (D175, Cell Signaling Technology, Beverly, Massachusets) at 1:1000. Following overnight incubation in primary antiserum at 4°C sections were thoroughly rinsed in PBS and incubated for 1 hr in complementary secondary antibodies (Vector Labs, Burlingame, CA, 1:200 dilution in 1% BSA in 0.01M PBS at pH 7.4). Sections were then rinsed and reacted in the dark with streptavidin-peroxidase reagent (standard Vectastain ABC Elite Kit, Vector Labs, Burlingame, CA). Finally immunoreacive product was visualized by VIP kit (Vector). Stained sections were mounted on glass slides, air-dried, dehydrated and coversilpped. In addition, representative immunostained sections were counterstainded by hematoxylin or Nissl stain to better reveal histological detail.
DeOlmos silver staining procedure
The DeOlmos cupric silver staining procedure was used to mark dying neurons. While it is not useful for distinguishing between apoptotic and non-apoptotic cell death the staining quite specifically reveals regions that undergo massive cell death. Pups were perfused with a fixative composed of 4% paraformaldehyde in cacodylate buffer. After removal from the skull, the brains were postfixed at 4°C in the same solution for at least 2 days before sectioning. Brains were then cut into 5 mm thick blocks, embedded in agar and cut on a vibratome into 50 µm sections. Free floating sections were washed in distilled water and stained with the cupric silver protocol according to DeOlmos and Ingram (1971).
Plastic sections for combined light and electron microscopy
For combined light and electron microscopy studies, rat pups were anesthetized and perfused transcardially with a fixative composed of 1.5% glutaraldehyde and 1% paraformaldehyde in cacodylate buffer, pH 7.4. The brains were sliced coronally into 1 mm slabs, yielding serial slabs containing all portions of the brain. These slabs were osmicated overnight (1% osmium tetroxide), dehydrated in graded ethanols, cleared in toluene and embedded flat in araldite. Thin sections, 1 µm thick, were cut at selected rostro-caudal levels of the brain, using glass knives (1/2 inch wide) and an MT-2B Sorval ultratome. These sections were heat dried on glass slides and stained with azure II and methylene blue for evaluation by light microscopy. This approach allows any given portion of any given brain to be evaluated by either light or electron microscopy. For electron microscopy, areas of special interest from a given block were trimmed to a smaller size, ultrathin sections were cut and suspended over a formvar coated slot grid (1 × 2mm opening), stained with uranyl acetate and lead citrate and viewed in a JEOL 100C transmission electron microscope. Slot grids were used because they permit a continuous viewing field (1 × 2 mm) uninterrupted by grid mesh bars.
ACKNOWLEDGEMENTS
This study was supported in part by NIH grants HD 37100 and NS 045237.
REFERENCES
- Aggleton JP, Brown MW. Episodic memory, amnesia, and the hippocampal-anterior thalamic axis. Behav Brain Sci. 1999;22:425–489. [PubMed] [Google Scholar]
- Aggleton JP, Hunt PR, Nagle S, Neave N. The effects of selective lesions within the anterior thalamic nuclei on spatial memory in the rat. Behav Brain Res. 1996;81:189–198. doi: 10.1016/s0166-4328(96)89080-2. [DOI] [PubMed] [Google Scholar]
- Aggleton JP, Neave N, Nagle S, Hunt PR. A comparison of the effects of anterior thalamic, mammillary body and fornix lesions on reinforced spatial alternation. Behav Brain Res. 1995;68:91–101. doi: 10.1016/0166-4328(94)00163-a. [DOI] [PubMed] [Google Scholar]
- Bain AC, Meaney DF. “Tissue-level thresholds for axonal damage in an experimental model of central nervous system white matter injury.”. J Biomech Engrg. 2000;122:615–622. doi: 10.1115/1.1324667. [DOI] [PubMed] [Google Scholar]
- Bain AC, Raghupathi R, Meaney DF. Dynamic stretch correlates to both morphological abnormalities and electrophysiological impairment in a model of traumatic axonal injury. J Neurotrauma. 2001;18(5):499–511. doi: 10.1089/089771501300227305. [DOI] [PubMed] [Google Scholar]
- Bayly PV, Ji S, Song SK, Okamoto RJ, Massouros P, Genin GM. Measurement of strain in physical models of brain injury: A method based on HARP analysis of tagged magnetic resonance images. J Biomech Engrg. 2004;126:523–528. doi: 10.1115/1.1785811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayly PV, Cohen TS, Leister EP, Ajo D, Leuthardt EC, Genin GM. Deformation of the human brain induced by mild acceleration. J. Neurotrauma. 2005;22(8):845–856. doi: 10.1089/neu.2005.22.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayly PV, Black EE, Pedersen RC, Leister EP, Genin GM. In vivo imaging of deformation and strain in an animal model of traumatic brain injury. J Biomech. 2006 doi: 10.1016/j.jbiomech.2005.02.014. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittigau P, Sifringer M, Pohl D, Stadthaus D, Ishimaru M, Shimizu H, Ikeda M, Lang D, Speer A, Olney JW, Ikonomidou C. Apoptic neurodegeneration following trauma is markedly enhanced in the immature brain. Ann Neurol. 1999;45:724–735. doi: 10.1002/1531-8249(199906)45:6<724::aid-ana6>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Buki A, Povlishck JT. All roads lead to disconnection? - Traumatic axonal injury revisited. Acta Neurochir (Wien) 2006;148(2):181–194. doi: 10.1007/s00701-005-0674-4. [DOI] [PubMed] [Google Scholar]
- DeOlmos JS, Ingram WR. An improved cupric-silver method for impregnation of axonal and terminal degeneration. Brain Res, 29. 1971;33(2):523–529. doi: 10.1016/0006-8993(71)90130-2. [DOI] [PubMed] [Google Scholar]
- Dikranian K, Ishimaru MJ, Tenkova T, Labruyere J, Qin YQ, Ikonomidou C, Olney JW. Apoptosis in the in vivo mammalian forebrain. Neurobiol Dis. 2001;8:359–379. doi: 10.1006/nbdi.2001.0411. [DOI] [PubMed] [Google Scholar]
- Geddes JF, Hackshaw AK, Vowles GH, Nickols CD, Whitwell HL. Neuropathology of inflicted head injury in children. I. Patterns of brain damage. Brain. 2001a;124:1290–1298. doi: 10.1093/brain/124.7.1290. [DOI] [PubMed] [Google Scholar]
- Geddes JF, Vowles GH, Hackshaw AK, Nickols CD, Scott IS, Whitwell HL. Neuropathology of inflicted head injury in children. II. Microscopic brain injury in infants. Brain. 2001b;124:1299–1306. doi: 10.1093/brain/124.7.1299. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Qin Y, Labruyere J, Kirby C, Olney J. Prevention of trauma-induced neurodegeneration in infant rat brain. Pediatric Research. 1996;39(6):1020–1027. doi: 10.1203/00006450-199606000-00015. [DOI] [PubMed] [Google Scholar]
- Kerr JFR, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wideranging implications in tissue kinetics. Brit J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskiniemi M, Kykka T, Nybo T, Jarho L. Long-term outcome after severe brain injury in preschoolers is worse than expected. Arch Pediatr Adolesc Med. 1995;49:249–254. doi: 10.1001/archpedi.1995.02170150029004. [DOI] [PubMed] [Google Scholar]
- Krazem A, Beracochea D, Jaffard R. Effects of mammillary bodies and mediodorsal thalamic lesions on the acquisition and retention of a learning set in mice: paradoxical effect of the intersession interval. Behav Brain Res. 1995;67:51–58. doi: 10.1016/0166-4328(94)00103-m. [DOI] [PubMed] [Google Scholar]
- Mitchell AS, Dalrymple-Alford JC, Christie MA. Spatial working memory and the brainstem cholinergic innervation to the anterior thalamus. J Neurosci. 2002;22:1922–1926. doi: 10.1523/JNEUROSCI.22-05-01922.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW, Wozniak DF, Jevtovic-Todorovic V, Farber NB, Bittigau P, Ikonomidou C. Drug-induced Neurodegeneration in the Developing Brain. Brain Pathology. 2002a;12:1–11. doi: 10.1111/j.1750-3639.2002.tb00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW, Tenkova T, Dikranian K, Muglia LJ, Jermakowicz WJ, D’Sa C, Roth KA. Ethanol-induced caspase-3 activation in the in vivo developing mouse brain. Neurobiol of Disease. 2002b;9:205–219. doi: 10.1006/nbdi.2001.0475. [DOI] [PubMed] [Google Scholar]
- Osman NF, McVeigh ER, Prince JL. Imaging heart motion using harmonic phase MRI. IEEE Transactions on Medical Imaging. 2000;19(3):186–202. doi: 10.1109/42.845177. [DOI] [PubMed] [Google Scholar]
- Pohl D, Bittigau P, Ishimaru M, Stadthaus D, Hubner C, Olney JW, Turski L, Ikonomidou C. N-methyl-D-aspartate antagonists and apoptotic cell death triggered by head trauma in developing rat brain. Proc Natl Acad Sci USA. 1999;96:2508–2513. doi: 10.1073/pnas.96.5.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL. Thalamus. In: Paxinos G, editor. The Rat Nervous System. London: Acad Press; 1995. [Google Scholar]
- Shibata H. Efferent projections from the anterior thalamic nuclei to the cingulate cortex in the rat. J Comp Neurol. 1993;330:533–542. doi: 10.1002/cne.903300409. [DOI] [PubMed] [Google Scholar]
- Reeves TM, Phillips LL, Povlishock JT. Myelinated and unmyelinated axons of the corpus callosum differ in vulnerability and functional recovery following traumatic brain injury. Exp Neurol. 2005;196(1):126–137. doi: 10.1016/j.expneurol.2005.07.014. [DOI] [PubMed] [Google Scholar]
- Stone JR, Okonkwo DO, Dialo AO, Rubin DG, Mutlu LK, Povlishock JT, Helm GA. Impaired axonal transport and altered axolemmal permability occur in distinct populations of damaged axons following traumatic brain injury. Exp Neurol. 2004;190:59–69. doi: 10.1016/j.expneurol.2004.05.022. [DOI] [PubMed] [Google Scholar]
- Sziklas V, Petrides M. Memory impairments following lesions to the mammillary region of the rat. Eur J Neurosci. 1993;5:525–540. doi: 10.1111/j.1460-9568.1993.tb00518.x. [DOI] [PubMed] [Google Scholar]
- van Groen T, Kadish I, Wyss JM. Role of the anterodorsal and anteroventral nuclei of the thalamus in spatial memory in the rat. Behav Brain Res. 2002;132:19–28. doi: 10.1016/s0166-4328(01)00390-4. [DOI] [PubMed] [Google Scholar]
- Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, Young C, Olney JW, Muglia LJ. Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol Disease. 2004;17:403–414. doi: 10.1016/j.nbd.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Young C, Klocke BJ, Tenkova T, Choi J, Labruyere J, Qin YQ, Holtzman DM, Roth KA, Olney JW. Ethanol-induced neuronal apoptosis in vivo requires BAX in the developing mouse brain. Cell Death Differ. 2003;10(10):1148–1155. doi: 10.1038/sj.cdd.4401277. [DOI] [PubMed] [Google Scholar]
- Young C, Tenkova T, Dikranian K, Labruyere J, Olney JW. Excitotoxic versus apoptotic mechanisms of neuronal cell death in perinatal hypoxia/ischemia. Current Molecular Medicine. 2004;4:73–81. doi: 10.2174/1566524043479158. [DOI] [PubMed] [Google Scholar]
- Zerhouni EA, Parish DM, Rogers WJ, Yang A, Shapiro EP. Human heart tagging with MR imaging - a method for noninvasive assessment of myocardial motion. Radiology. 1988;169(1):59–63. doi: 10.1148/radiology.169.1.3420283. [DOI] [PubMed] [Google Scholar]